3.3. Synthetic Procedures
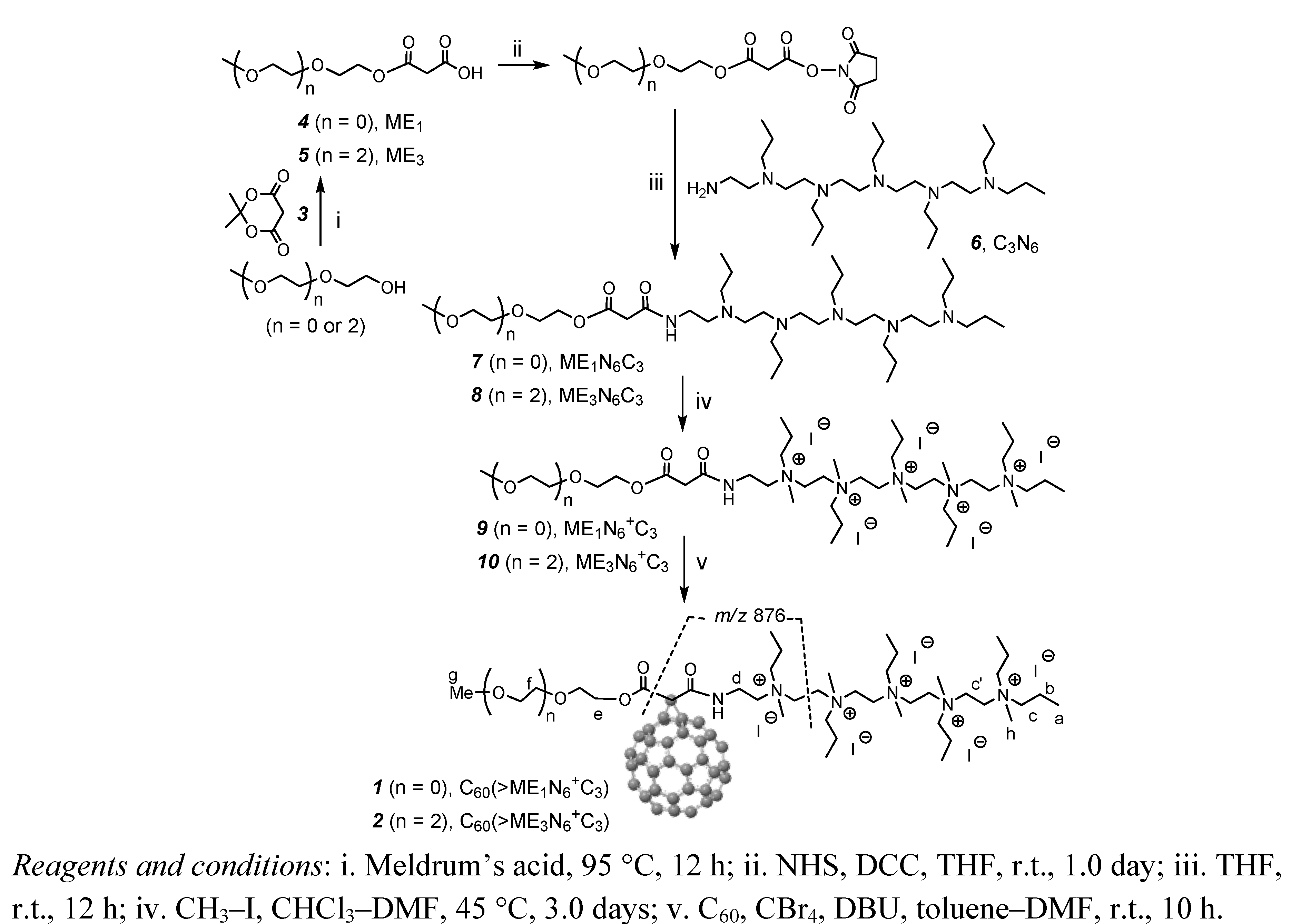
3.3.1. Synthesis of Malonic Acid Methoxyethyleneglycol Ester, ME1 (4)
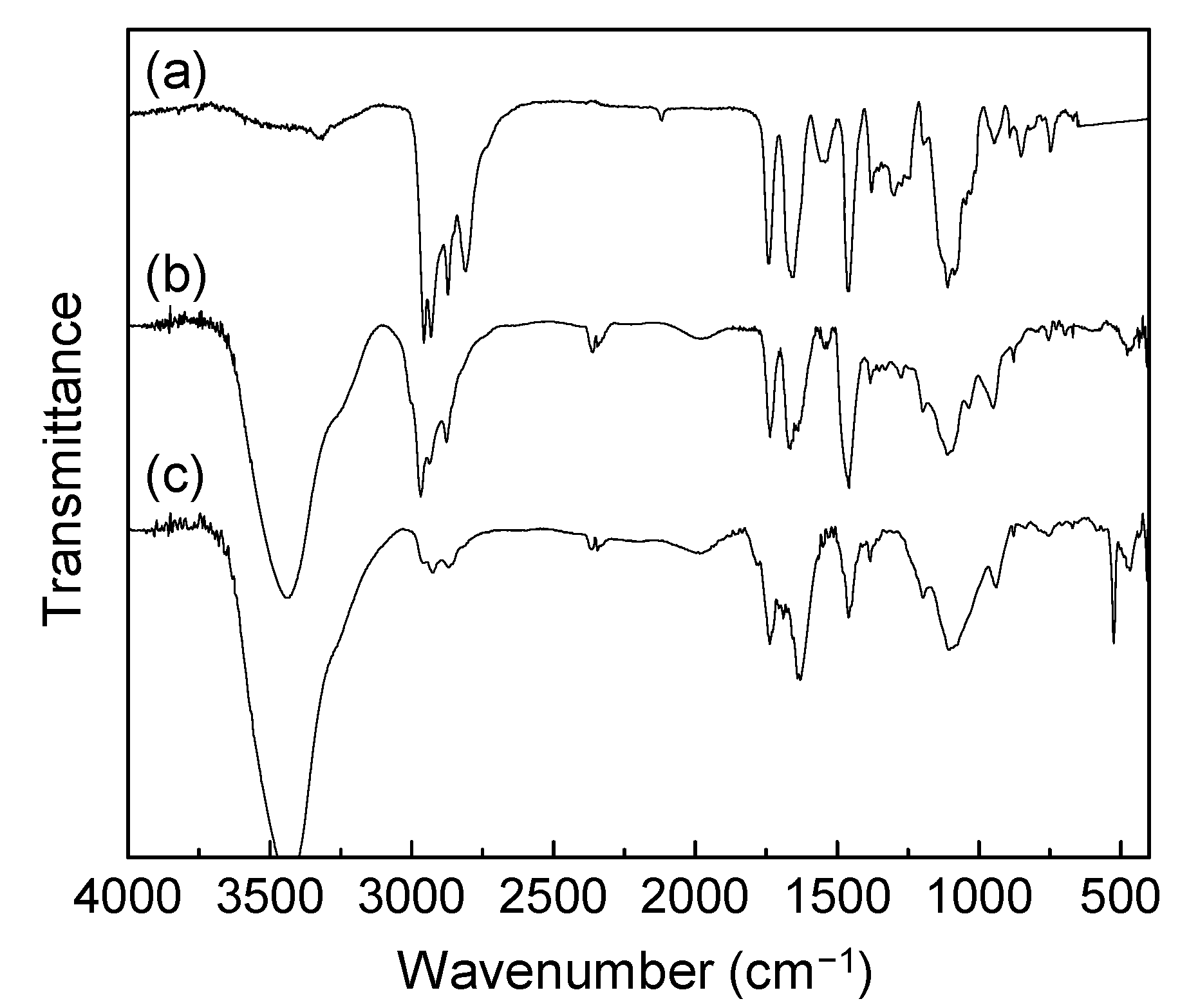
A mixture of 2-methoxyethanol (1.0 g, 13.1 mmol) and 2,2-dimethyl-1,3-dioxane-4,6-dione (3, Meldrum’s acid, 1.9 g, 13.2 mmol) was stirred under an Ar atmosphere over a period of 12 h at 95 °C. The reaction mixture was cooled to room temperature, treated with aqueous sodium carbonate solution (5%), and washed with diethyl ether. The resulting aqueous layer was subsequently treated with dil. HCl (2.0 N) and extracted with ethyl acetate (50 mL). The ethyl acetate solution was dried over Na2SO4 and concentrated on rotary evaporator to give the product ME1 (4) in 95% yield (2.0 g). Spectroscopic data: FT-IR (KBr) υmax 3060 (w), 2880 (w), 2814 (w), 1727 (vs), 1458 (w), 1399 (m), 1318 (s), 1249 (s), 1134 (s), 1100 (s), 1039 (m), 952 (w), 848 (s), and 755 (m) cm−1; 1H-NMR (500 MHz, CDCl3, ppm) δ 10.60 (s, br, 1H), 4.31 (t, J = 4.63 Hz, 2H), 3.62 (t, J = 4.63 Hz, 2H), 3.45 (s, 2H), and 3.38 (s, 3H).
