New Trifluoromethyl Triazolopyrimidines as Anti-Plasmodium falciparum Agents

Abstract
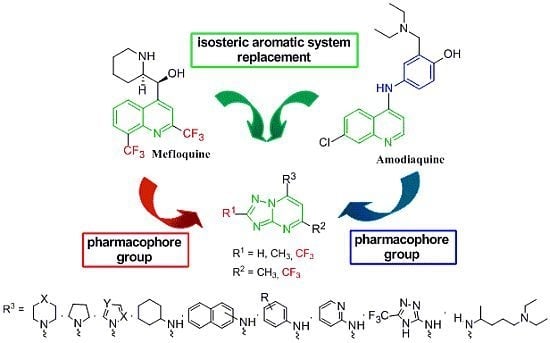
:
1. Introduction


2. Results and Discussion
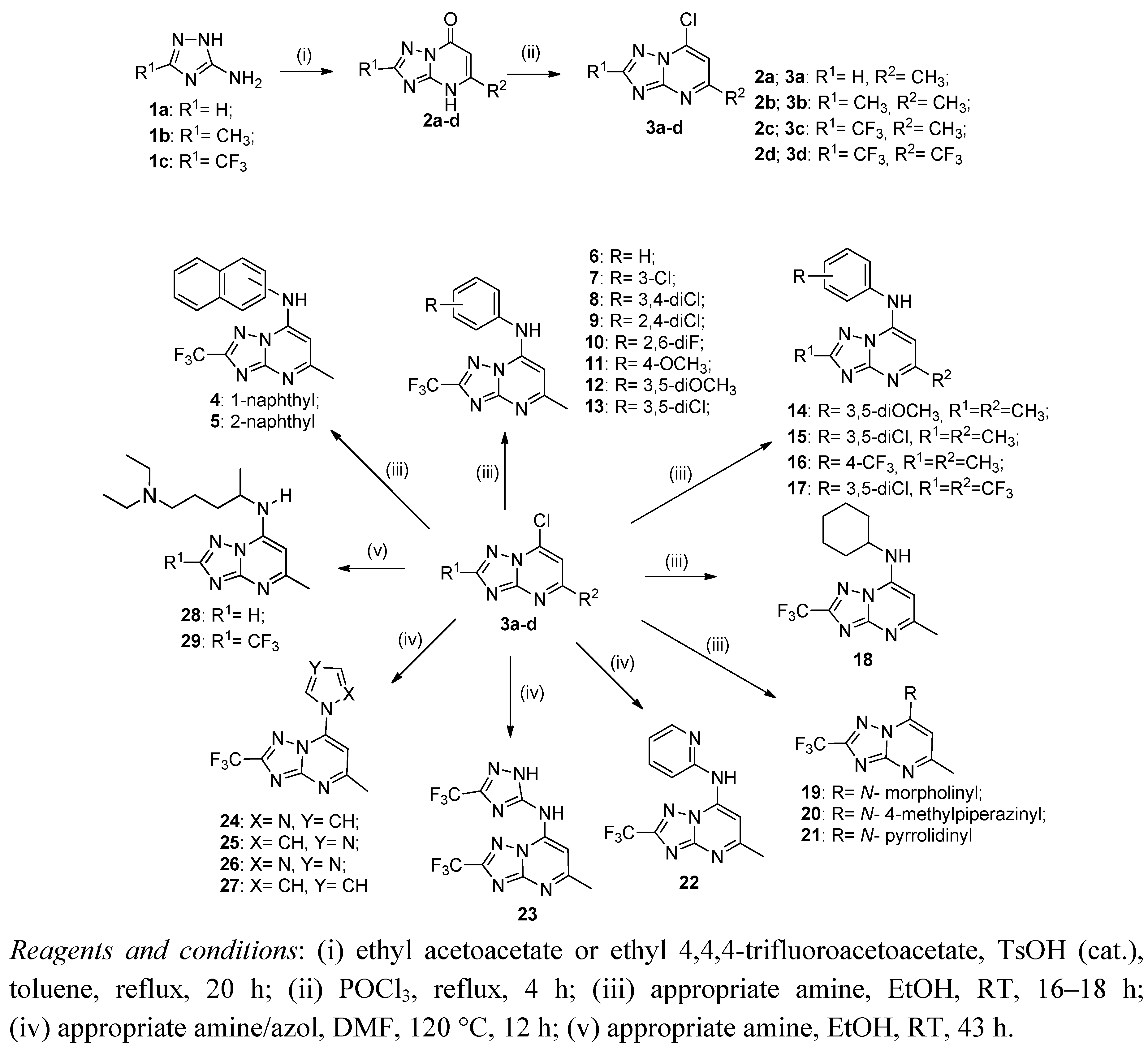
2.1. Synthesis of Compounds

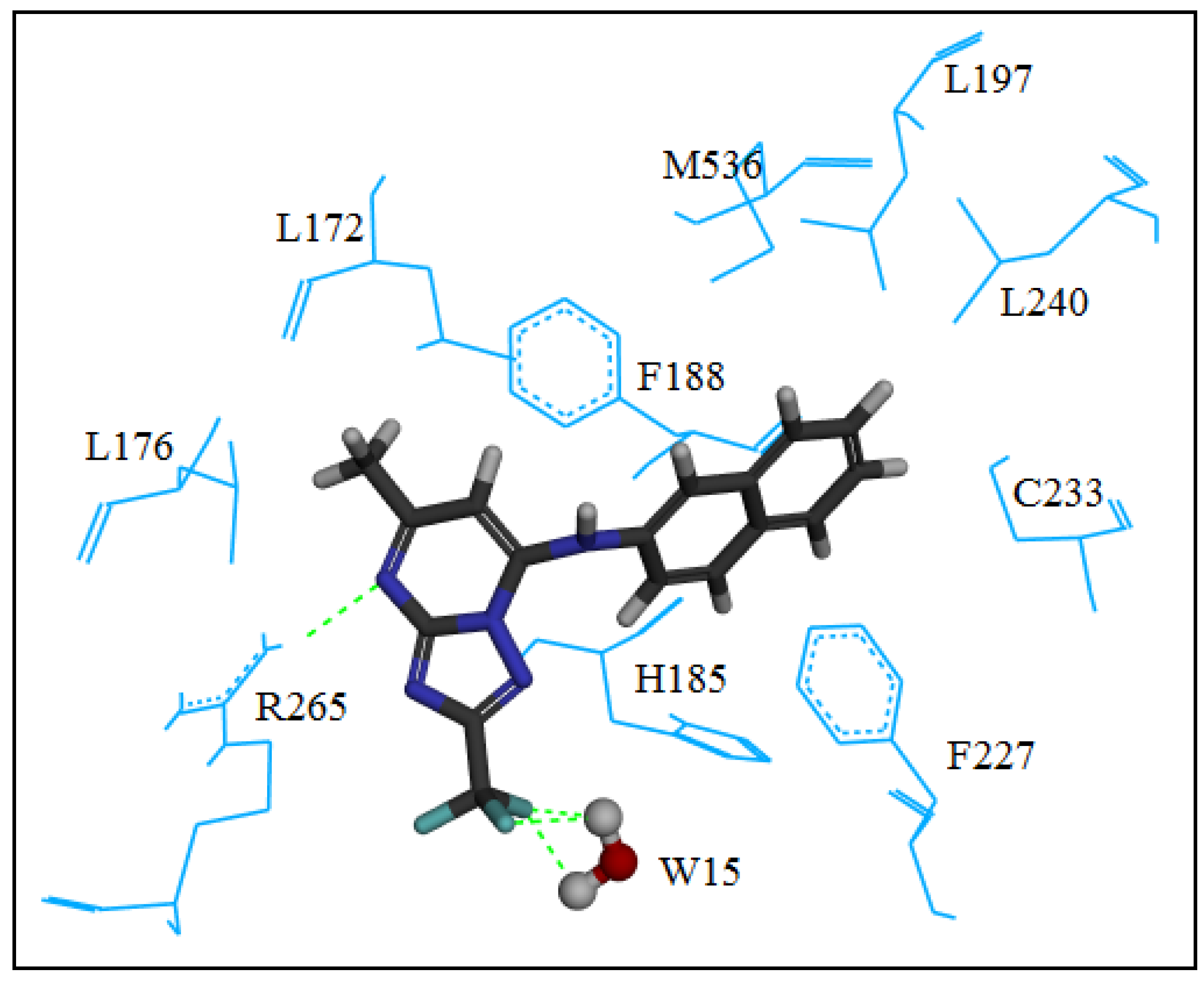
2.2. Molecular Modeling

2.3. Continuous Cultures and in Vitro Assays with P. falciparum-Infected Erythrocytes
2.4. Cell Cultures and Cytotoxicity Tests

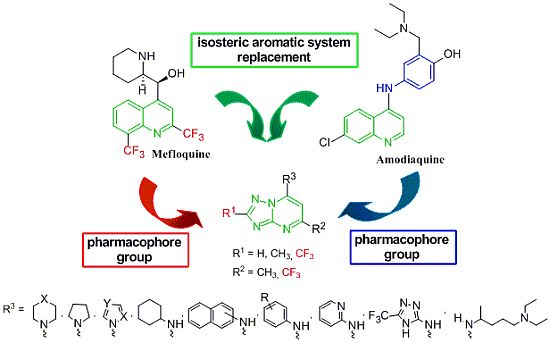{kind=link}
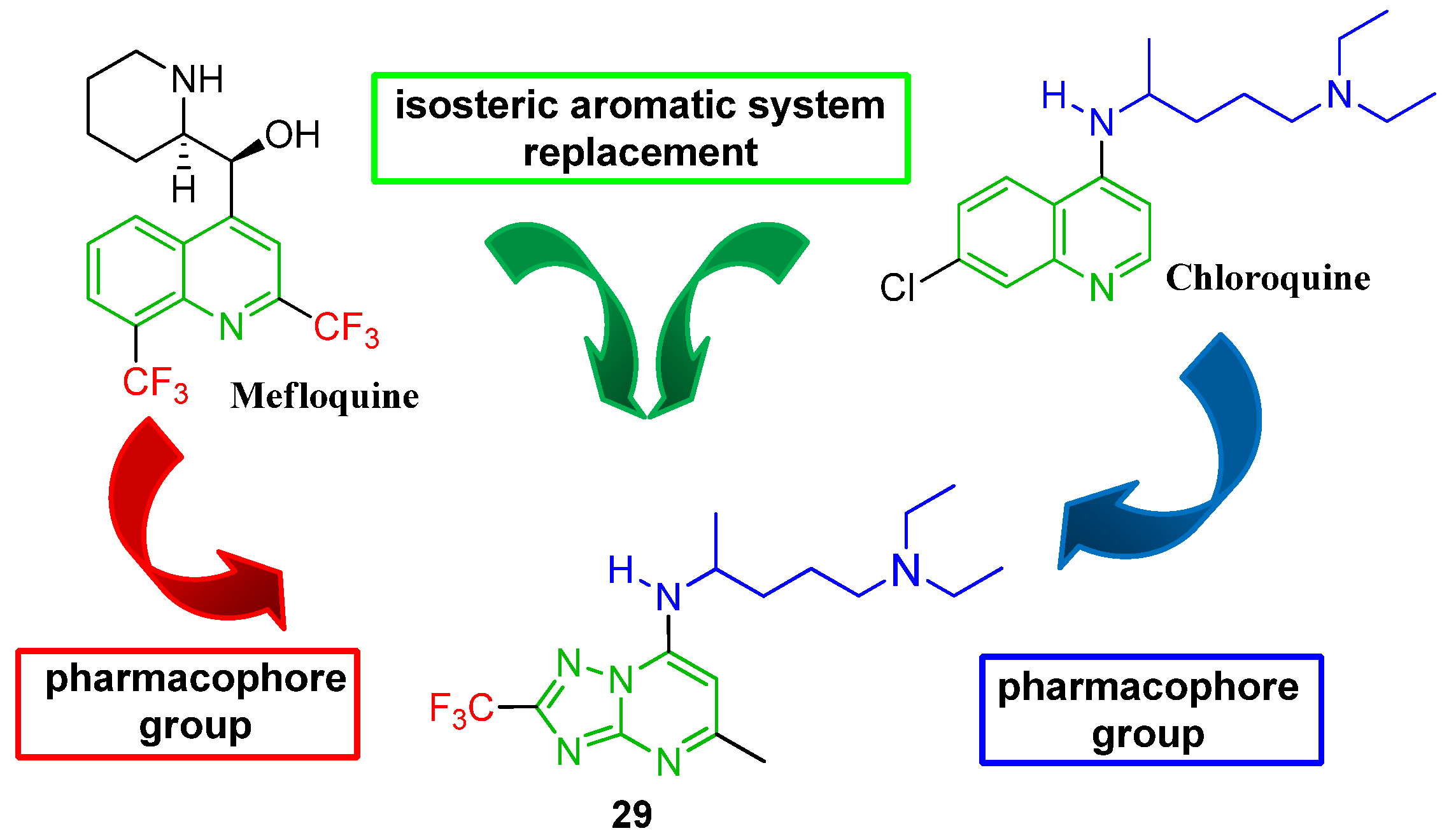{kind=link}
