Novel Synthesis of 8-Deaza-5,6,7,8-tetrahydroaminopterin Analogues via an Aziridine Intermediate

Abstract
:
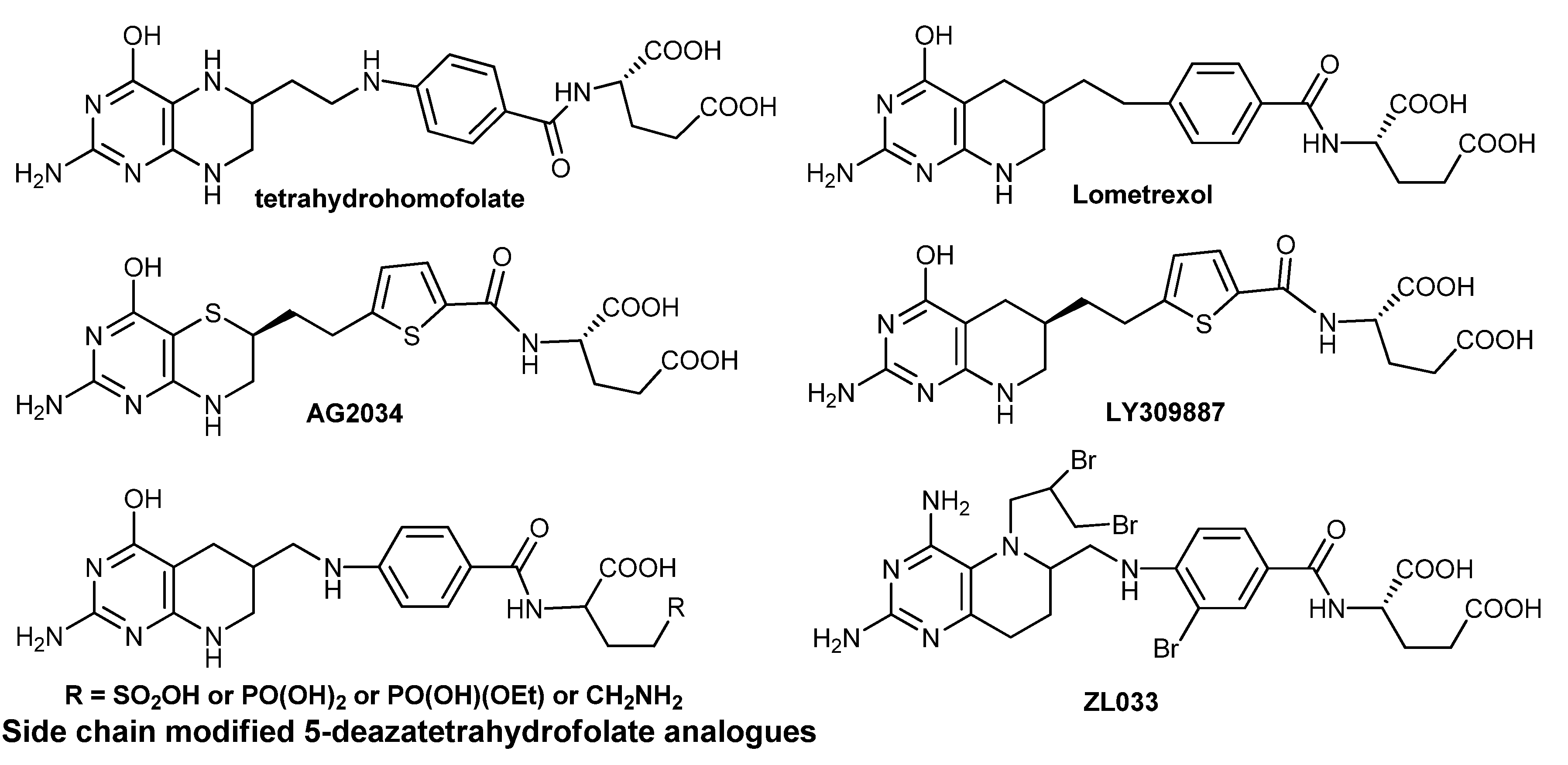
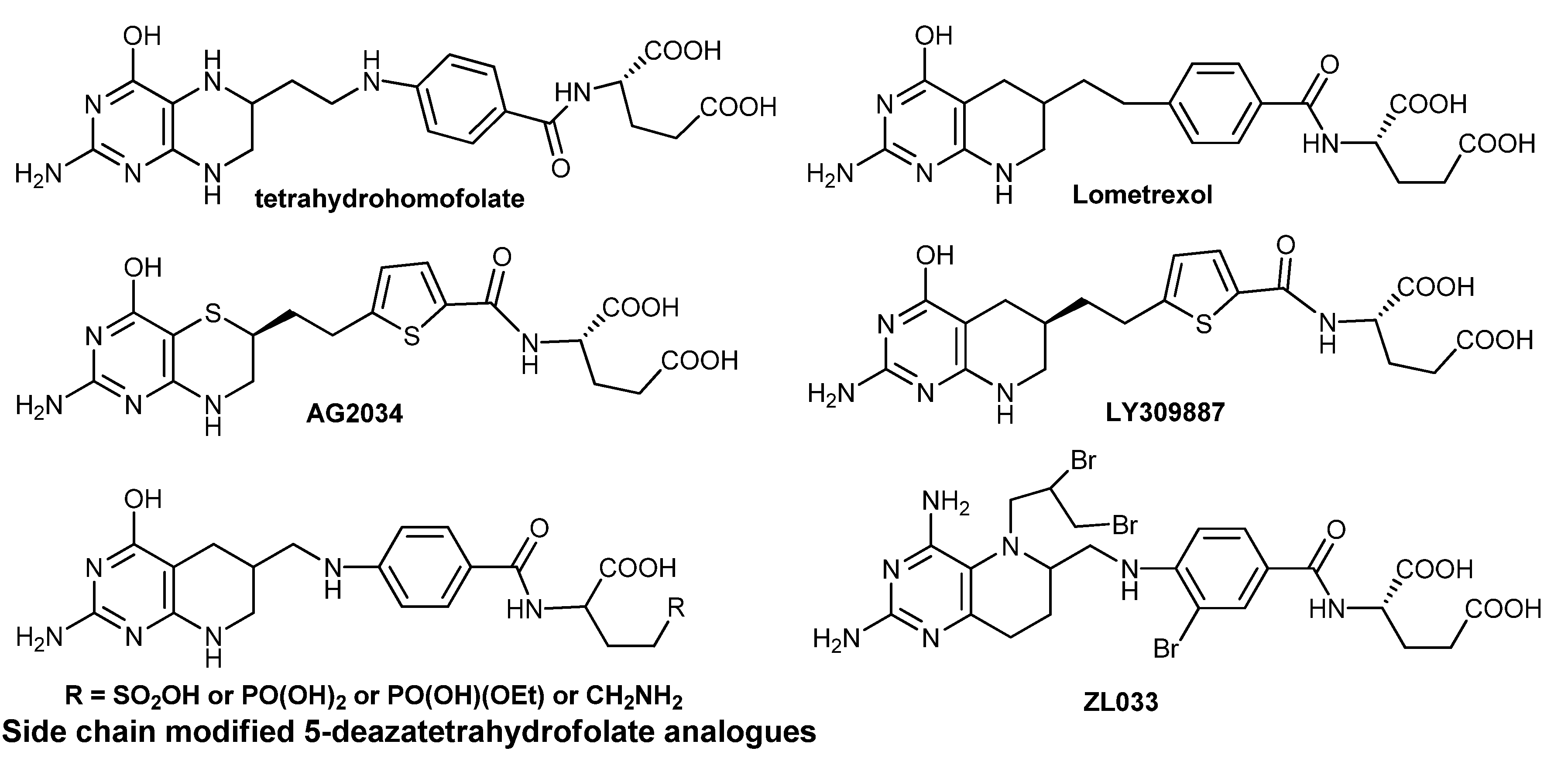
1. Introduction

2. Results and Discussion
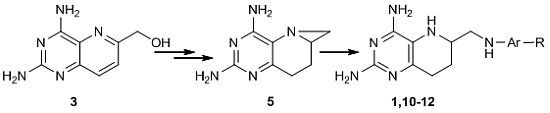
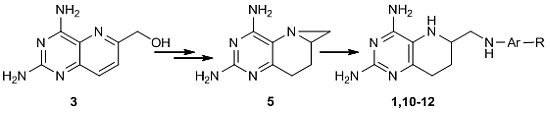
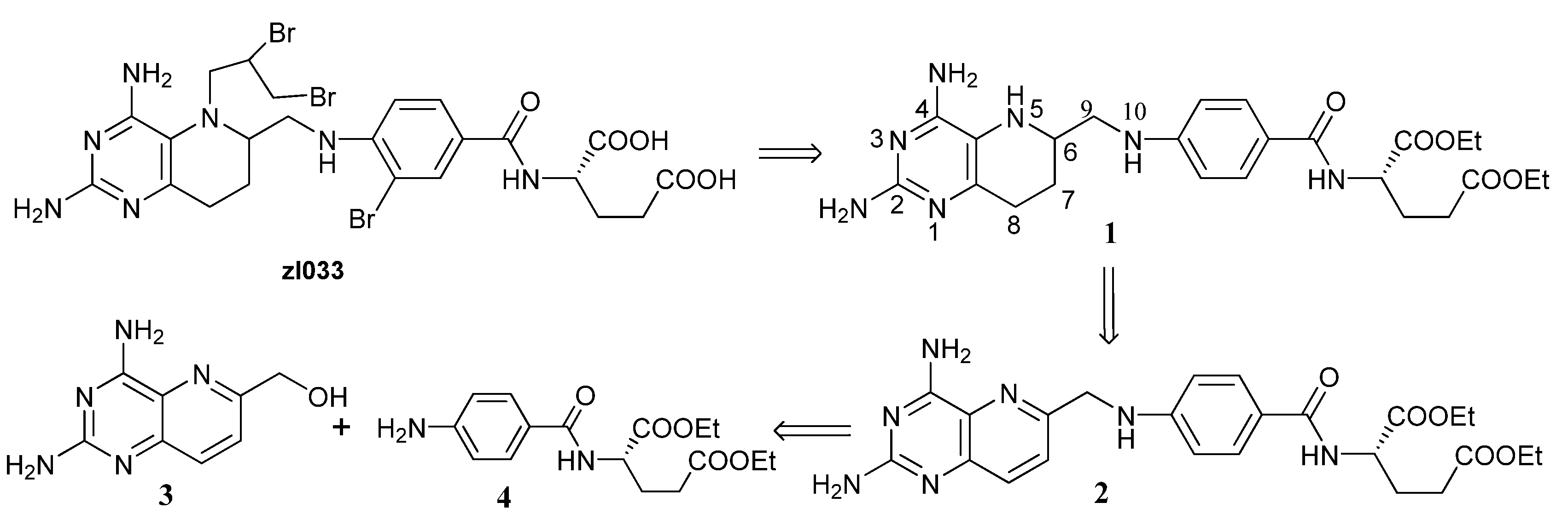
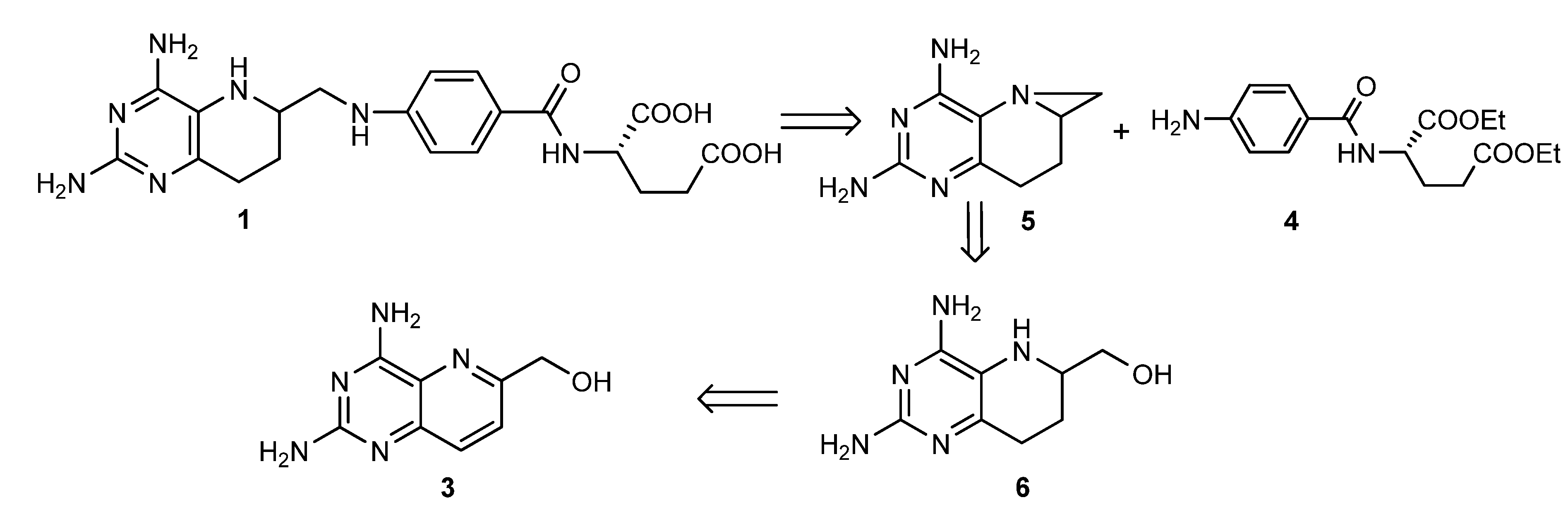
2.1. Retrosynthetic Analysis of 1

2.2. Catalytic Hydrogenation of 3


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Acid | Equiv. | Solvent | Time (h) | Yield (%) b |
|---|---|---|---|---|---|
| 1 | - | - | Ethanol | 120 | - |
| 2 | AcOH | 4 | Ethanol | 60 | 54 |
| 3 | AcOH | 8 | Ethanol | 60 | 47 |
| 4 | AcOH | excess | AcOH | 60 | 59 |
| 5 | HCl c | 4 | Ethanol | 24 | 82 |
| 6 | HCl c | 8 | Ethanol | 24 | 93 |
| 7 | HCl c | 16 | Ethanol | 24 | 78 |
| 8 | HCl c | 8 | Methanol | 8 | 21 |
| 9 | HCl c | 8 | Acetone | 48 | 46 |
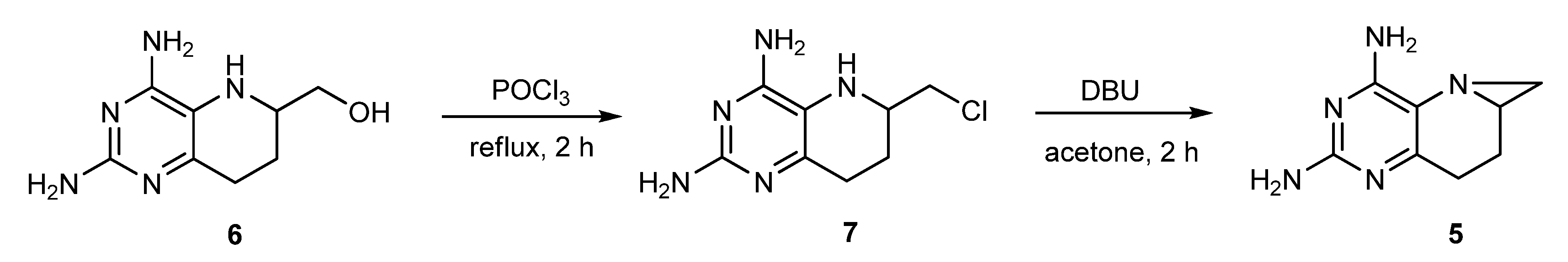
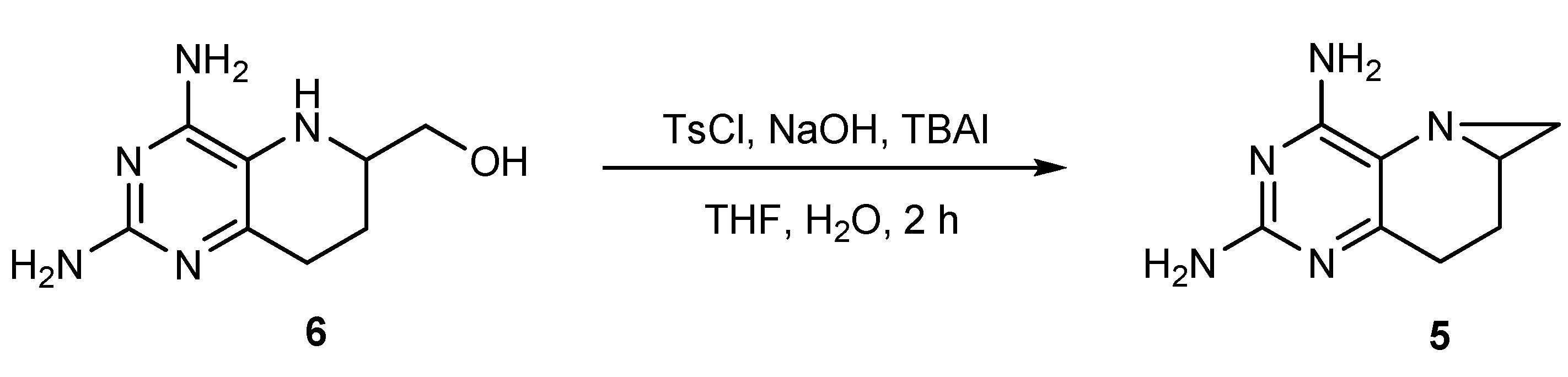
2.3. Optimized Synthesis of 5


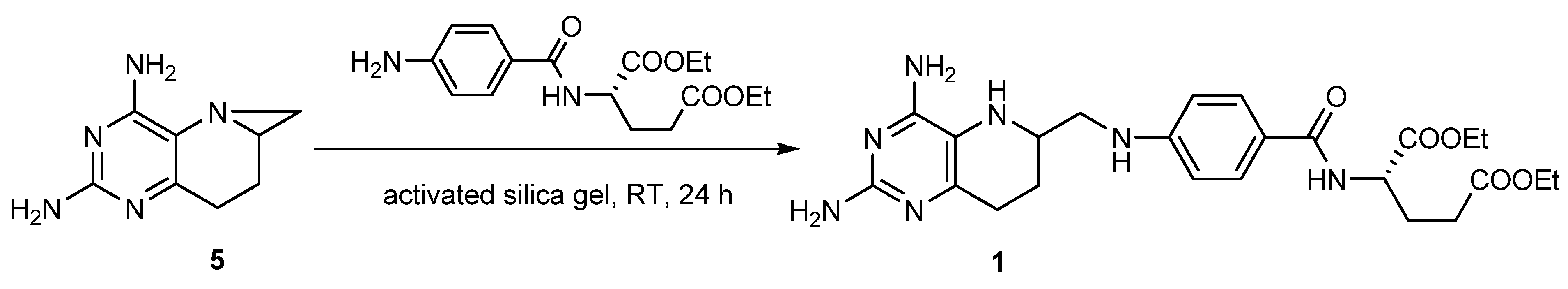
2.4. Synthesis of 1

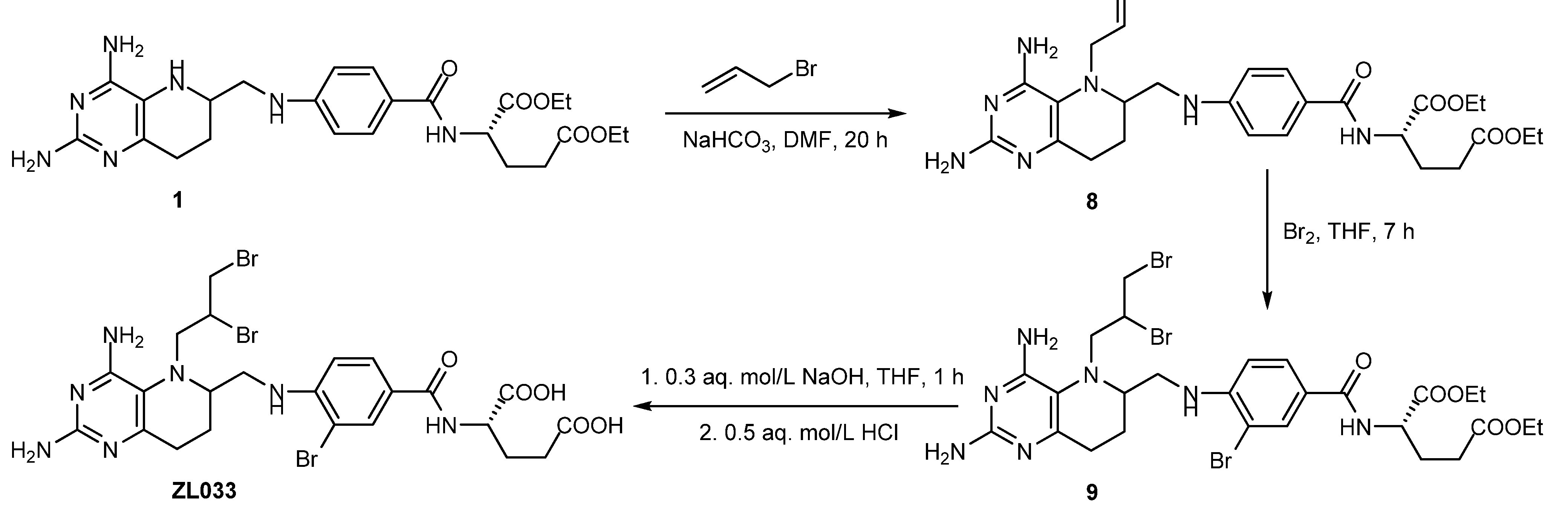