3.3.2. Synthesis of Malonic Acid Methoxytriethyleneglycol Ester, ME3 (5)
A mixture of triethylene glycol monomethyl ether (1.0 g, 6.1 mmol) and 2,2-dimethyl-1,3-dioxane-4,6-dione (3, Meldrum’s acid, 0.9 g, 6.2 mmol) was stirred under Ar atmosphere over a period of 12 h at 90 °C. The reaction mixture was cooled to room temperature, treated with aqueous sodium carbonate solution (5%), and washed with diethyl ether. The resulting aqueous layer was subsequently treated with dil. HCl (2.0 N) and extracted with ethyl acetate (50 mL). The ethyl acetate solution was dried over Na2SO4 and concentrated on rotary evaporator to give the product ME3 (5) in 90% yield (1.37 g). Spectroscopic data: FT-IR (KBr) υmax 3056 (w), 2880 (w), 2817 (w), 1728 (vs), 1455 (w), 1394 (m), 1318 (s), 1251 (s), 1134 (vs), 1098 (vs), 1034 (m), 952 (w), 847 (s), and 753 (m) cm−1; 1H-NMR (500 MHz, CDCl3, ppm) δ 9.65 (s, br, 1H), 4.32 (t, J = 4.50 Hz, 2H), 3.70 (t, J = 4.50 Hz, 2H), 3.65–3.55 (m, 8H), 3.42 (s, 3H), and 3.41 (s, 2H).
3.3.3. Synthesis of Methoxyethyleneglycol-[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amino]-malonamide Ester, ME1N6C3 (7)
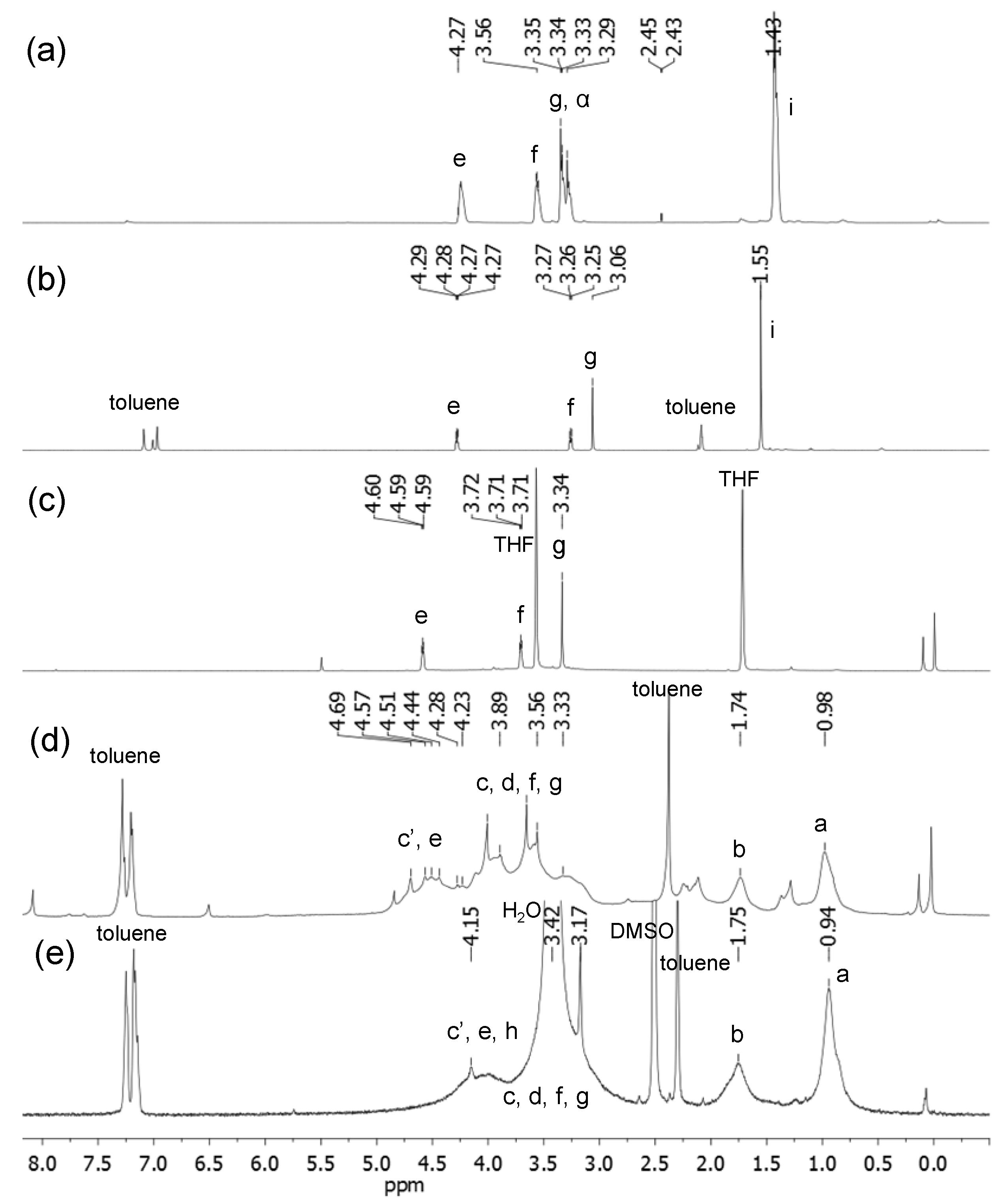
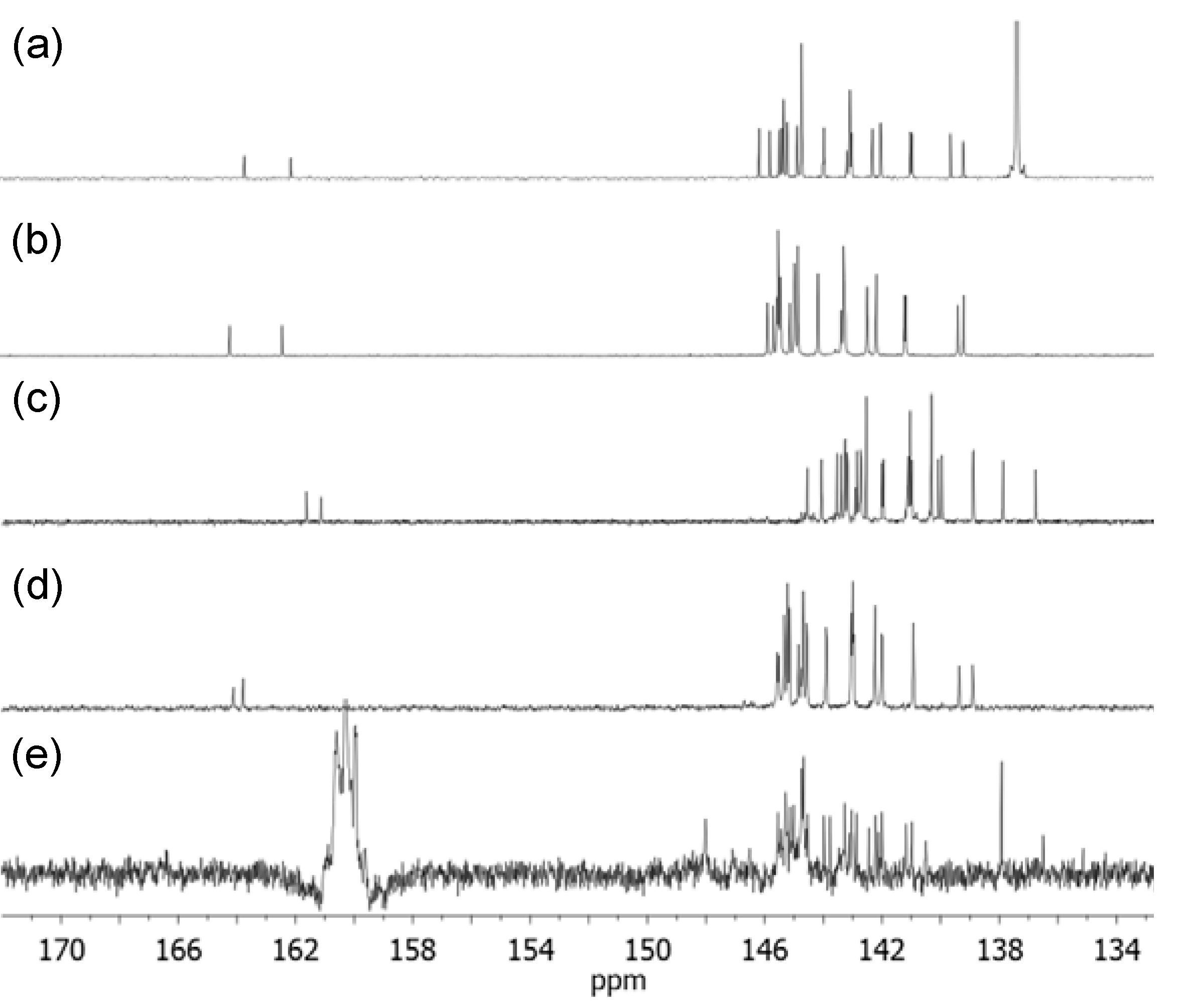
A mixture of malonic acid methoxyethyleneglycol ester 4 (0.5 g, 3.08 mmol), N-hydroxysuccinamide (0.35 g, 3.08 mmol), and N,N′-dicyclohexyl carbodiimide (DCC, 0.63 g, 4.0 mmol) in anyhydrous tetrahydrofuran (20 mL) were stirred under N2 atmosphere over a period of 12 h at ambient temperature. The resulting white solids of N,N′-dicyclohexyl urea byproduct were filtered off