A Facile One-Pot Process for the Formation of Hindered Tertiary Amines

Abstract
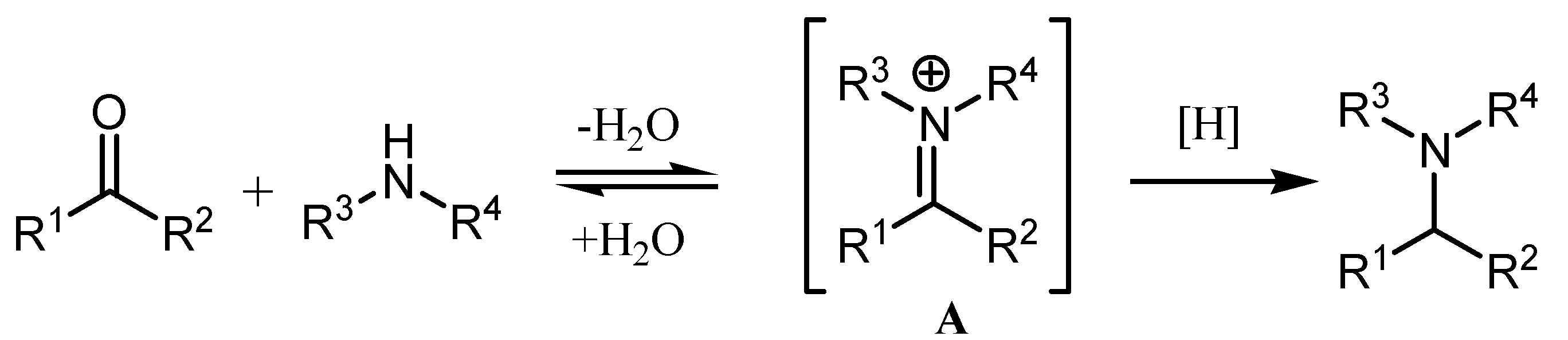
:1. Introduction

2. Results and Discussion


| Entry | Solvent | Activator (equiv.) | Yield (%) b |
|---|---|---|---|
| 1 | DCM | - | 19 |
| 2 | DCM | DMF (1.0) | 63 |
| 3 | DCM | DMAc (1.0) | 70 |
| 4 | DCM | Lutidine (1.0) | 78 |
| 5 | DCM | Pyridine (1.0) | 88 |
| 6 | DCM | DIEA (1.0) | 84 |
| 7 | DCM | TEA (1.0) | 43 |
| 8 | DCM | HMPA (1.0) | 68 |
| 9 | DCM | TMEDA (1.0) | 92 |
| 10 | DCM | TMEDA (0.5) | 85 |
| 11 | DCM | TMEDA (0.2) | 80 |
| 12 | Toluene | TMEDA (1.0) | 60 |
| 13 | CH3CN | TMEDA (1.0) | 76 |
| 14 | CHCl3 | TMEDA (1.0) | 74 |
| 15 | ClCH2CH2Cl | TMEDA (1.0) | 73 |
| 16 | THF | TMEDA (1.0) | 24 |

{kind=link}
{kind=link}
| Entry | Ketone | Yield (%) b | Entry | Ketone | Yield (%) b |
|---|---|---|---|---|---|
| 1 |  (1a) (1a) | 92 | 10 |  (1j) (1j) | 62 |
| 2 |  (1b) (1b) | 76 | 11 |  (1k) (1k) | 82 |
| 3 |  (1c) (1c) | 83 | 12 |  (1l) (1l) | 89 |
| 4 |  (1d) (1d) | 86 | 13 |  (1m) (1m) | 88 |
| 5 |  (1e) (1e) | 85 | 14 |  (1n) (1n) | 61 |
| 6 |  (1f) (1f) | 89 | 15 |  (1o) (1o) | 90 |
| 7 |  (1g) (1g) | 60 | 16 |  (1p) (1p) | 88 |
| 8 |  (1h) (1h) | 87 | 17 |  (1q) (1q) | 76 |
| 9 |  (1i) (1i) | 79 | 18 |  (1r) (1r) | 66 |

3. Experimental
3.1. General
3.2. General Procedure for the Direct Reductive Amination of Ketones with N-Methylaniline
4. Conclusions
Supplementary Materials
Acknowledgments
References and Notes
- Birtill, J.J.; Chamberlain, M.; Hall, J.; Wilson, R.; Costello, I. Optimization of reaction conditions in single-stage reductive amination of aldehydes and ketones. In Catalysis of Organic Reactions; Herkers, F.E., Ed.; Dekker: New York, NY, USA, 1998; pp. 255–271. [Google Scholar]
- Baxter, E.W.; Reitz, A.B. Reductive aminations of carbonyl compounds with borohydride and borane reducing agents. In Organic Reactions; Denmark, S.E., Ed.; Wiley: New York, NY, USA, 2002; Volume 59, pp. 1–57. [Google Scholar]
- Gomez, S.; Peters, J.A.; Maschmeyer, T. The reductive amination of aldehydes and ketones and the hydrogenation of nitriles: mechanistic aspects and selectivity control. Adv. Syn. Catal. 2002, 344, 1037–1057. [Google Scholar] [CrossRef]
- Tararov, V.I.; Kadyrov, R.; Riermeier, T.H.; Fischer, C.; Börner, A. Direct reductive amination versus hydrogenation of intermediates—A comparison. Adv. Syn. Catal. 2004, 346, 561–565. [Google Scholar] [CrossRef]
- Ohkuma, T.; Noyori, R. Hydrogenation of imino groups. In Comprehensive Asymmetric Catalysis; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: New York, NY, USA, 2004; Volume 1, pp. 199–242. [Google Scholar]
- Nugent, T.C.; El-Shazly, M. Chiral amine synthesis—Recent developments and trends for enamide reduction, reductive amination, and imine reduction. Adv. Synth. Catal. 2010, 352, 753–819. [Google Scholar] [CrossRef]
- Tripathi, R.P.; Verma, S.S.; Pandey, J.; Tiwari, V.K. Recent development on catalytic reductive amination and applications. Curr. Org. Chem. 2008, 12, 1093–1115. [Google Scholar] [CrossRef]