2.5. Synthesis of ZL033

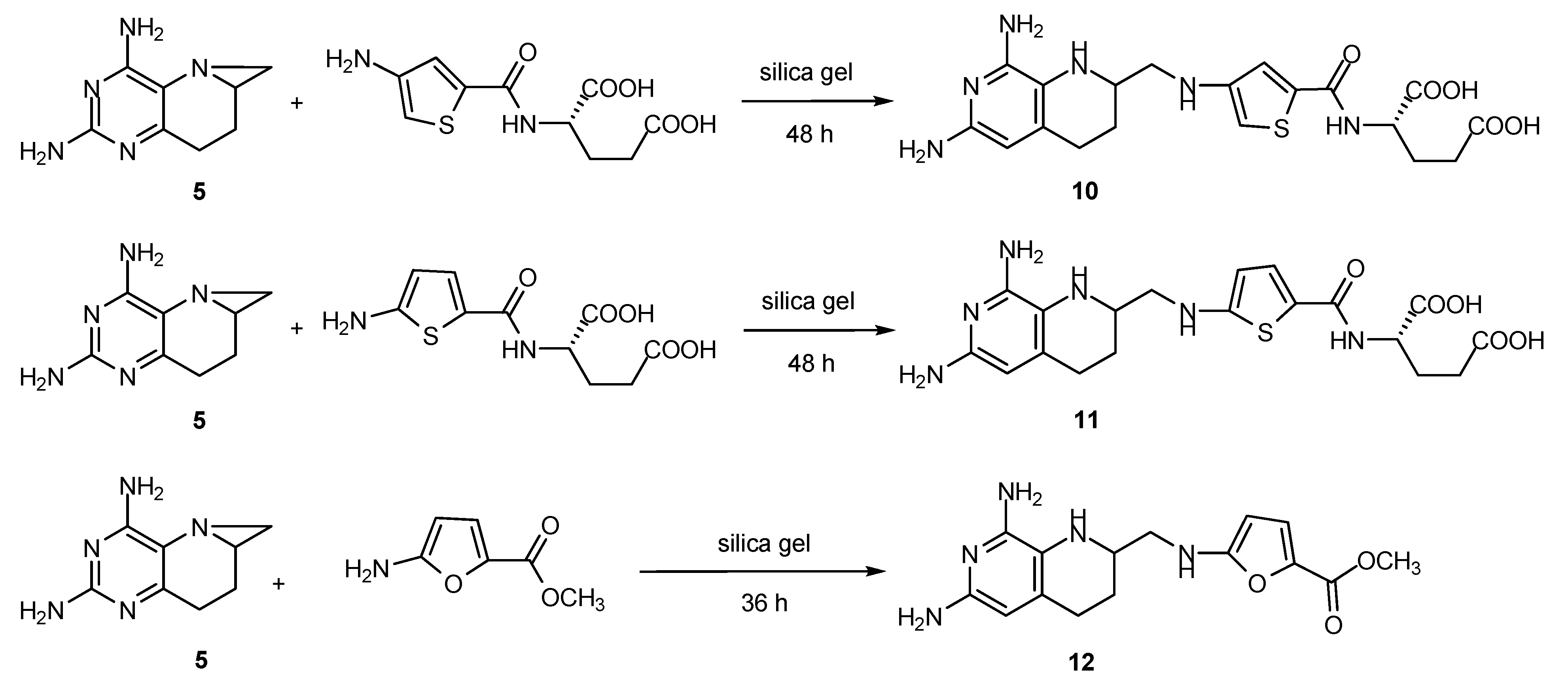
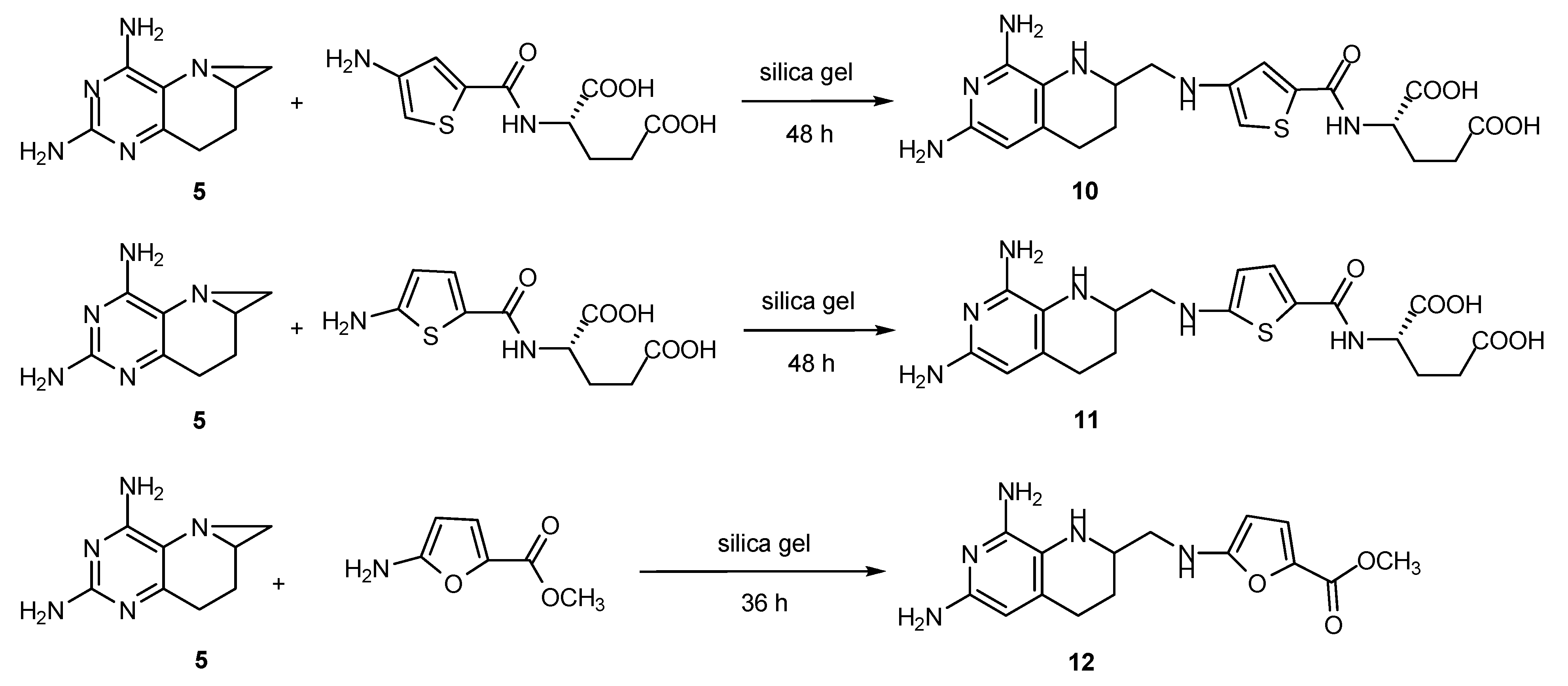
2.6. Synthesis of Other Tetrahydrofolate Analogues
3. Experimental
3.1. General
4. Conclusions
Acknowledgments
References and Notes
- Christopherson, R.I.; Lyons, S.D. Potent inhibitors of de novo pyrimidine and purine biosynthesis as chemotherapeutic agents. Med. Res. Rev. 1990, 10, 505–548. [Google Scholar] [CrossRef]
- Appling, D.R. Compartmentation of folate-mediated one-carbon metabolism in eukaryotes. FASEB J. 1991, 9, 482–487. [Google Scholar]
- Daidone, F.; Florio, R.; Rinaldo, S.; Contestabile, R.; di Salvo, M.L.; Cutruzzola, F.; Bossa, F.; Paiardini, A. In silico and in vitro validation of serine hydroxymethyltransferase as a chemotherapeutic target of the antifolate drug pemetrexed. Eur. J. Med. Chem. 2011, 46, 1616–1621. [Google Scholar] [CrossRef]
- Berger, S.H.; Pittman, D.L.; Wyatt, M.D. Uracil in DNA: Consequences for arcinogenesis and chemotherapy. Biochem. Pharmacol. 2008, 76, 697–706. [Google Scholar] [CrossRef]