and the filtrate was taken into a second round-bottom flask containing N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amine 6 (1.49 g, 3.08 mmol). The mixture was stirred under N2 atmosphere for an additional period of 12 h. At the end of the reaction, the solvent was removed on rotavap. To this residue, ice-cold hexane–dichloromethane (1:1, 15 mL) was added followed by filtration to remove further white solids of N-hydroxysuccinamide. The filtrate was washed with aqueous sodium carbonate (5%) solution (10 mL). The organic layer was then dried and concentrated to give ME1N6C3 (7) as yellow liquid in 82% yield (1.59 g). Spectroscopic data: ESI–MS (rel. intensity) m/z 568, 570 (M–CH3OCH2CH2, 75%), 571, 629 (M+), 630 (MH+, 100%), 631, 646, 666, 670, 688, 700, 715 [MH++CH2CH2N(CH2CH2CH3), 48%], 716, 755, 757, 772, 773, 775, 800 (MH+2[CH2CH2N(CH2CH2CH3)], 15%), 801, 832, 860, 1150, 1210, 1212 (the dimer mass–CH3OCH2), 1236, and 1252 (from the dimer ion mass); FT-IR (KBr) υmax 3259 (w), 2956 (vs), 2932 (s), 2872 (m), 2808 (m), 1742 (s), 1660 (s), 1550 (w), 1459 s), 1273 (m), 1128 (m), 1074 (s), 1031 (m), and 742 (w) cm−1; 1H-NMR (500 MHz, CDCl3, ppm) δ 4.29 (t, J = 4.25 Hz, 2H), 3.61 (t, J = 4.25 Hz, 2H), 3.40 (s, 3H), 3.34–3.32 (m, 4H), 2.38–2.57 (m, 30H), 1.47–1.44 (m, 12H), and 0.90–0.87 (t, J = 7.15 Hz, 18H); 13C-NMR (500 MHz, CDCl3, ppm) δ 168.78 (–C=O), 164.65 (O=C-NH–), 70.06, 64.15, 58.90, 57.35, 57.29, 57.13, 56.83 (4C), 56.72, 56.55, 53.65, 53.00, 52.81, 52.58, 52.46, 52.00, 41.60, 37.58, 20.41, 20.39, 20.37, 20.32 (2C), 20.18, 11.90 (3C), 11.87 (2C), and 11.74.