- Kadyrov, R.; Riermeier, T.H. Highly enantioselective hydrogen-transfer reductive amination: Catalytic asymmetric synthesis of primary amines. Angew. Chem. Int. Ed. 2003, 42, 5472–5474. [Google Scholar] [CrossRef]
- Storer, R.I.; Carrera, D.E.; Ni, Y.; MacMillan, D.W.C. Enantioselective organocatalytic reductive amination. J. Am. Chem. Soc. 2006, 128, 84–86. [Google Scholar] [CrossRef]
- Höhne, M.; Kühl, S.; Robins, K.; Bornscheuer, U.T. Efficient asymmetric synthesis of chiral amines by combining transaminase and pyruvate decarboxylase. ChemBioChem 2008, 9, 363–365. [Google Scholar] [CrossRef]
- Koszelewski, D.; Lavandera, I.; Clay, D.; Guebitz, G.M.; Rozzell, D.; Kroutil, W. Formal asymmetric biocatalytic reductive amination. Angew. Chem. Int. Ed. 2008, 47, 9337–9340. [Google Scholar]
- Nugent, T.C.; Negru, D.E.; El-Shazly, M.; Hu, D.; Sadiq, A.; Bibi, A.; Umar, M.N.J. Sequential reductive amination hydrogenolysis: Aone-pot synthesis of challenging chiral primary amines. Adv. Synth. Catal. 2011, 353, 2085–2092. [Google Scholar] [CrossRef]
- Villa-Marcos, B.; Li, C.; Mulholland, K.R.; Hogan, P.J.; Xiao, J. Bifunctional catalysis: Direct reductive amination of aliphatic ketones with an Iridium-phosphate catalyst. Molecules 2010, 15, 2453–2472. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F.; Carson, K.G.; Harris, B.D.; Maryanoff, C.A.; Shah, R.D. Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride. Studies on direct and indirect reductive amination procedures. J. Org. Chem. 1996, 61, 3849–3862. [Google Scholar]
- Alinezhad, H.; Tajbakhsh, M.; Zamani, R. One-pot reductive amination of aldehydes and ketones using N-methylpiperidine zinc borohydride (ZBNMPP) as a new reducing agent. Synlett 2006, 431–434. [Google Scholar]
- Menche, D.; Böhm, S.; Li, J.; Rudolph, S.; Zander, W. Synthesis of hindered tertiary amines by a mild reductive amination procedure. Tetrahedron Lett. 2007, 48, 365–369. [Google Scholar] [CrossRef]
- Lee, O.Y.; Law, K.L.; Ho, C.Y.; Yang, D. Highly chemoselective reductive amination of carbonyl compounds promoted by InCl3/Et3SiH/MeOH system. J. Org. Chem. 2008, 73, 8829–8837. [Google Scholar] [CrossRef]
- Jung, Y.J.; Bae, J.W.; Park, E.S.; Chang, Y.M.; Yoon, C.M. An efficient conversion of nitroaromatics and aromatic amines to tertiary amines in one-pot way. Tetrahedron 2003, 59, 10333–10338. [Google Scholar]
- Wang, C.; Pettman, A.; Basca, J.; Xiao, J. A Versatile Catalyst for Reductive Amination by Transfer Hydrogenation. Angew. Chem. Int. Ed. 2010, 49, 7548–7552. [Google Scholar]
- Kobayashi, S.; Yasuda, M.; Hachiya, I. Trichlorosilane-dimethylformamide (Cl3SiH-DMF) as an efficient reducing agent. Reduction of aldehydes and imines and reductive amination of aldehydes under mild conditions using hypervalent hydridosilicates. Chem. Lett. 1996, 407–408. [Google Scholar]
- Guizzetti, S.; Benaglia, M. Trichlorosilane-mediated stereoselective reduction of C=N bonds. Eur.J. Org. Chem. 2010, 5529–5541. [Google Scholar] [CrossRef]
- Jones, S.; Warner, C.J.A. Trichlorosilane mediated asymmetric reductions of the C=N bond. Org. Biomol. Chem. 2012, 10, 2189–2200. [Google Scholar] [CrossRef]
