2. Results and Discussion
In the first phase of the current effort, we observed that reaction of the 3-oxo-3-phenyl-2-(
p-tolylhydrazono)propanal (
1a) with ethyl cyanoacetate in acetic acid for 30 min in the presence of a catalytic amount of ammonium acetate leads to formation of the ethyl 2-hydroxy-6-phenyl-5-
p-tolylazonicotinate (
6a;
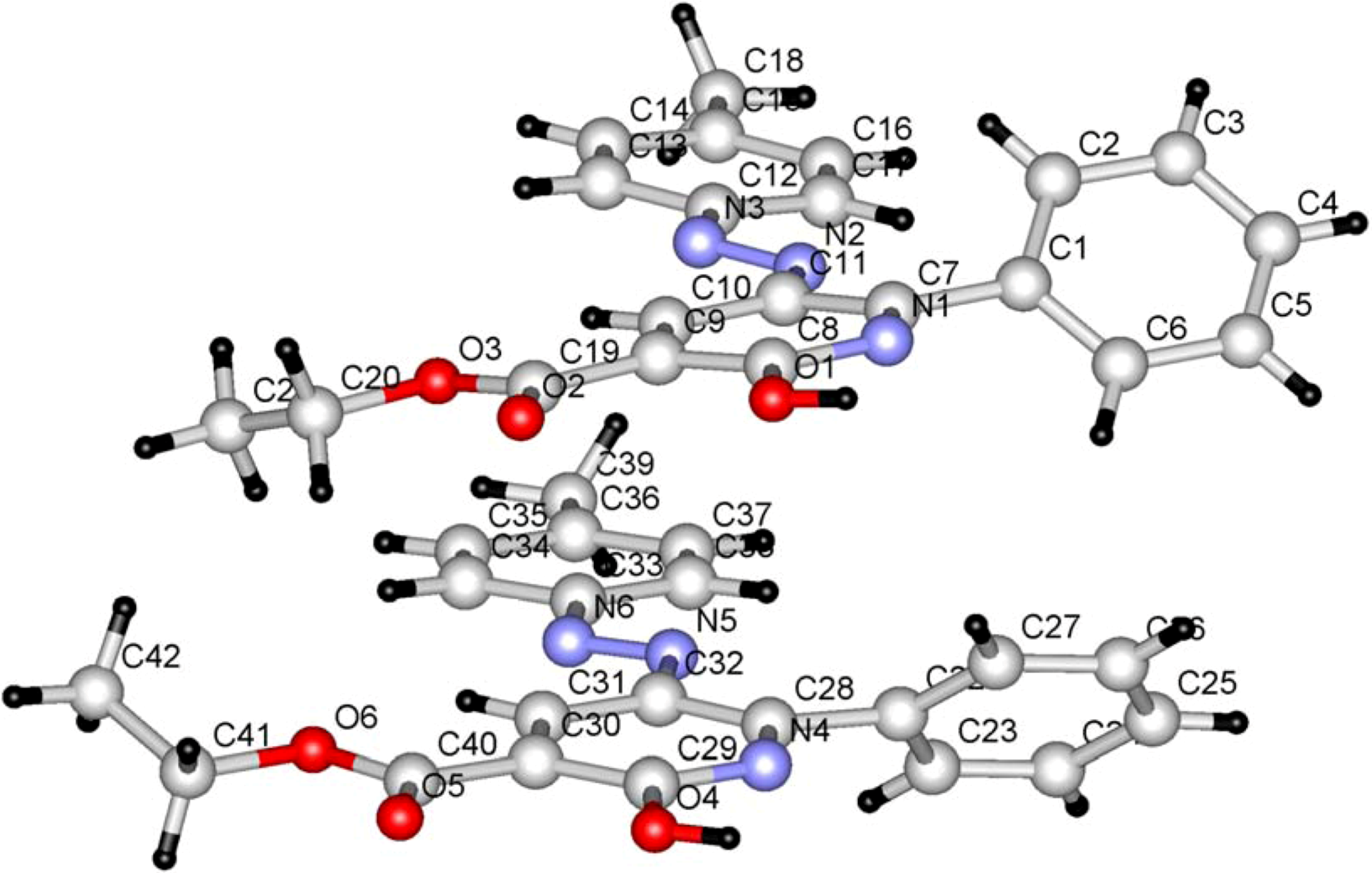
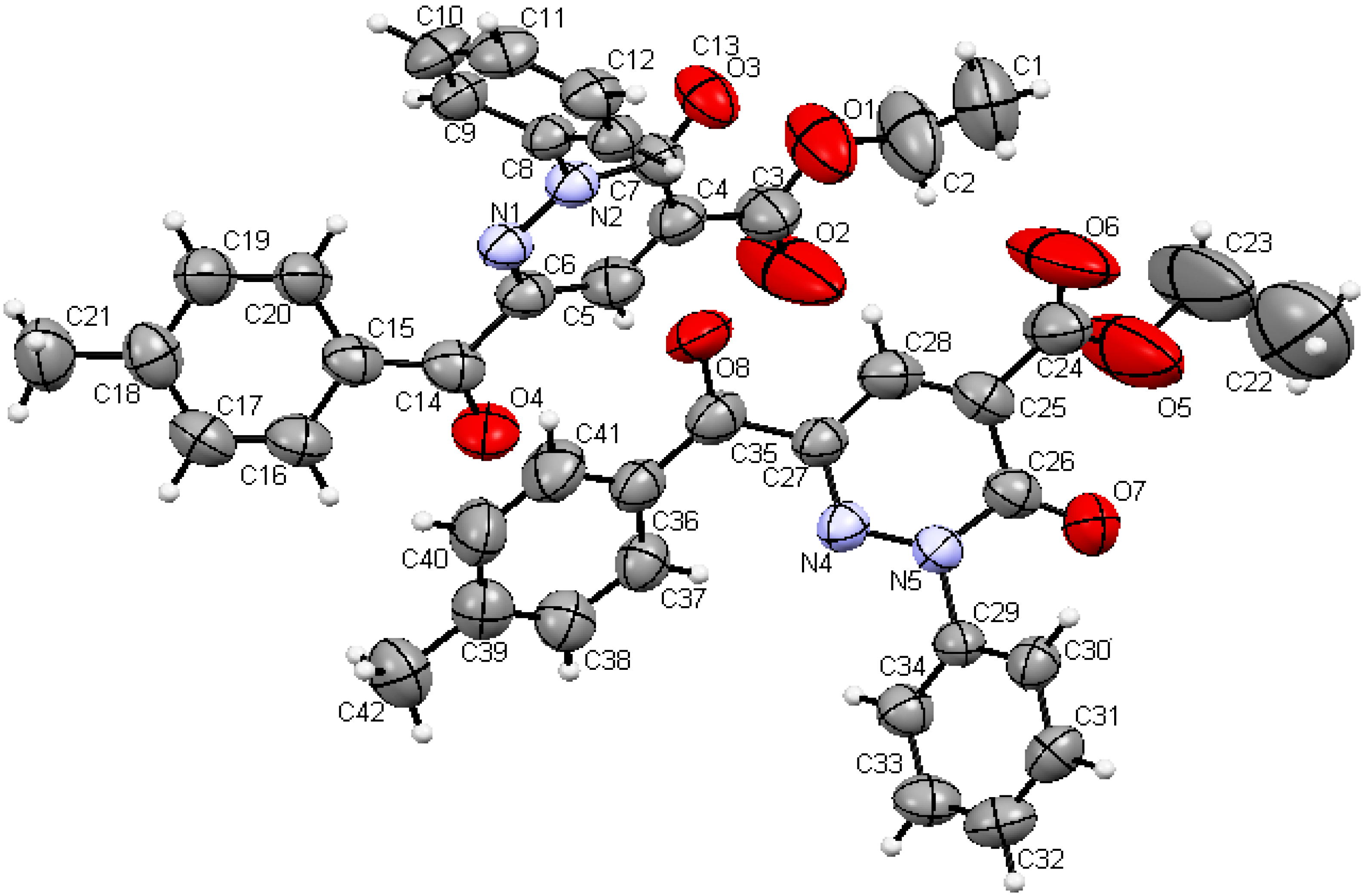
Scheme 1) whose structure was established by X-ray crystallographic analysis (
Figure 1) [
12].
In contrast, when the condensation reaction of