3.3.4. Synthesis of Methoxy-tri(ethyleneglycol)-[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)-amino]-malonamide Ester, ME3N6C3 (8)
A mixture of malonic acid methoxytriethyleneglycol ester 5 (1.0 g, 4.0 mmol), N-hydroxysuccinamide (0.45 g, 4.0 mmol), and N,N′-dicyclohexyl carbodiimide (0.82 g, 4.0 mmol) in anyhydrous tetrahydrofuran (30 mL) were stirred under N2 atmosphere for a period of 12 h at ambient temperature. The resulting white solids of N,N′-dicyclohexyl urea byproduct were filtered off and the filtrate was taken into a second round-bottom flask containing N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amine 6 (1.94 g, 3.9 mmol). The mixture was stirred under N2 atmosphere for an additional period of 12 h. At the end of the reaction, the solvent was removed on rotavap. To this residue, ice-cold hexane–dichloromethane (1:1, 20 mL) was added followed by filtration to remove white solids of N-hydroxysuccinamide. The filtrate was washed with aqueous sodium carbonate (5%) solution (10 mL). The organic phase was then dried and concentrated to give ME3N6C3 (8) as yellow liquid in 80% yield (2.29 g). Spectroscopic data: ESI–MS (rel. intensity) m/z 512, 568 [M–CH3O(CH2CH2O)2CH2, 65%], 660 (M–CH3OCH2CH2, 55%), 716, 718 (MH+, 100%), 734, 776, 803 [MH++CH2CH2N(CH2CH2CH3) from the dimer mass, 46%], 819, 860, 888 (MH++2[CH2CH2N(CH2CH2CH3)] from the dimer mass, 8%), 944, 951, 1008, 1090, 1226 (the dimer mass–[MeO(EG)3-CO], 3%), 1238, 1282, 1340, 1430, and 1434 (the dimer ion); FT-IR (KBr) υmax 3328 (w), 2952 (m), 2928 (m), 2862 (m), 2805 (m), 1736 (vs), 1667 (s), 1533 (m), 1456 (m), 1400 (m), 1379 (m), 1245 (m), 1104 (vs), 1032 (w), 938 (w), and 732 (m) cm−1; 1H-NMR (500 MHz, CDCl3, ppm) δ 4.22 (t, J = 4.68 Hz,2H), 3.65 (t, J = 4.68 Hz, 2H), 3.58–3.60 (m, 6H), 3.48 (t, J = 4.60 Hz, 2H), 3.32 (s, 5H), 3.26 (s, br, 2H), 2.31−2.48 (m, 30H), 1.40–1.38 (m, 12H), and 0.82−0.79 (t, J = 6.95 Hz, 18H); 13C-NMR (500 MHz, CDCl3, ppm) δ 168.83 (–C=O), 164.70 (O=C-NH–), 71.86 (2C), 70.53, 70.51, 68.74, 64.30, 58.95, 57.32 (2C), 57.14, 56.83 (4C), 56.72, 56.53, 53.64, 53.41, 52.98 (2C), 52.80, 52.45, 41.54, 37.55, 20.40, 20.37, 20.31 (3C), 20.24, 11.91 (3C), 11.88 (2C), and 11.76.
3.3.5. Synthesis of Methoxyethyleneglycol-[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amino]-malonamide Ester Quaternary Methyl Ammonium Salt, ME1N6+C3 (9)
A solution of malonamide ester 7 (1.0 g, 1.6 mmol) in anhydrous chloroform–DMF (10:1) was added iodomethane (6.0 mL, excess, in portions) and stirred at 45 °C for a period of 3.0 days. At the end of quaternization, the solvent was evaporated to afford ME1N6+C3 (9) in 94% yield (2.0 g). Spectroscopic data: FT-IR (KBr) υmax 3444 (s), 3236 (m), 2968 (vs), 2937 (vs), 2877 (m), 1735 (s), 1667 (s), 1536 (m), 1459 (vs), 1331 (w), 1126 (m), 1032 (m), 948 (m), 872 (w), 750 (vs), and 661 (m) cm−1; 1H-NMR (500 MHz, DMSO-d6, ppm) δ 3.80–4.25 (m, br, 16H), 3.22–3.80 (m, br, 41H), 1.50–1.85 (m, br, 12H), and 0.88-0.99 (m, br, 18H); 13C-NMR (500 MHz, DMSO-d6, ppm) δ 168.09 (-C=O), 166.69 (O=C-NH–), 79.90, 79.70, 70.01, 64.28 (4C), 63.59 (5C), 58.54, 55.54 (4C), 53.96 (2C), 49.37 (3C), 48.85 (2C), 42.62, 16.27 (2C), 15.95 (4C), 10.94 (2C), 10.82 (2C), and 10.72 (2C) (one peak is covered by DMSO peaks). Anal. Calcd for C34H72N6O4∙4.5CH3I∙2H2O (based on 90% quarternization on average): C, 35.47; H, 6.92; N, 6.45; I, 43.80; O, 7.36%. Found: C, 34.72; H, 6.82; N, 6.46; I, 42.31%.
3.3.6. Synthesis of Methoxy-tri(ethyleneglycol)-[N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)-amino]-malonamide Ester Quaternary Methyl Ammonium Salt, ME3N6+C3 (10)
A solution of malonamide ester 8 (0.50 g, 0.70 mmol) in anhydrous chloroform–DMF (10:1) was added iodomethane (3.0 mL, excess, in portions) and stirred at 45 °C for a period of 3.0 days. At the end of quaternization, the solvent was evaporated to afford ME3N6+C3 (10) in 92% yield (0.92 g). Spectroscopic data: ESI–MS (rel. intensity) m/z 448 (100%), 503, 588 (40%), 731 (M+–5I–4CH3, 6%), 760, 815, 872, 873 (M+–4I–3CH3, 8%), 930, 1014, 1016 (M+–3I–2CH3, 13%), 1074, 1099, 1157 (M+–2I–CH3, 37%), 1198, 1216, 1299, 1300 (M+–I, 22%), 1385, 1391, 1392, 1442, 1443, and 1444 (M++H2O); FT-IR (KBr) υmax 3436 (vs), 3255 (m), 3006 (m), 2968 (s), 2935 (s), 2877 (m), 1736 (s), 1665 (s), 1634 (s), 1545 (w), 1459 (s), 1383 (w), 1351 (w), 1332 (w), 1271 (w), 1200 (m), 1110 (s), 1034 (m), 950 (m), 877 (w), 757 (w), and 600 (m) cm−1; 1H-NMR (500 MHz, DMSO-d6, ppm) δ 3.80–4.25 (m, br, 16H), 2.90–3.80 (m, br, 49H), 1.50–1.80 (m, br, 12H), and 0.85–0.99 (m, br, 18H); 13C-NMR (500 MHz, DMSO-d6, ppm) δ 168.13 (–C=O), 166.69 (O=C-NH–), 71.72, 70.23, 70.18, 70.03, 68.57, 63.56 (9C), 54.79 (6C), 49.05 (5C), 42.62, 16.18 (2C), 15.97 (4C), 10.95 (2C), 10.82 (3C), and 10.70 (one peak is covered by DMSO peaks). Anal. Calcd for C38H80N6O6∙4CH3I∙3H2O (based on 80% quarternization on average): C, 37.67; H, 7.38; N, 6.28; I, 37.91; O, 10.76%. Found: C, 36.10; H, 7.03; N, 6.81; I, 37.90%.