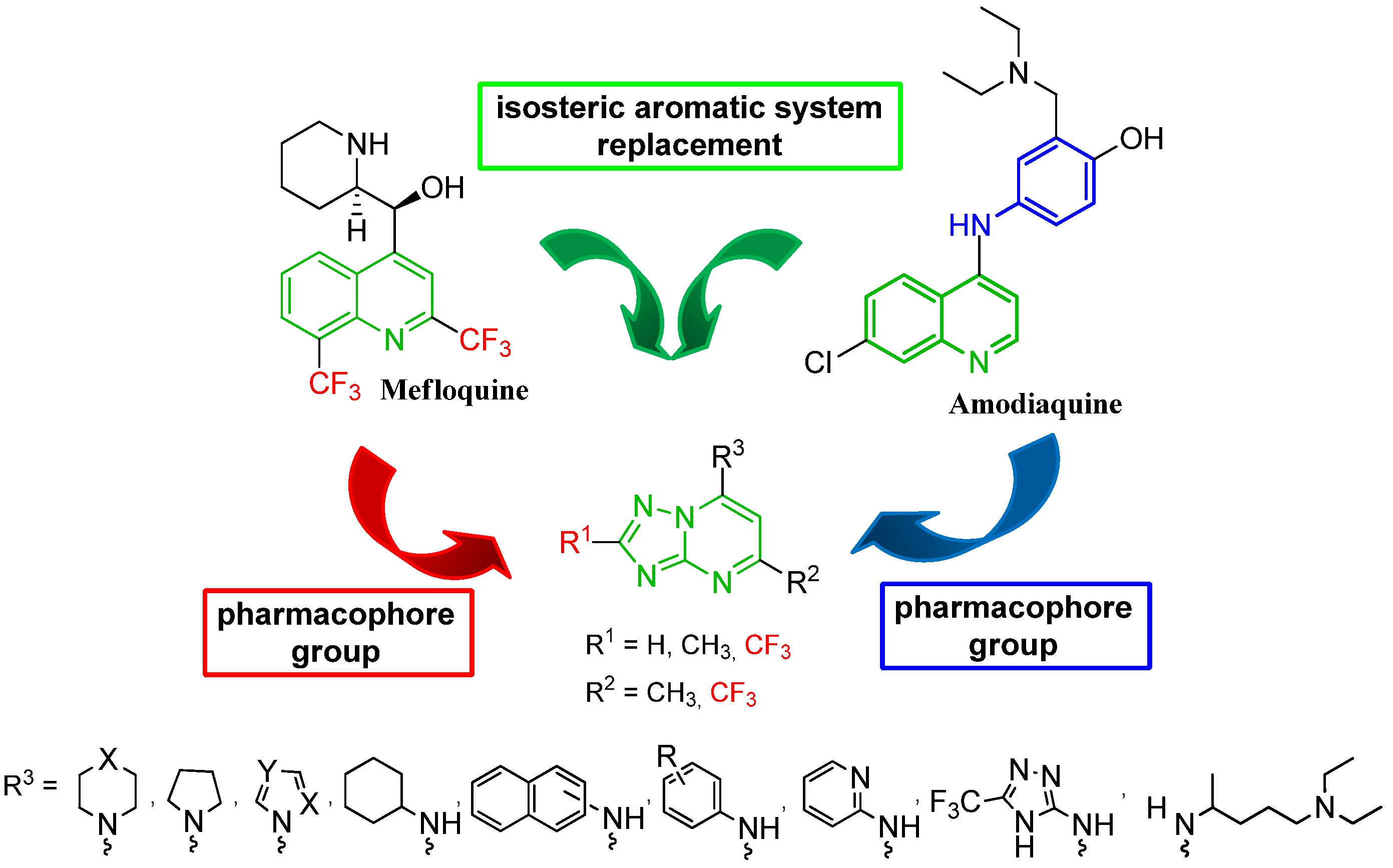{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (µM) * Plasmodium Falciparum | MDL50 HepG2 (µM) | SI MDL50/IC50 | |
|---|---|---|---|---|
| Anti-HRP2 | Hypoxanthine | |||
| 4 | 2.2 | Nd | 326 | 148 |
| 5 | 0.023 ± 0.002 | Nd | 425 | 18,478 |
| 6 | 3 ± 2 | 10.2 ± 5 | >682 | >227 |
| 7 | 1.83 ± 1.10 | 1.22 | 373 | 203 |
| 8 | 0.55 ± 0.66 | 0.11 ± 0.05 | >552 | 1,003 |
| 9 | 2.7 ± 2.21 | 3.32 ± 1.10 | 320 | 118 |
| 10 | >69.9 | >148.9 | 337 | Inactive |
| 11 | 0.4 ± 0.09 | 1.5 ± 1.2 | >619 | >1,547 |
| 12 | 2.5 ± 0.05 | 0.36 ± 0.1 | 498 | >199 |