- Goodman, L.; Kisliuk, R.L.; Pastore, E.J.; Plante, L.T.; Al-Nahas, A.; Morningstar, J.F.; Kwok, G.; Wilson, L.; Donovan, E.F.; Ratzan, J. Tetrahydrohomofolate, a specific inhibitor of thymidylate synthetase. J. Am. Chem. Soc. 1964, 86, 308–309. [Google Scholar]
- Taylor, E.C.; Harrington, P.J.; Fletcher, S.R.; Beardsley, G.P.; Moran, R.G. Synthesis of the antileukemic agents 5,10-dideazaaminopterin and 5,10-dideaza-5,6,7,8-tetrahydroaminopterin. J. Med. Chem. 1985, 28, 914–921. [Google Scholar] [CrossRef]
- Boritzki, T.J.; Barlett, C.A.; Zhang, C.; Howland, E.F.; Margosiak, S.A.; Palmer, C.L.; Romines, W.H.; Jackson, R.C. AG2034: A novel inhibitor of glycinamide ribonucleotide formyltransferase. Invest. New Drugs 1996, 14, 295–303. [Google Scholar] [CrossRef]
- Barnett, C.J.; Wilson, T.M.; Evans, D.A.; Somers, T.C. Asymmetric synthesis of dideazafolate antitumor agents via amidomethylation of nonracemic oxazolidinone imidates. Synthesis of LY309887, a cytotoxic dideazafolate analog related to lometrexol. Tetrahedron Lett. 1997, 38, 735–738. [Google Scholar]
- Rosowsky, A.; Forsch, R.A.; Reich, V.E.; Freisheim, J.H.; Moran, R.G. First use of the Taylor pteridine synthesis as a route to polyglutamate derivatives of antifolates. 