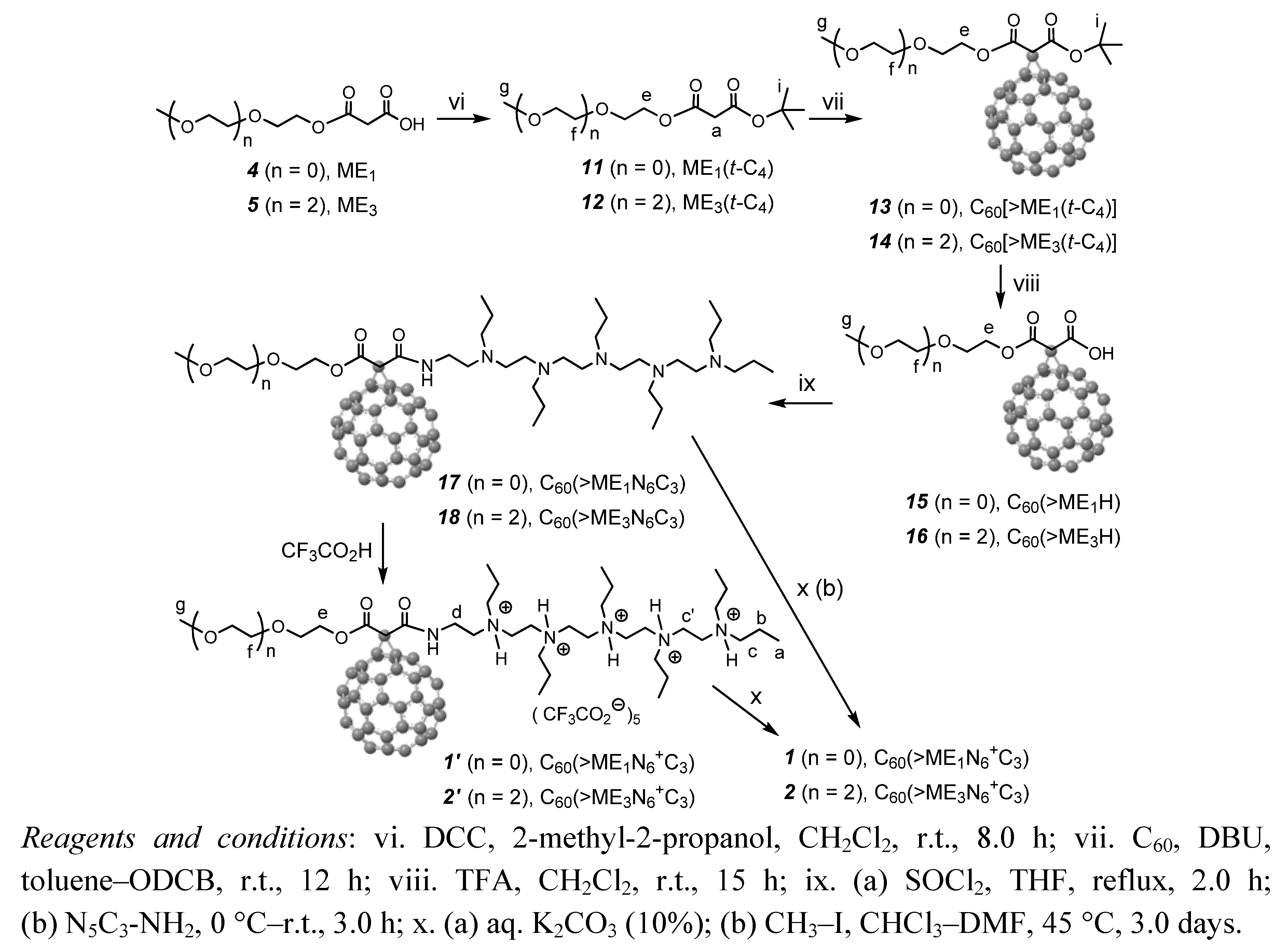
3.3.7. Synthesis of Pentacationic Methoxyethyleneglycol-(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13, 16,19-hexaaza-nonadecan-19-yl)[60]fullerenyl Malonate Quaternary Methyl Ammonium Salt, C60(˃ME1N6+C3) (1)
Finely divided [60]fullerene (0.94 g, 1.30 mmol, more than two-fold excess to allow the formation of monoadduct only) was taken into a round bottom flask and added anhydrous toluene (700 mL) under nitrogen. The solution was stirred for 12 h at ambient temperature to ensure complete dissolution of C60. To the resulting purple-colored solution added carbon tetrabromide (0.19 g, 0.57 mmol) followed by a solution of the compound 9 (0.70 g, 0.52 mmol) in anhydrous DMF (100 mL). The solution mixture was stirred for an additional 30 min and added slowly 1.8-diazabicyclo[5.4.0]-undec-7-ene (DBU, 0.17 g, 1.15 mmol) over a period of 45 min. The color of solution slowly turns into brown in a reaction period of 8.0 h. The solution was then concentrated on rotavap to roughly 100 mL. Upon the addition of methanol to this concentrated solution, the crude product was precipitated as brown solids which were collected via centrifugation. Unreacted C60 in the crude solids was removed by repeated washings with toluene (5 × 100 mL) until no color in the washing solution or filtrate. The remaining product of C60(>ME1N6+C3) (1) was obtained as brown solids in 55% yield (0.59 g, after recovered C60). Spectroscopic data: FT-IR (KBr) υmax 3383 (vs), 2963 (m), 2932 (m), 2870 (m), 2814 (w), 1739 (s), 1686 (s), 1625 (s), 1455 (vs), 1426 (s), 1373 (w), 1240 (w), 1067 (s), 1031 (s), 947 (m), 728 (m), 575 (m), and 525 (vs, a characteristic band of C60monoadduct) cm−1; UV-Vis (DMF, cutoff at 268 nm, 2.0 × 10–5 M) λmax 323 nm (shoulder peak); 1H-NMR [500 MHz, DMSO-d6–toluene-d8 (2:1), ppm] δ 3.80–4.25 (m, br, 16H), 2.90–3.80 (m, br, 39H), 1.50–1.80 (m, br, 12H), and 0.88–0.99 (m, br, 18H). We found that electronic interferences of iodide anions in a high quantity with the fullerene cage or possible partial electron-transfer events prohibited the detection of fullerenyl carbon peaks (in low signal intensity).
3.3.8. Synthesis of Pentacationic Methoxy-tri(ethyleneglycol)-(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-nonadecan-19-yl)[60]fullerenyl Malonate Methyl Quaternary Ammonium Salt, C60(˃ME1N6+C3) (2)
Finely divided [60]fullerene (1.0 g, 1.40 mmol, more than two-fold excess to allow the formation of monoadduct only) was taken into a round bottom flask and added anhydrous toluene (700 mL) under nitrogen. The solution was stirred for 12 h at ambient temperature to ensure complete dissolution of C60. To the resulting purple-colored solution added carbon tetrabromide (0.17 g, 0.51 mmol) followed by a solution of compound 10 (0.65 g, 0.45 mmol) in anhydrous DMF (100 mL). The solution mixture was stirred for an additional 30 min of stirring and added slowly 1.8-diazabicyclo[5.4.0]-undec-7-ene (DBU, 0.15 g, 0.98 mmol) over a period of 45 min. The color of solution slowly turns into brown in a reaction period of 8 h. The solution was then concentrated on rotavap to roughly 100 mL. Upon the addition of methanol to this concentrated solution, the crude product was precipitated as brown solids which were collected via centrifugation. Unreacted C60 in the crude solids was removed by repeated washings with toluene (5 × 100 mL) until no color in the washing solution or filtrate. The remaining product of C60(>ME3N6+C3) (2) was obtained as brown solids in 50% yield (0.343 g, after recovered C60). Spectroscopic data: FT-IR (KBr) υmax 3433 (vs), 3262 (s), 2963 (m), 2925 (m), 2868 (m), 2809 (w), 1736 (s), 1695 (m), 1630 (s), 1459 (s), 1384 (w), 1197 (m), 1107 (s), 1071 (s), 938 (m), 757 (w), and 525 (s, a characteristic band of C60monoadduct) cm−1; UV-Vis (DMF, cutoff at 268 nm, 2.0 × 10−5 M) λmax 323 nm (shoulder peak); 1H-NMR [500 MHz, DMSO-d6–toluene-d8 (2:1), ppm] δ 3.80–4.20 (m, br, 16H), 2.90–3.80 (m, br, 47H), 1.50–1.75 (m, br, 12H), and 0.88–0.99 (m, br, 18H). We found that electronic interferences of iodide anions in a high quantity with the fullerene cage or possible partial electron-transfer events prohibited the detection of fullerenyl carbon peaks (in low signal intensity).