- Iwasaki, F.; Onomura, O.; Mishima, K.; Kanematsu, T.; Maki, T.; Matsumura, Y. First chemo- and stereoselective reduction of imines using trichlorosilane activated with N-formylpyrrolidine derivatives. Tetrahedron Lett. 2001, 42, 2525–2527. [Google Scholar]
- Matsumura, Y.; Ogura, K.; Kouchi, Y.; Iwasaki, F.; Onomura, O. New efficient organic activators for highly enantioselective reduction of aromatic ketones by trichlorosilane. Org. Lett. 2006, 8, 3789–3792. [Google Scholar] [CrossRef]
- Malkov, A.V.; Mariani, A.; MacDougall, K.N.; Kočovský, P. Role of noncovalent interactions in the enantioselective reduction of aromatic ketimines with trichlorosilane. Org. Lett. 2004, 6, 2253–2256. [Google Scholar] [CrossRef]
- Malkov, A.V.; Liddon, A.; Ramirez-Lopez, P.; Bendova, L.; Haigh, D.; Kočovský, P. Remote chiral induction in the organocatalytic hydrosilylation of aromatic ketones and ketimines. Angew. Chem. Int. Ed. 2006, 45, 1432–1435. [Google Scholar]
- Malkov, A.V.; Stoncius, S.; Vrankova, K.; Arndt, M.; Kocovsky, P. Dynamic kinetic resolution in the asymmetric synthesis of β-amino acids by organocatalytic reduction of enamines with trichlorosilane. Chem. Eur. J. 2008, 14, 8082–8085. [Google Scholar] [CrossRef]
- Malkov, A.V.; Vrankova, K.; Stoncius, S.; Kocovsky, P. Asymmetric reduction of imines with trichlorosilane, catalyzed by sigamide, an amino acid-derived formamide: Scope and limitations. J. Org. Chem. 2009, 74, 5839–5849. [Google Scholar] [CrossRef]
- Figlus, M.; Caldwell, S.T.; Walas, D.; Yesilbag, G.; Cooke, G.; Kocovsky, P.; Malkov, A.V.; Sanyal, A. Dendron-anchored organocatalysts: The asymmetric reduction of imines with trichlorosilane, catalysed by an amino acid-derived formamide appended to a dendron. Org. Biomol. Chem. 2010, 8, 137–141. [Google Scholar]
- Wang, Z.Y.; Ye, X.X.; Wei, S.Y.; Wu, P.C.; Zhang, A.J.; Sun, J. A highly enantioselective Lewis Basic organocatalyst for reduction of N-aryl imines with unprecedented substrate spectrum. Org. Lett. 2006, 8, 999–1001. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Cheng, M.N.; Wu, P.C.; Wei, S.Y.; Sun, J. L-piperazine-2-carboxylic acid derived N-formamide as a highly enantioselective Lewis basic catalyst for hydrosilylation of N-aryl imines with an unprecedented substrate profile. Org. Lett. 2006, 8, 3045–3048. [Google Scholar] [CrossRef]
- Pei, D.; Wang, Z.Y.; Zhang, Y.; Wei, S.Y.; Sun, J. S-chiral sulfinamides as highly enantioselective organocatalysts. Org. Lett. 2006, 8, 5913–5915. [Google Scholar]
- Zhou, L.; Wang, Z.Y.; Wei, S.Y.; Sun, J. Evolution of chiral Lewis basic N-formamide as highly effective organocatalyst for asymmetric reduction of both ketones and ketimines with an unprecedented substrate scope. Chem. Commun. 2007, 2977–2979. [Google Scholar]
- Pei, D.; Zhang, Y.; Wei, S.Y.; Wang, M.; Sun, J. Rationally-designed S-chiral bissulfinamides as highly enantioselective organocatalysts for reduction of ketimines. Adv. Synth. Catal. 2008, 350, 619–623. [Google Scholar] [CrossRef]