46. Side chain modified 5-deazafolate and 5-deazatetrahydrofolate analogs as mammalian folylpolyglutamate synthetase and glycinamide ribonucleotide formyl transferase inhibitors: Synthesis and in vitro biological evaluation. J. Med. Chem. 1992, 35, 1578–1588. [Google Scholar] [CrossRef]
- Tang, C.; Zhang, Z.L.; Xu, B.; Li, M.; Liu, J.Y.; Cui, J.R. Two newly synthesized 5-methyltetrahydrofolate-like compounds inhibit methionine synthase activity accompanied by cell cycle arrest in G1/S phase and apoptosis in vitro. Anti-Cancer Drugs 2008, 19, 697–704. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Wu, J.; Ran, F.X.; Guo, Y.; Tian, R.; Zhou, S.X.; Wang, X.W.; Liu, Z.M.; Zhang, L.R.; Cui, J.R.; et al. Novel 8-deaza-5,6,7,8-tetrahydroaminopterin derivatives as dihydrofolate inhibitor: Design, synthesis and antifolate activity. Eur. J. Med. Chem. 2009, 44, 764–771. [Google Scholar] [CrossRef]
- Temple, C., Jr.; Kussner, C.L.; Rose, J.D.; Smithers, D.L.; Bennett, L.L., Jr.; Montgomery, J.A. New synthesis of N-[4-[[(2-amino-4(3H)-oxopyrido[3,2-d]pyrimidin-6-yl)methyl]amino]benzoyl]-L-glutamic acid (8-deazafolic acid) and the preparation of some 5,6,7,8-tetrahydro derivatives. J. Med. Chem. 1981, 24, 1254–1258. [Google Scholar] [CrossRef]