3.3.9. Synthesis of tert-Butyl(2-methoxyethyl)malonate, ME1(t-C4) (11)
A mixture of malonic acid methoxyethyleneglycol ester 4 (1.0 g, 6.16 mmol), 2-methyl-2-propanol (0.54 g, 7.39 mmol), and N,N′-dicyclohexyl carbodiimide (DCC, 1.27 g, 6.16 mmol) in anyhydrous dichloromethane (20 mL) were stirred under atmospheric pressure of N2 over a period of 8.0 h at ambient temperature. The resulting white solid of N,N′-dicyclohexyl urea was filtered and the filtrate was washed with aqueous sodium carbonate solution (5%, 10 mL). The organic layer was then dried over sodium sulfate and concentrated on rotavap to give ME1(t-C4) (11) in 74% yield (1.0 g) as light yellow liquid. Spectroscopic data: FT-IR (KBr) υmax 2980 (w), 2930 (w), 2880 (w), 2824 (w), 1748 (s), 1726 (vs), 1455 (w), 1406 (w), 1393 (w), 1368 (m), 1330 (m), 1281 (m), 1250 (m), 1199 (m), 1127 (vs), 1100 (m), 1037 (s), 966 (m), 864 (m), 838 (m), 759 (w), and 738 (w) cm−1; 1H-NMR (500 MHz, CDCl3, ppm) δ 4.27 (t, J = 3.94 Hz, 2H), 3.56 (t, J = 3.94 Hz, 2H), 3.34 (s, 3H), 3.29 (s, 2H, Hα), and 1.43 (s, 9H).
3.3.10. Synthesis of tert-Butyl(methoxy-triethyleneglycol)malonate, ME3(t-C4) (12)
A mixture of malonic acid methoxytriethyleneglycol ester 5 (3 g, 11.98 mmol), 2-methyl-2-propanol (1.06 g, 14.38 mmol), and N,N′-dicyclohexyl carbodiimide (2.47 g, 11.98 mmol) in anyhydrous dichloromethane (20 mL) were stirred under atmospheric pressure of N2 over a period of 8.0 h at ambient temperature. The resulting white solid of N,N′-dicyclohexyl urea was filtered and the filtrate was washed with aqueous sodium carbonate solution (5%, 10 mL). The organic layer was then dried over sodium sulfate and concentrated on rotavap to give ME3(t-C4) (12) in 77% yield (2.85 g) as light yellow liquid. Spectroscopic data: FT-IR (KBr) υmax 2973 (w), 2930 (w), 2876 (w), 2817 (w), 1748 (s), 1727 (vs), 1455 (w), 1393 (w), 1368 (m), 1330 (m), 1282 (m), 1250 (m), 1198 (m), 1134 (vs), 1104 (vs), 1040 (m), 966 (m), 864 (m), 759 (w), and 735 (w) cm−1; 1H-NMR (500 MHz, CDCl3, ppm) δ 4.26 (t, J = 4.50 Hz, 2H), 3.68 (t, J = 4.50 Hz, 2H), 3.63 (m, 6H), 3.52 (t, J = 4.50 Hz, 2H), 3.35 (s, 3H), 3.29 (s, 2H, α-H), and 1.44 (s, 9H).
3.3.11. Synthesis of tert-Butyl(2-methoxyethyl)[60]fullerenyl Malonate, C60[˃ME1(t-C4)] (13)
Finely divided [60]fullerene (1.23g, 1.70 mmol) was taken into a round bottom flask and added anhydrous toluene (850 mL) and 1,2-dichlorobenzene (30 mL) under nitrogen. The solution was stirred for 1.0 h at ambient temperature to ensure complete dissolution of C60. To the resulting purple-colored solution was added carbon tetrabromide (0.50 g, 1.51 mmol) followed by a solution of tert-butyl(2-methoxyethyl)malonate (11, 0.30 g, 1.37 mmol) in anhydrous toluene (10 mL). The solution mixture was stirred for an additional 30 min and added slowly 1.8-diazabicyclo[5.4.0]-undec-7-ene (DBU, 0.44 g, 2.88 mmol) over a period of 1.0 h. The color of solution slowly turned into brown in a reaction period of 12 h. The solution was then concentrated on a rotavap. The resulting crude product was purified using column chromatography with silica gel as the stationary phase and toluene–ethyl acetate (20:1) as eluent, giving the isolation of tert-butyl(2-methoxyethyl)[60]fullerenyl malonate (13), C60[>ME1(t-C4)], as brown solids in 78% yield (1.00 g). Spectroscopic data: FT-IR (KBr) υmax 3442 (br, s), 2972 (w), 2917 (w), 2873 (w), 2814 (w), 1739 (vs), 1645 (m), 1426 (m), 1390 (w), 1366 (m), 1268 (s), 1232 (vs), 1179 (m), 1151 (s), 1110 (m), 1059 (m) 1026 (m), 828 (w), 738 (w), 704 (w), 577 (m), 551 (m), and 525 (vs, a characteristic band of C60 monoadduct) cm−1; 1H-NMR (500 MHz, toluene-d8, ppm) δ 4.28 (t, J = 4.55 Hz, 2H), 3.26 (t, J = 4.55 Hz, 2H), 3.06 (s, 3H), and 1.55 (s, 9H); 13C-NMR (500 MHz, toluene-d8, ppm) δ 163.45 (O=C-O-tert-butyl), 161.86 (O=C-O–), 145.89 (2C), 145.53 (2C), 145.22 (2C), 145.13 (2C), 145.06 (3C), 144.95 (2C), 144.93 (2C), 144.60 (2C), 144.59 (2C), 144.46 (3C), 144.45 (4C), 143.71 (2C), 143.68 (2C), 142.91 (C), 142.89 (C), 142.83 (2C), 142.81 (3C), 142.80 (3C), 142.74 (2C), 142.03 (2C), 142.02 (2C), 141.76 (2C), 141.74 (2C), 140.74 (2C), 140.68 (2C), 139.37 (2C), 138.93 (2C), 84.13, 72.07 (fullerenyl sp3 carbons, 2C), 69.74, 65.49, 58.00, 53.45, and 27.49 (3C).