1a with ethyl cyanoacetate is conducted in the presence of excess of ammonium acetate, ethyl 2-amino-6-phenyl-5-
p-tolylazonicotinate (
8) is produced. The structure of
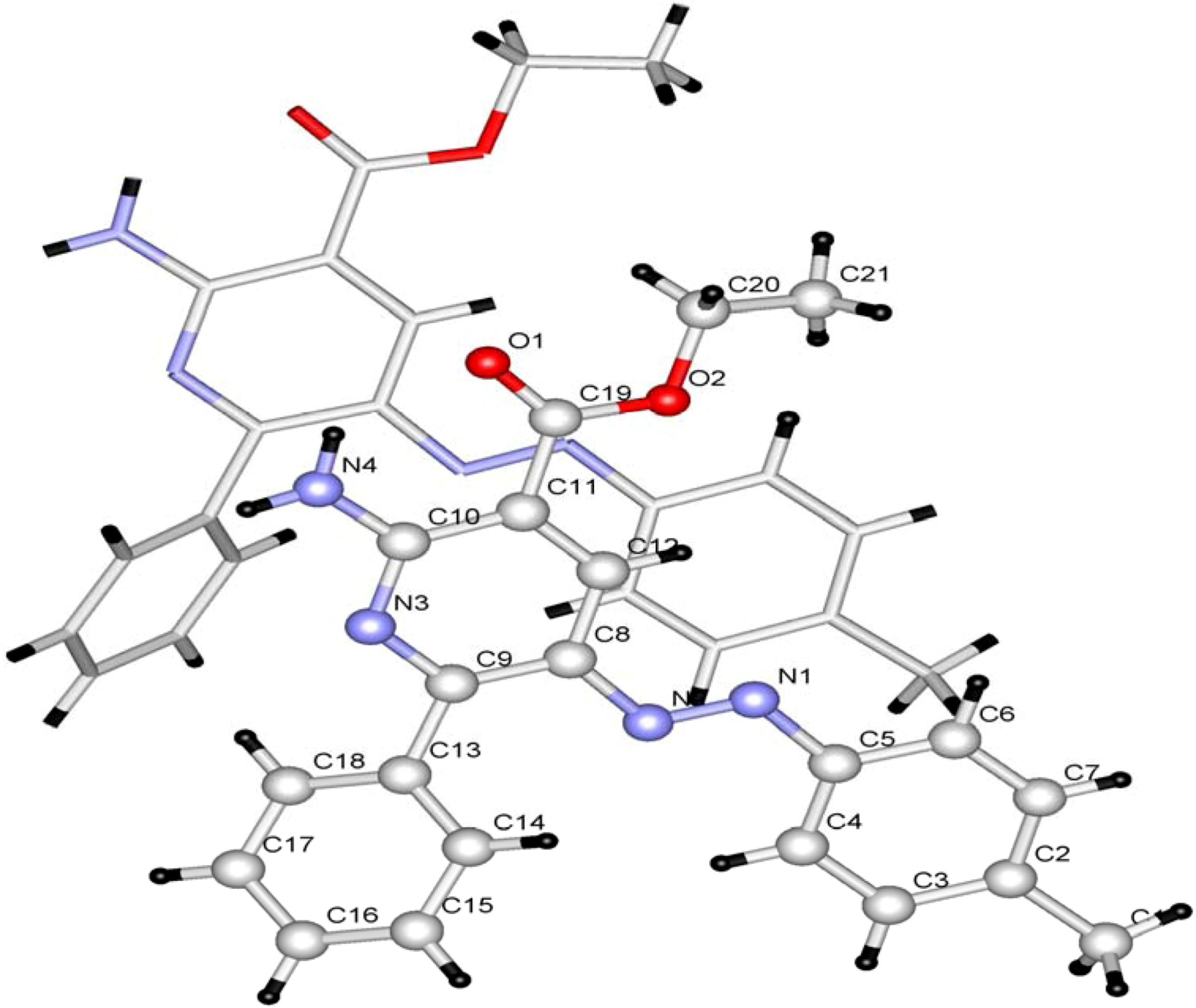
8 was also assigned by using X-ray crystallographic methods (
Figure 2) [
12].
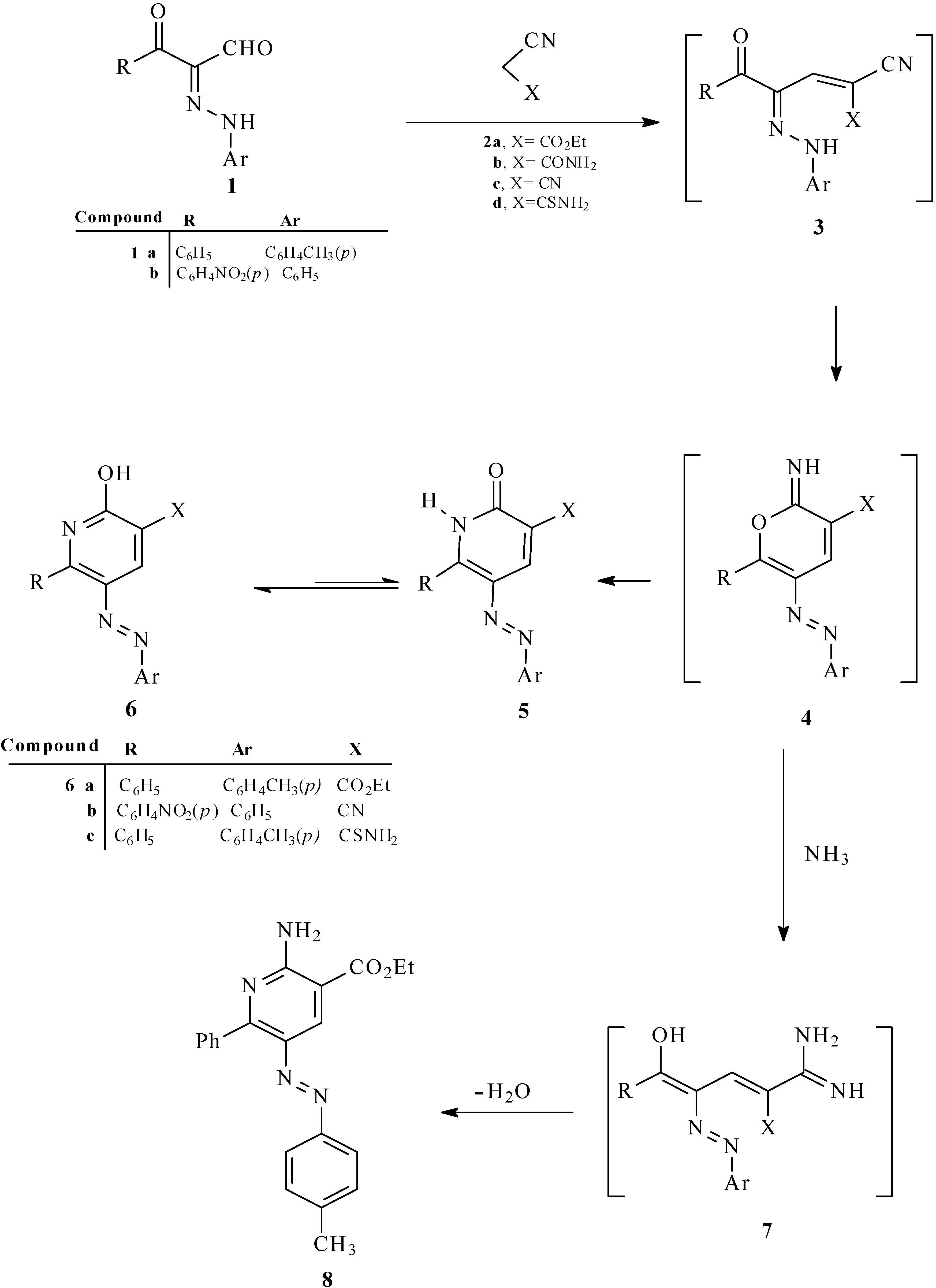
It is believed that these processes involve initial reaction of 1a with ethyl cyanoacetate to yield the hydrazono-enone 3 that then cyclizes to generate the pyran-imine 4. In the absence of ammonium ion, 4 undergoes a Dimroth type rearrangement to yield 6a. However, in the presence of a high concentration of ammonium