| 13 | 1.47 ± 0.11 | 0.83 ± 0.58 | 320 | 218 |
| 14 | 3.84 ± 0.40 | 15.05 ± 10.03 | 515 | 134 |
| 15 | >162.3 | 149.3 | 415 | Inactive |
| 16 | 0.3 | 0.4 ± 0.06 | 446 | 1486 |
| 17 | 12.31 | 23.42 | 269 | Inactive |
| 18 | 8.69 ± 0.46 | 10.03 | 515 | 64 |
| 19 | >174.0 | >174.0 | >697 | Inactive |
| 20 | 39 ± 17 | 57 ± 13 | >666 | Inactive |
| 21 | >184.5 | >184.5 | >738 | Inactive |
| 22 | >156.4 | >170.0 | 394 | Inactive |
| 23 | 36.93 ± 11.36 | 31.25 ± 17.04 | <71 | Inactive |
| 24 | 2.1 | Nd | >746 | >355 |
| 25 | 21 ± 2 | 15 ± 0.7 | <93 | Inactive |
| 26 | 29.73 | 33.45 ± 14.86 | <93 | Inactive |
| 27 | 97.37 ± 22.47 | 78.65 ± 7.49 | >749 | Inactive |
| 28 | >172.4 | >172.4 | >689 | Inactive |
| 29 | 73 ± 17 | 39.1 | >558 | Inactive |
| Chloroquine | 0.22 | 0.23 | 490 | 4,200 |
3. Experimental
3.1. General Procedure for Preparing [1,2,4]Triazolo[1,5-a]pyrimidin-7(4H)-ones (2a–d)
3.2. General Procedure for Preparing 7-Chloro[1,2,4]triazolo[1,5-a]pyrimidines 3a–d
3.3. General Procedure for Preparing 5-Methyl-7-aryl/cycloalkylamine[1,2,4]triazolo[1,5-a]pyrimidines 4–21
3.4. General Procedure for Preparing 5-Methyl-7-substituted[1,2,4]triazolo[1,5-a]pyrimidines 22–27
3.5. General Procedure for Preparing 5-Methyl-7-N'-(N,N-diethylpentane-1,4-diamine)[1,2,4]triazolo [1,5-a]pyrimidines 28, 29
4. Conclusions
Acknowledgments
References
- 10 Facts on Malaria. Available online: http://www.who.int/features/factfiles/malaria/en/index.html (accessed on 22 March 2011).
- World Health Organization (WHO). Global report on antimalarial drug efficacy and drug resistance: 2000–2010. 2010. Available online: http://www.who.int/malaria/en (accessed on 22 March 2011).