3.3.12. Synthesis of tert-Butyl(methoxy-triethyleneglycol)[60]fullerenyl Malonate, C60[˃ME3(t-C4)] (14)
Finely divided [60]fullerene (1.86 g, 2.59 mmol) was taken into a round bottom flask and added anhydrous toluene (850 mL) and 1,2-dichlorobenzene (50 mL) under nitrogen. The solution was stirred for 1.0 h at ambient temperature to ensure complete dissolution of C60. To the resulting purple-colored solution was added carbon tetrabromide (0.75 g, 2.27 mmol) followed by a solution of tert-butyl(methoxy-triethyleneglycol)malonate 12 (0.63 g, 2.07 mmol) in anhydrous toluene (10 mL). The solution mixture was stirred for an additional 30 min and added slowly 1.8-diazabicyclo[5.4.0]-undec-7-ene (DBU, 0.66 g, 4.35 mmol) over a period of 1.0 h. The color of solution slowly turned into brown in a reaction period of 12 h. The solution was then concentrated on rotavap and the resulting crude product was purified by column chromatography with silica gel as the stationary phase and toluene−ethyl acetate (9:1) as eluent to afford C60[>ME3(t-C4)] (14) as brown solids in 75% yield (1.60 g). Spectroscopic data: FT-IR (KBr) υmax 3421 (br, s), 2967 (w), 2914 (w), 2864 (w), 2814 (w), 1740 (s), 1634 (m), 1456 (m), 1426 (m), 1392 (m), 1368 (m), 1269 (s), 1253 (s), 1226 (s), 1180 (m), 1154 (s), 1108 (m), 1029 (m), 845 (m), 737 (m), 702 (m), 669 (m), 576 (m), 550 (m), and 525 (vs) cm−1; 1H-NMR (500 MHz, CDCl3, ppm) δ 4.65 (t, J = 4.60 Hz, 2H), 3.90 (t, J = 4.60 Hz, 2H), 3.73 (t, J = 4.60 Hz, 2H), 3.65–3.72 (m, 4H), 3.56 (t, J = 4.60 Hz, 2H), 3.39 (s, 3H), and 1.70 (s, 9H); 13C-NMR (500 MHz, CDCl3, ppm) δ 163.95 (O=C-O-tert-butyl), 162.16 (O=C-O–), 145.61 (2C), 145.41 (2C), 145.29 (2C), 145.25 (4C), 145.22 (2C), 145.16 (3C), 144.85 (2C), 144.71 (C), 144.69 (3C), 144.66 (2C), 144.57 (4C), 143.89 (3C), 143.09 (2C), 143.08 (2C), 143.01 (4C), 142.99 (2C), 142.97 (2C), 142.23 (2C), 142.21 (2C), 141.91 (2C), 141.90 (2C), 140.94 (2C), 140.89 (2C), 139.11 (2C), 138.92 (2C), 85.16, 71.95 (fullerenyl sp3 carbons, 2C), 71.84, 70.68, 70.67, 70.65, 68.84, 66.07, 59.10, 53.06, and 28.07 (3C).
3.3.13. Synthesis of 2-[60]Fullerenyl-3-(2-methoxyethoxy)-3-oxopropanoic Acid, C60(˃ME1H) (15)
The compound of [60]fullerenyl malonate 13 (0.5 g, 0.64 mmol) was taken into a round bottom flask containing anhydrous dichloromethane (50 mL) and purged with N2 for 15 minutes at ambient temperature. To this reaction mixture was added trifluoroacetic acid (30 mL, excess) and stirred for overnight at room temperature. At the end of the reaction, dichloromethane was removed on rotavap. Additional dichloromethane (3 × 15 mL) was added and removed on rotavap in order to fully eliminate an excessive amount of trifluoroacetic acid. The resulting residue was then washed with diethyl ether (2 × 15 mL) to afford C60(>ME1H) (15) as brown solids in 89% yield (0.50 g). Spectroscopic data: FT-IR (KBr) υmax 3670 (br, s), 2977 (w), 2917 (w), 2878 (w), 2814 (w), 1792 (w), 1750 (s), 1708 (s), 1681 (m), 1541 (w), 1427 (m), 1390 (w), 1366 (m), 1267 (s), 1250 (s), 1231 (s), 1186 (m), 1114 (w), 1059 (w) 1020 (w), 864 (w), 743 (w), 701 (m), 572 (m), 552 (m), and 525 (vs, a characteristic band of C60 monoadduct) cm−1; 1H-NMR (500 MHz, THF-d8, ppm) δ 4.59 (t, J = 4.50 Hz,2H), 3.71 (t, J = 4.50 Hz, 2H), and 3.34 (s, 3H); 13C-NMR [500 MHz, THF-d8–CS2 (2:1), ppm] δ 161.62 (O=C-O–), 161.13 (O=C-OH), 144.54 (2C), 144.06 (2C), 143.53 (2C), 143.39 (2C), 143.26 (2C), 143.25 (2C), 143.19 (2C), 143.16 (2C), 142.91 (C), 142.85 (2C), 142.78 (C), 142.73 (2C), 142.70 (2C), 142.53 (4C), 141.99 (2C), 141.95 (2C), 141.14 (C), 141.12 (C), 141.10 (2C), 141.04 (4C), 140.98 (2C), 140.31 (4C), 140.07 (2C), 139.95 (2C), 138.89 (2C) , 138.87 (2C), 137.87 (2C), 136.76 (2C), 70.48 (fullerenyl sp3 carbons, 2C), 68.14, 63.98, 56.30, and 51.46
3.3.14 Synthesis of 2-[60]Fullerenyl-3-(methoxy-triethyleneglycol)-3-oxopropanoic Acid, C60(˃ME3H) (16)
The compound of [60]fullerenyl malonate 14 (0.75 g, 0.73 mmol) was taken into a round bottom flask containing anhydrous dichloromethane (50 mL) and purged with N2 for a period of 15 minutes at ambient temperature. To this reaction mixture was added trifluoroacetic acid (50 mL) and stirred for overnight at room temperature. At the end of the reaction, dichloromethane was removed on rotavap. Additional dichloromethane (3 × 20 mL) was added and removed on rotavap in ordered to fully eliminate an excessive amount of trifluoroacetic acid. The resulting residue was then washed with diethyl ether (3 × 20 mL) to afford C60(>ME3H) (16) as brown solids in 87% yield (0.62 g). Spectroscopic data: FT-IR (KBr) υmax 3673 (br, s), 2917 (w), 2894 (w), 2870 (w), 2814 (w), 1788 (w), 1741 (vs), 1578 (w), 1532 (w), 1426 (m), 1383 (w), 1266 (m), 1230 (s), 1203 (m), 1180 (w), 1095 (m), 1060 (w) 845 (w), 698 (m), 579 (m), 549 (m), and 525 (vs) cm−1; 1H-NMR (500 MHz, CDCl3, ppm) δ 4.70 (t,J = 4.55 Hz, 2H), 3.95 (t, J = 4.55 Hz, 2H), 3.80 (m, 6H), 3.76 (t, J = 4.60 Hz, 2H), and 3.59 (s, 3H); 13C-NMR (500 MHz, CDCl3, ppm) δ 164.12 (O=C-O–), 163.79 (O=C-OH), 145.57 (2C), 145.52 (2C), 145.33 (3C), 145.32 (3C), 145.22 (4C), 145.15 (3C), 144.84 (2C), 144.75 (C), 144.68 (4C), 144.57 (2C), 144.54 (2C), 143.90 (2C), 143.88 (2C), 143.04 (3C), 142.99 (4C), 142.96 (2C), 142.23 (3C), 142.01 (2C), 141.97 (2C), 140.93 (3C), 140.92 (3C), 139.36 (2C), 138.90 (2C), 72.19, 71.88 (fullerenyl sp3 carbon, 2C), 70.89, 70.64, 70.33, 69.70, 68.34, 65.99, 58.78, and 52.51.