- Gangjee, A.; Shi, J.; Queener, S.F.; Barrows, L.R.; Kisliuk, R.L. Synthesis of 5-methyl-5-deaza nonclassical antifolates as inhibitors of dihydrofolate reductases and as potential antipneumocystis, antitoxoplasma, and antitumor agents. J. Med. Chem. 1993, 36, 3437–3443. [Google Scholar] [CrossRef]
- Taylor, E.C.; Hamby, J.M.; Shih, C.; Grindey, G.B.; Rinzel, S.M.; Beardsley, G.P.; Moran, R.G. Synthesis and antitumor activity of 5-deaza-5,6,7,8-tetrahydrofolic acid and its N10-substituted analogs. J. Med. Chem. 1989, 32, 1517–1522. [Google Scholar] [CrossRef]
- Singh, S.K.; Singer, S.C.; Ferone, R.; Waters, K.A.; Mullin, R.J.; Hynes, J.B. Synthesis and biological evaluation of N.alpha.-(5-deaza-5,6,7,8-tetrahydropteroyl)-L-ornithine. J. Med. Chem. 1992, 35, 2002–2006. [Google Scholar] [CrossRef]
- Pomel, V.; Gaillard, P.; Desforges, G.; Quattropani, A.; Montagne, C. 4-Morpholino-pyrido[3,2-d]pyrimidines. WO 2010037765 2010. [Google Scholar]
- Srinivasan, A.; Broom, A.D. Pyridopyrimidines. 12. Synthesis of 8-deaza analogs of aminopterin and folic acid. J. Org. Chem. 1981, 46, 1777–1781. [Google Scholar] [CrossRef]