acetate, pyran-imine 4 participates in a ring opening to yield amidine 7 that then cyclizes followed by water elimination to yield 8.
In a similar manner, the 3-oxo-3-aryl-2-arylhydrazonopropanals
1a,
b undergo condensation reactions with other active methylene nitriles
2c,
d (
Scheme 1) to yield the corresponding arylazonicotinates
6b,
c (catalytic ammonium acetate).
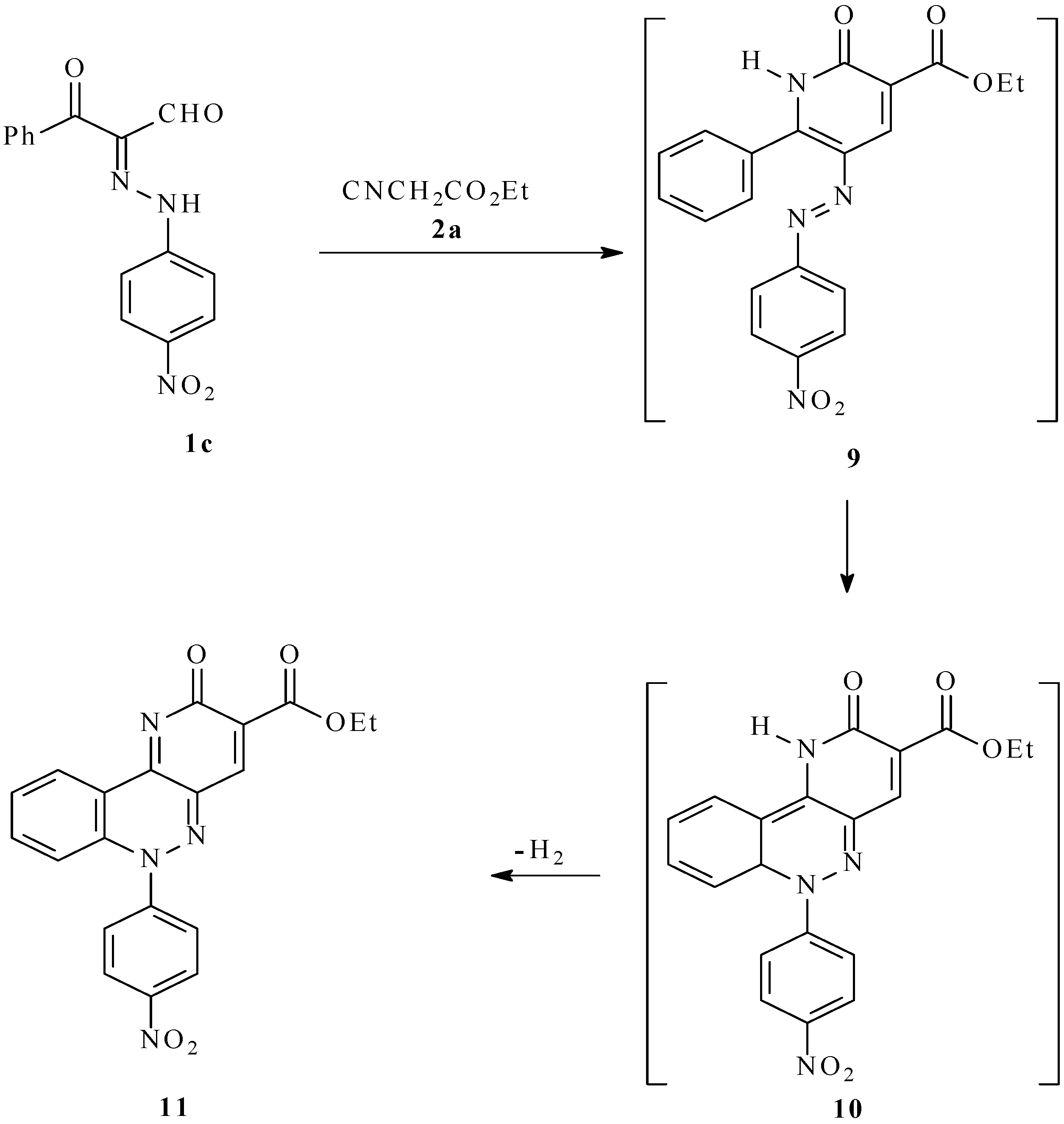
In contrast to the reactivity profiles displayed by its analogs, the 2-[(4-nitrophenyl)-hydrazono]-3-oxo-3-phenylpropanal (
1c) reacts with ethyl cyanoacetate (90 min) to afford the novel ethyl 6-(4-nitrophenyl)-2-oxo-2,6-dihydropyrido[3,2-c]cinnoline-3-carboxylate (
11;
Scheme 2). This substance is believed to be formed via a 6π-electrocyclization reaction of the initially formed arylazonicotinate
9 (analogous to
5 in
Scheme 1) that generates the tricyclic intermediate product
10, which then aromatizes to produce the pyrido[3,2–c]cinnoline
11. The high electrocyclization reactivity of
9 appears to be a consequence of the presence of the electron-withdrawing nitro substituent that apparently alters in a favorable way the frontier orbital interactions involved in the pericyclic process.