- Wang, C.; Wu, X.; Zhou, L.; Sun, J. A highly enantioselective organocatalytic method for reduction of aromatic N-alkyl ketimines. Chem. Eur. J. 2008, 14, 8789–8792. [Google Scholar] [CrossRef]
- Baudequin, C.; Chaturvedi, D.; Tsogoeva, S.B. Organocatalysis with chiral formamides: asymmetric allylation and reduction of imines. Eur.J. Org. Chem. 2007, 2623–2629. [Google Scholar]
- Guizzetti, S.; Benaglia, M.; Rossi, S. Highly stereoselective metal-free catalytic reduction of imines: An easy entry to enantiomerically pure amines and natural and unnatural α-amino esters. Org. Lett. 2009, 11, 2928–2931. [Google Scholar] [CrossRef]
- Xiao, Y.C.; Wang, C.; Yao, Y.; Sun, J.; Chen, Y.C. Direct asymmetric hydrosilylation of indoles: Combined Lewis base and brønsted acid activation. Angew. Chem. Int. Ed. 2011, 50, 10661–10664. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Wang, C.; Zhou, L.; Lu, X.; Sun, J. Chiral Lewis base catalyzed highly enantioselective reduction of N-alkyl β-enamino esters with trichlorosilane and water. Chem. Eur. J. 2011, 17, 2846–2848. [Google Scholar] [CrossRef]
- Zheng, H.J.; Chen, W.B.; Wu, Z.J.; Deng, J.G.; Lin, W.Q.; Yuan, W.C.; Zhang, X.M. Highly enantioselective synthesis of β-Amino acid derivatives by the Lewis base catalyzed hydrosilylation of β-enamino esters. Chem. Eur. J. 2008, 14, 9864–9867. [Google Scholar]
- Xue, Z.Y.; Jiang, Y.; Peng, X.Z.; Yuan, W.C.; Zhang, X.M. The first general, highly enantioselective Lewis base organocatalyzed hydrosilylation of benzoxazinones and quinoxalinones. Adv. Synth. Catal. 2010, 352, 2132–2136. [Google Scholar] [CrossRef]
- Chen, X.; Zheng, Y.; Shu, C.; Yuan, W.; Liu, B.; Zhang, X. Enantioselective synthesis of 4-substituted 4,5-dihydro-1H-[1,5]benzodiazepin-2(3H)-onesby the Lewis base-catalyzed hydrosilylation. J. Org. Chem. 2011, 76, 9109–9115. [Google Scholar] [CrossRef]
- Xue, Z.Y.; Liu, L.X.; Jiang, Y.; Yuan, W.C.; Zhang, X.M. Highly enantioselective Lewis base organocatalyzed hydrosilylation of γ-imino esters. Eur. J. Org. Chem. 2012, 251–255. [Google Scholar]
- We examined the DRA of acetophenone 1a with aliphatic secondary amine and cyclic secondary amine using one equivalent of DMF as the Lewis base activator in dichloromethane at room temperature. No desired product was observed.
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, Z.; Pei, D.; Zhang, Y.; Wang, C.; Sun, J. A Facile One-Pot Process for the Formation of Hindered Tertiary Amines. Molecules 2012, 17, 5151-5163. https://doi.org/10.3390/molecules17055151
Wang Z, Pei D, Zhang Y, Wang C, Sun J. A Facile One-Pot Process for the Formation of Hindered Tertiary Amines. Molecules. 2012; 17(5):5151-5163. https://doi.org/10.3390/molecules17055151
Chicago/Turabian StyleWang, Zhouyu, Dong Pei, Yu Zhang, Chao Wang, and Jian Sun. 2012. "A Facile One-Pot Process for the Formation of Hindered Tertiary Amines" Molecules 17, no. 5: 5151-5163. https://doi.org/10.3390/molecules17055151
APA StyleWang, Z., Pei, D., Zhang, Y., Wang, C., & Sun, J. (2012). A Facile One-Pot Process for the Formation of Hindered Tertiary Amines. Molecules, 17(5), 5151-5163. https://doi.org/10.3390/molecules17055151