- Anand, R.V.; Pandey, G.; Singh, V.K. Silica gel induced cleavage of aziridines by aromatic amines under solvent free conditions. Tetrahedron Lett. 2002, 43, 3975–3976. [Google Scholar] [CrossRef]
- Sweeney, J.B. Aziridines: Epoxides’ ugly cousins? Chem. Soc. Rev. 2002, 31, 247–258. [Google Scholar] [CrossRef]
- Hu, X.E. Nucleophilic ring opening of aziridines. Tetrahedron 2004, 60, 2701–2743. [Google Scholar] [CrossRef]
- Watson, I.D.G.; Yu, L.; Yudin, A.K. Advances in nitrogen transfer reactions involving aziridines. Acc. Chem. Res. 2006, 39, 194–206. [Google Scholar] [CrossRef]
- Eis, M.J.; Ganem, B. BF3-etherate promoted alkylation of aziridines with organocopper reagents: A new synthesis of amines. Tetrahedron Lett. 1985, 26, 1153–1156. [Google Scholar] [CrossRef]
- Nishikawa, T.; Ishikawa, M.; Wade, K.; Isobe, M. Total synthesis of α-C-mannosyltryptophan, a naturally occurring C-glycosyl amino acid. Synlett 2001, 10, 945–947. [Google Scholar]
- Yavad, J.S.; Reddy, B.V.S.; Rao, K.V.; Raj, K.S.; Prasad, A.R. Indium tribromide catalyzed aminolysis of aziridines: An efficient synthesis of vicinal-diamines. Synthesis 2002, 13, 1061–1064. [Google Scholar]