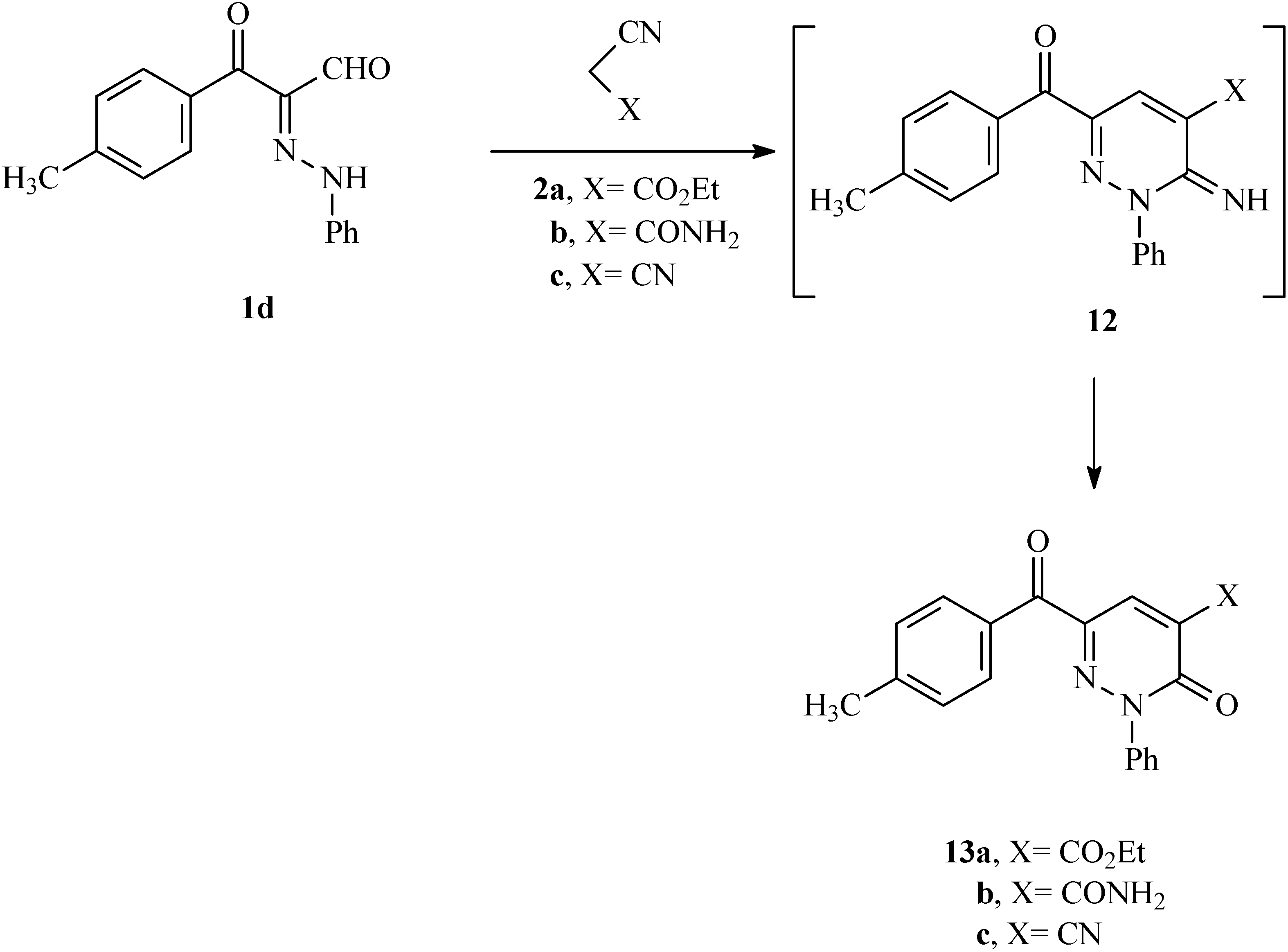
In the final phase of the current effort, we observed that reactions of the 3-oxo-2-(phenylhydrazono)-3-
p-tolylpropanal (
1d) with active methylene nitriles
2a–c in the presence of catalytic amounts of ammonium acetate in acetic acid for 30 min lead to the respective pyridazinones
13a–c, which are likely formed via the intermediacy of the readily hydrolyzed imine analogs
12 (
Scheme 3). The structure of
13a was established by X-ray crystallography (
Figure 3) [
12].