3.3.15. Synthesis of Methoxyethyleneglycol-(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-nonadecan-19-yl)[60]fullerenyl Malonate, C60(˃ME1N6C3) (17) and C60(>ME1N6+C3) (1′)
The compound of 2-[60]fullerenyl-3-(2-methoxyethoxy)-3-oxopropanoic acid 15 (0.15 g, 0.17 mmol) was taken into a round bottom flask containing anhydrous tetrahydrofuran (20 mL). To this reaction mixture was added thionyl chloride (0.03 g, 2.55 mmol) under N2 atmosphere and refluxed for a period of 2.0 h. An excessive amount of thionyl chloride was removed on rotavap. Fresh anhydrous tetrahydrofuran (20 mL) was added. To this reaction solution was added slowly N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amine 6 (0.08 g, 0.17 mmol) at 0 °C. It was warmed gradually to room temperature and stirred at this temperature for a period of 3.0 h. The resulting solution was concentrated on rotavap with the residue washed sequentially with hexane (10 mL), methanol (2 × 10 mL), and toluene (3 × 10 mL) to fully remove unreacted N,N′,N,N,N,N-hexapropyl-hexa(aminoethyl)amine and decarboxylated C60 byproducts. A relatively pure C60(>ME1N6C3) (17) was obtained in 47% yield (0.11 g). It was subsequently treated with CF3COOH to result in pentacationic quaternary ammonium−trifluoroacetate salt, C60(>ME1N6+C3) (1′), specifically, for NMR measurements. Spectroscopic data of the compound 17: FT-IR (KBr) υmax 3670 (br, s), 2977 (w), 2917 (w), 2878 (w), 2814 (w), 1792 (w), 1750 (s), 1708 (s), 1681 (m), 1541 (w), 1427 (m), 1390 (w), 1366 (m), 1267 (s), 1250 (s), 1231 (s), 1186 (m), 1114 (w), 1059 (w), 1020 (w), 864 (w), 743 (w), 701 (m), 572 (m), 552 (m), and 525 (vs, a characteristic band of C60monoadduct) cm−1. Spectroscopic data of the compound 1′:1H-NMR [500 MHz, CDCl3–toluene-d8–TFA (3:1:1), ppm] δ 4.22–4.68 (m, br, 2H), 3.90–4.05 (m, br, 2H), 3.22–3.75 (m, br, 35H), 1.28–1.36 (m, br, 12H), and 0.97 (m, br, 18H); 13C-NMR [500 MHz, CDCl3–toluene-d8–TFA (3:1:1), ppm] d 167.72 (O=C-O–), 166.46 (O=C-NH–), 148.00 (2C), 145.54 (2C), 145.30 (2C), 145.29 (2C), 145.24 (2C), 145.14 (2C), 145.02 (2C), 144.73 (4C), 144.68 (4C), 144.53 (2C), 143.98 (2C), 143.77 (2C), 143.27 (2C), 143.11 (C), 143.04 (2C), 143.01 (2C), 142.98 (C), 142.87 (2C), 142.44 (2C), 142.22 (2C), 142.12 (2C), 142.01 (2C), 141.17 (2C), 140.98 (2C), 140.51 (2C), 137.92 (4C), 136.50 (2C), 70.47, 70.23 (fullerenyl sp3 carbon, 2C), 70.08, 69.73, 64.65, 64.40, 58.94, 58.68, 38.43, 31.84, 22.04, 21.40, 16.66, and 10.10 (quaternary aminocarbon peaks were low in intensity).
3.3.16. Synthesis of Pentacationic Methoxyethyleneglycol-(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-nonadecan-19-yl)[60]fullerenyl Malonate Quaternary Methyl Ammonium Salt, C60(˃ME1N6+C3) (1)
A solution of [60]fullerenyl malonate quaternary ammonium−trifluoroacetate salt 1′ (100 mg) in chloroform (50 mL) was neutralized with aqueous potassium carbonate (10%, 50 mL). The resulting [60]fullerenyl malonate 17 was then dissolved in a mixture of anhydrous chloroform (30 mL) and dimethylformamide (15 mL). To this reaction, an excess amount of iodomethane was added in several portions over the reaction period and stirred at 45 °C for 3.0 days. At the end of the reaction, the solvent was removed on rotavap to yield pentacationic C60(>ME1N6+C3) (1). Spectroscopic data: MALDI–TOF–MS (sinapic acid as the matrix, rel. intensity) m/z 673 (20%), 697 (50%), 721 (C60H+, 100%), 734 (80%), 746 (15%), 761 (10%), 772 (10%), 874 [C60(>H(C=O)NHCH2CH2N+-propylMe2), 20%], 1442 [(C60H)2+ cluster], 1565, 1634, 1709, 1769, 1851, 1930 (M+–I−); FT-IR (KBr) υmax 3688 (br, s), 2918 (s), 2870 (m), 2840 (m), 2807 (w), 1784 (w), 1736 (vs), 1663 (s), 1574 (w), 1433 (m), 1383 (w), 1252 (w), 1187 (m), 1163 (s), 1126 (m), 1090 (m), 1031 (s), 842 (w), 762 (w), 704 (w), 661 (w), 569 (w), and 524 (vs, a characteristic band of C60 monoadduct) cm−1; UV-Vis (DMF, cutoff at 268 nm, 2.0 × 10–5 M) λmax 323 nm (shoulder peak); 1H-NMR [500 MHz, DMSO-d6–toluene-d8 (2:1), ppm] δ 3.80–4.25 (m, br, 16H), 2.90–3.80 (m, br, 39H), 1.50–1.80 (m, br, 12H), and 0.88–0.99 (m, br, 18H). We found that electronic interferences of iodide anions in a high quantity with the fullerene cage or possible partial electron-transfer events prohibited the detection of fullerenyl carbon peaks (in low signal intensity).
3.3.17. Synthesis of Pentacationic Methoxy-tri(ethyleneglycol)-(20-oxo-4,7,10,13,16-pentapropyl-4,7,10,13,16,19-hexaaza-nonadecan-19-yl)[60]fullerenyl Malonate Methyl Quaternary Ammonium Salt, C60(˃ME3N6+C3) (2)
Synthesis of the compound 2 was carried out by using a similar procedure as that of 1 except methoxytriethyleneglycol ester was applied instead. Spectroscopic data: MALDI–TOF–MS (sinapic acid as the matrix, rel. intensity)m/z 698 (10%), 721 (C60H+, 100%), 735 (C60>H2, 20%), 749, 773, 782, 789, 809, 874 [C60(>H(C=O)NHCH2CH2N+-propylMe2)], 914, 940, 995, 1027, 1054, 1278 (w), 1395 (w), 1417 (vw), 1499 (vw), 1792 (vw), and 2019 (vw, MH+–I−); FT-IR (KBr) υmax 3424 (vs), 2967 (m), 2925 (m), 2874 (m), 2824 (w), 1738 (s), 1681 (s), 1628 (s), 1454 (vs), 1429 (s), 1383 (m), 1064 (s), 1028 (s), 941 (m), 727 (m), 572 (m), and 524 (vs, a characteristic band of C60monoadduct) cm−1; UV-vis (DMF, cutoff at 268 nm, 2.0 × 10−5 M) λmax 323 nm (shoulder peak); 1H-NMR [500 MHz, DMSO-d6–toluene-d8 (2:1), ppm] δ 3.80–4.20 (m,br, 16H), 2.90–3.80 (m, br, 47H), 1.50–1.75 (m, br, 12H), and 0.88–0.99 (m, br, 18H). We found that electronic interferences of iodide anions in a high quantity with the fullerene cage or possible partial electron-transfer events prohibited the detection of fullerenyl carbon peaks (in low signal intensity).

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