- Watson, I.D.G.; Yudin, A.K. Ring-opening reactions of nonactivated aziridines catalyzed by tris(pentafluorophenyl)borane. J. Org. Chem. 2003, 68, 5160–5167. [Google Scholar] [CrossRef]
- Kotake, Y.; Okauchi, T.; Iijima, A.; Yoshimatsu, K.; Nomura, H. Novel 6-5 fused ring heterocycle antifolates with potent antitumor activity: Bridge modifications and heterocyclic benzoyl isosters of 2,4-diamino-6,7-dihydro-5H-cyclopenta[d]pyrimidine Antifolate. Chem. Pharm. Bull. 1995, 43, 829–841. [Google Scholar] [CrossRef]
- Jackman, A.L.; Marsham, P.R.; Moran, R.G.; O’Connor, B.M.; Hughes, L.R.; Calverts, A.H. Thymidylate synthase inhibitors: The in vitro activity of a series of heterocyclic benzoyl ring modified 2-desamino-2-methyl-N10-substituted-5,8-dideazafolates. Adv. Enzyme Regul. 1991, 31, 13–27. [Google Scholar] [CrossRef]
- Jackman, A.L.; Calvert, A.H. Folate-based thymidylate synthase inhibitors as anticancer drugs. Ann. Oncol. 1995, 6, 871–881. [Google Scholar]