The differences in the reactivity profiles of 1a–c vs. 1d may be a result of the decreased nucleophilicity of the aroyl carbon in the later substance, enabling cyclization of the hydrazone moiety to predominate.
Scheme 1.
Synthesis of 2-hydroxy-6-substituted-5-arylazonicotinates derivatives 6a–c and 2-amino-6-phenyl-5-p-tolylazo-nicotinate 8.
Scheme 1.
Synthesis of 2-hydroxy-6-substituted-5-arylazonicotinates derivatives 6a–c and 2-amino-6-phenyl-5-p-tolylazo-nicotinate 8.
Figure 1.
Ball and stick drawing of 6a.
Figure 1.
Ball and stick drawing of 6a.
Figure 2.
Ball and stick drawing of 8a.
Figure 2.
Ball and stick drawing of 8a.
Scheme 2.
Synthesis of ethyl 6-(4-nitrophenyl)-2-oxo-2,6-dihydropyrido[3,2-c]cinnoline-3-carboxylate (11).
Scheme 2.
Synthesis of ethyl 6-(4-nitrophenyl)-2-oxo-2,6-dihydropyrido[3,2-c]cinnoline-3-carboxylate (11).
Scheme 3.
Synthesis of 6-(4-methylbenzoyl)-3-oxo-2-phenyl-2,3-dihydropyridazine derivatives 13a–c.
Scheme 3.
Synthesis of 6-(4-methylbenzoyl)-3-oxo-2-phenyl-2,3-dihydropyridazine derivatives 13a–c.
Figure 3.
ORTEP drawing of 13a.
Figure 3.
ORTEP drawing of 13a.
Currently, we are utilizing the 2-hydroxy-and 2-aminoazonicotinates dyes as disperse dyes and applying them to polyester fabrics by using high temperature dyeing method. We are also inspecting the biological activity of these disperse dyes against Gram positive bacteria, Gram negative bacteria and yeast.
3. Experimental
3.1. General
Melting points were recorded on a Gallenkamp apparatus. IR spectra were recorded using KBr pellets on a Jasco FTIR-6300 FT-IR spectrophotometer. 1H- and 13C-NMR spectra were recorded on Bruker DPX 400 MHz or AvanceII 600 MHz super-conducting NMR spectrometers (proton spectra measured at 400, 600 MHz and carbon spectra at 100 and 150 MHz, respectively). Mass spectra were measured on a high resolution GC/MS DFS-Thermo. Microanalyses were performed on Elementar-Vario Micro cube Analyzer. X-Ray analyses were performed using a Rigaku Rapid II diffractometer.
3.2. General Procedure for the Preparation of Compounds 6a–c
Independent mixtures of 1a,b (0.01 mol), active methylenenitrile derivatives 2a or 2c or 2d (0.01 mol), and ammonium acetate (0.5 g) in acetic acid (10 mL) were stirred at reflux for 30 min (progress of the reactions was monitored by using TLC using 1:1 ethyl acetate-petroleum ether as eluent). The mixtures were cooled and then poured into ice-water. The solids that formed were collected by using filtration and crystallized from proper solvents to give 6a–c.
2-Hydroxy-6-phenyl-5-p-tolylazonicotinic acid ethyl ester (6a). This compound was obtained as dark brown crystals (83%); mp 180–182 °C; IR (KBr): = 3401 (OH), 1692 (CO) cm−1; 1H-NMR (DMSO-d6): δ = 1.35 (t, 3H, J = 7.2 CH3), 2.50 (s, 3H, CH3), 4.36 (q, 2H, J = 7.2 CH2), 7.32 (d, 2H, J = 8.0 Hz arom-H), 7.48–7.50 (m, 3H, arom-H), 7.60 (d, 2H, J = 8.0 Hz arom-H), 7.78–7.79 (m, 2H, arom-H), 7.98 (s, 1H, OH, D2O exchangeable). 8.54 (s, 1H, arom-H); 13C-NMR (DMSO-d6): δ = 166.5, 161.0, 158.5, 154.5, 150.6, 142.9, 138.3, 136.5, 131.0, 130.8, 129.8, 127.4, 122.3, 105.2, 61.0, 20.9, 14.1; MS, m/z (%), 359 ([M-2]+, 100), 331 (7), 241 (10), 213 (23), 196 (12), 168 (8), 148.1 (17), 115 (14), 91 (35). HRMS: m/z (EI) for C21H19N3O3; calcd. 361.1417; found: 361.1417.