- Hodson, S.J.; Bigham, E.C.; Duch, S.D.; Smith, G.K.; Ferone, R. Thienyl and thiazolyl acyclic analogues of 5-deazatetrahydrofolic acid. J. Med. Chem. 1994, 37, 2112–2115. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, S.; Tian, C.; Li, C.; Guo, Y.; Wang, X.; Liu, J.; Zhang, Z. Novel Synthesis of 8-Deaza-5,6,7,8-tetrahydroaminopterin Analogues via an Aziridine Intermediate. Molecules 2012, 17, 5604-5614. https://doi.org/10.3390/molecules17055604
Zhou S, Tian C, Li C, Guo Y, Wang X, Liu J, Zhang Z. Novel Synthesis of 8-Deaza-5,6,7,8-tetrahydroaminopterin Analogues via an Aziridine Intermediate. Molecules. 2012; 17(5):5604-5614. https://doi.org/10.3390/molecules17055604
Chicago/Turabian StyleZhou, Shouxin, Chao Tian, Chao Li, Ying Guo, Xiaowei Wang, Junyi Liu, and Zhili Zhang. 2012. "Novel Synthesis of 8-Deaza-5,6,7,8-tetrahydroaminopterin Analogues via an Aziridine Intermediate" Molecules 17, no. 5: 5604-5614. https://doi.org/10.3390/molecules17055604
APA StyleZhou, S., Tian, C., Li, C., Guo, Y., Wang, X., Liu, J., & Zhang, Z. (2012). Novel Synthesis of 8-Deaza-5,6,7,8-tetrahydroaminopterin Analogues via an Aziridine Intermediate. Molecules, 17(5), 5604-5614. https://doi.org/10.3390/molecules17055604