2-Hydroxy-6-(4-nitrophenyl)-5-phenylazonicotinonitrile (6b). This compound was obtained as a dark brown powder (91%); mp 276–278 °C; IR (KBr): = 3446 (OH), 2201 (CN) cm−1; 1H-NMR (DMSO-d6): 7.25–7.62 (m, 6H, arom-H, OH), δ = 7.27 (d, 1H, J = 8.8 Hz arom-H), 8.01–8.08 (m, 1H, arom-H), 8.20 (d, 1H, J = 8.8 Hz arom-H), 8.27–8.36 (m, 2H, arom-H), 13C-NMR (DMSO-d6): δ = 159.8, 152.2, 151.1, 147.6, 141.7, 139.6, 139.0, 131.1, 129.7, 129.2, 126.3, 123.4, 123.1, 114.0, MS, m/z (%), 343 ([M-2]+, 10), 313 (8), 284 (16), 207 (36), 193 (12), 150 (26), 93 (100), 77 (54). Anal. Calcd for C18H11N5O3: C, 62.61; H, 3.21; N, 20.28. Found: C, 62.78; H, 3.28; N, 20.20
2-Hydroxy-6-phenyl-5-p-tolylazothionicotinamide (6c). This compound was obtained as a dark brown powder (77%); mp 276–278 °C; IR (KBr): = 3441 (OH), 3385, 3291 (NH2), 1660 (CO) cm−1; 1H- NMR (DMSO-d6): δ = 2.26 (s, 3H, CH3), 7.02–8.01 (m, 9H, arom-H), 8.57 (s, 1H, OH), 8.76 (s, 1H, arom-H), 12.38 (s, 2H, NH2, D2O exchangeable). 13C-NMR (DMSO-d6): δ = 190.3, 162.2, 158.0, 152.7, 141.7, 138.8, 137.3, 135.8, 131.7, 130.6, 129.4, 129.0, 124.5, 94.0, 20.8; MS, m/z (%), 348 ([M]+, 6), 333 (7), 315 (42), 257 (4), 183 (6), 119 (100), 91 (50), 77 (20). Anal. Calcd for C19H16N4OS: C, 65.50; H, 4.63; N, 16.08; S, 9.20. Found: C, 65.60; H, 4.30; N, 16.43; S, 8.73.
3.3. 2-Amino-6-phenyl-5-p-tolylazonicotinic Acid Ethyl Ester (8)
Independent mixtures of 1a (0.01 mol), ethyl cyanoacetate 2a (0.01 mol), and ammonium acetate (3 g) in acetic acid (10 mL) were stirred at reflux for 30 min (progress of the reactions was monitored by using TLC using 1:1 ethyl acetate-petroleum ether as eluent). The mixtures were cooled and then poured into ice-water. The solids that formed were collected by using filtration and crystallized from proper solvents to give 8 as wine red crystals (83%) (mp 210–212 °C); IR (KBr): = 3402, 3275 (NH2) 1693 (CO) cm−1; 1H-NMR (DMSO-d6): δ = 1.37 (t, 3H, J = 7.2 CH3), 2.50 (s, 3H, CH3), 4.39 (q, 2H, J = 7.2 CH2), 7.31 (d, 2H, J = 8.4 Hz arom-H), 7.47–7.48 (m, 3H, arom-H), 7.98 (s, 2H, NH2, D2O exchangeable). 7.59 (d, 2H, J = 8.4 Hz arom-H), 7.80–7.82 (m, 2H, arom-H), 8.54 (s, 1H, arom-H); 13C-NMR (DMSO-d6): δ = 166.2, 161.3, 159.5, 150.4, 140.6, 137.2, 136.5, 130.8, 129.8, 129.2, 127.4, 127.2, 122.3, 104.9, 61.0, 20.9, 14.1; MS, m/z (%), 359 ([M-1]+, 100), 315 (9), 290 (12), 241 (6), 213 (20), 196 (9), 168 (5), 140 (9), 105 (26), 91 (36), 77 (16). Anal. Calcd for C21H20N4O2: C, 69.98; H, 5.59; N, 15.55. Found: C, 69.99; H, 5.50; N, 15.25.
3.4. Synthesis of Ethyl 6-(4-nitrophenyl)-2-oxo-2,6-dihydropyrido[3,2-c]cinnoline-3-carboxylate (11)
A mixture of 1c (0.01 mol), ethyl cyanoacetate (0.01 mol), and ammonium acetate (0.5 g) in acetic acid (10 mL) was stirred at reflux for 90 min (progress of the reaction was monitored by using TLC using 1:1 ethyl acetate: petroleum ether). The mixture was cooled and then poured into ice-water. The solid that formed was collected by using filtration and crystallized from dioxane to give 11. This compound was obtained as a dark brown powder (62%); mp 148–150 °C; IR (KBr): = 1694 (CO) cm−1; 1H-NMR (DMSO-d6): δ = 1.35 (t, 3H, J = 7.2 CH3), 4.37 (q, 2H, J = 7.8 CH2), 7.51–7.55 (m, 3H, arom-H), 7.78–8.04 (m, 3H, arom-H), 8.28–8.38 (m, 2H, arom-H), 8.62 (s, 1H, arom-H); 13C-NMR (DMSO-d6): δ = 189.4, 166.0, 163.8, 160.2, 155.7, 147.6, 142.2, 136.9, 131.0, 130.2, 129.8, 128.2, 127.4, 125.1, 123.2, 105.3, 61.2, 14.1; MS, m/z (%), 390 ([M]+, 100), 361 (9), 321 (7), 257 (5), 241 (8), 213 (24), 196 (14), 168 (11), 140 (22), 105 (31), 77 (16). Anal. Calcd for C20H14N4O5: C, 61.54; H, 3.62; N, 14.35. Found: 61.61; H, 3.57; N, 14.58.
3.5. General Procedure for the Preparation of Compounds 13a–c
Independent mixtures of 1d (0.01 mol), active methylenenitrile derivatives 2a–c (0.01 mol), and ammonium acetate (0.5 g) in acetic acid (10 mL) were stirred at reflux for 30 min (progress of the reactions was monitored by using TLC using 1:1 ethyl acetate-petroleum ether as eluent). The mixtures were cooled and then poured into ice-water. The solids that formed were collected by using filtration and crystallized from proper solvents to give 13a–c.
6-(4-Methylbenzoyl)-3-oxo-2-phenyl-2,3-dihydropyridazine-4-carboxylic acid ethyl ester (13a). This compound was obtained as buff crystals (55%); mp 108–110 °C; IR (KBr): = 1746 (CO) cm−1; 1H-NMR (DMSO-d6): δ = 1.32 (t, 3H, J = 7.2 CH3), 2.37 (s, 3H, CH3), 4.34 (q, 2H, J = 7.2 CH2), 7.33 (d, 2H, J = 8.0 Hz arom-H), 7.45–7.49 (m, 1H, arom-H), 7.51–7.55 (m, 2H, arom-H), 7.59–7.63 (m, 2H, arom-H), 7.93 (d, 2H, J = 8.4 Hz arom-H), 8.31 (s, 1H, arom-H); 13C-NMR (DMSO-d6): δ = 188.3, 162.6, 155.9, 144.4, 141.6, 141.1, 132.3, 132.1, 131.0, 130.7, 129.2, 128.8, 126.0, 115.2, 61.8, 21.2, 13.9; MS, m/z (%), 362 ([M]+, 48), 315 (18), 290 (16), 261 (6), 182 (12), 119 (100), 91 (46), 77 (26). HRMS: m/z (EI) for C21H18N2O4; calcd. 362.1261; found: 362.1261.
6-(4-Methylbenzoyl)-3-oxo-2-phenyl-2,3-dihydropyridazine-4-carboxylic acid amide (13b). This compound was obtained as a brown powder (52%); mp 243–245 °C; IR (KBr): = 3343, 3156 (NH2), 1698 (CO) cm−1; 1H-NMR (DMSO-d6): δ = 1.37 (s, 3H, CH3), 7.33 (d, 2H, J = 8.0 Hz arom-H), 7.47–7.56 (m, 3H, arom-H), 7.63 (d, 2H, J = 7.8 Hz arom-H), 7.93 (d, 2H, J = 8.0 Hz arom-H), 8.27 (s, 2H, NH2, D2O exchangeable). 8.56 (s, 1H, arom-H); 13C-NMR (DMSO-d6): δ = 188.4, 162.1, 159.5, 144.1, 142.6, 141.1, 132.5, 132.3, 130.9, 130.7, 129.0, 128.9, 128.8, 126.1, 21.2; MS, m/z (%), 333 ([M]+, 100), 318 (15), 261 (5), 214 (5), 182 (9), 119 (100), 91 (36), 77 (20). Anal. Calcd for C19H15N3O3: C, 68.46; H, 4.54; N, 12.61. Found: 68.93; H, 4.56; N, 13.02.
6-(4-Methylbenzoyl)-3-oxo-2-phenyl-2,3-dihydropyridazine-4-carbonitrile (13c). This compound was obtained as brown powder crystals (64%); mp 186–188 °C; IR (KBr): 2209 (CN), 1656 (CO) cm−1; 1H-NMR (DMSO-d6): δ = 2.32 (s, 3H, CH3), 7.25 (d, 2H, J = 8.0 Hz arom-H), 7.46–7.58 (m, 5H, arom-H), 7.85 (d, 2H, J = 8.0 Hz arom-H), 8.60 (s, 1H, arom-H); 13C-NMR (DMSO-d6): δ = 187.6, 162.0, 156.1, 144.4, 141.6, 140.5, 138.6, 132.0, 130.7, 130.2, 126.1, 125.6, 115.2, 113.7, 21.0, MS, m/z (%), 315 ([M]+, 60), 257 (6), 211 (8), 183 (14), 119 (100), 91 (42), 77 (20). HRMS: m/z (EI) for C19H13N3O2; calcd. 315.1001; found: 315.1001.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