- Oliveira-Ferreira, J.; Lacerda, M.V.G.; Brasil, P.; Ladislau, J.L.B.; Tauil, P.L.; Daniel-Ribeiro, C.T. Malaria in Brazil: An overview. Malaria J. 2010, 9, 115–130. [Google Scholar] [CrossRef]
- Agnandji, S.T.; Lell, B.; Soulanoudjingar, S.S.; Fernandes, J.F.; Abossolo, B.P.; Conzelmann, C.; Methogo, B.G.; Doucka, Y.; Flamen, A.; Mordmüller, B.; et al. First results of phase 3 trial of RTS,S/AS01 malaria vaccine in African children. N. Engl. J. Med. 2011, 365, 1863–1875. [Google Scholar]
- Krettli, A.U.; Adebayo, J.O.; Krettli, L.G. Testing of natural products and synthetic molecules aiming at new antimalarials. Curr. Drug Targets 2009, 10, 261–270. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Welch, J.T.; Eswarakrishnan, S. Fluorine in Bioorganic Chemistry; John Wiley & Sons: New York, NY, USA, 1991. [Google Scholar]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; John Wiley & Sons: Chichester, UK, 2009. [Google Scholar]
- Kirk, K.L. Fluorination in medicinal chemistry: Methods, strategies, and recent developments. Org. Process. Res. Dev. 2008, 12, 305–321. [Google Scholar] [CrossRef]
- Park, B.K.; Kitteringham, N.R.; O’Neill, P.M. Metabolism of fluorine containing drugs. Ann. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. [Google Scholar] [CrossRef]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Boechat, N.; Bastos, M.M. Trifluoromethylation of carbonyl compounds. Curr. Org. Synth. 2010, 7, 403–413. [Google Scholar] [CrossRef]
- Milner, E.; McCalmont, W.; Bhonsle, J.; Caridha, D.; Cobar, J.; Gardner, S.; Gerena, L.; Goodine, D.; Lanteri, C.; Melendez, V.; et al. Anti-malarial activity of a non-piperidine library of next-generation quinoline Methanols. Malaria J. 2010, 9, 51–61. [Google Scholar] [CrossRef]
- Biot, C.; Chibale, K. Novel approaches to antimalarial drug discovery. Infect. Dis. Drug Targets 2006, 6, 173–204. [Google Scholar] [CrossRef]
- Blackie, M.A.L.; Yardley, V.; Chibale, K. Synthesis and evaluation of phenylequine for antimalarial activity in vitro and in vivo. Bioorg. Med. Chem. Lett. 2010, 20, 1078–1080. [Google Scholar] [CrossRef]
- Iwaniuk, D.P.; Whetmore, E.D.; Rosa, N.; Ekoue-Kovi, K.; Alumasa, J.; Dios, A.C.; Roepe, P.D.; Wolf, C. Synthesis and antimalarial activity of new chloroquine analogues carrying a multifunctional linear side chain. Bioorg. Med. Chem. 2009, 17, 6560–6566. [Google Scholar] [CrossRef]
- Delarue-Cochin, S.; Paunescu, E.; Mães, L.; Mouray, E.; Sergheraert, C.; Grellier, P.; Melnyk, P. Synthesis and antimalarial activity of new analogues of amodiaquine. Eur. J. Med. Chem. 2008, 43, 252–260. [Google Scholar] [CrossRef]
- Casagrande, M.; Basilico, N.; Parapini, S.; Romeo, S.; Taramelli, D.; Sparatore, A. Novel amodiaquine congeners as potent antimalarial agents. Bioorg. Med. Chem. 2008, 16, 6813–6823. [Google Scholar] [CrossRef]
- Guglielmo, S.; Bertinaria, M.; Rolando, B.; Crosetti, M.; Fruttero, R.; Yardley, V.; Croft, S.L.; Gasco, A. A new series of amodiaquine analogues modified in the basic side chain with in vitro antileishmanial and antiplasmodial activity. Eur. J. Med. Chem. 2009, 44, 5071–5079. [Google Scholar]
- Jonet, A.; Dassonville-Klimpt, A.; da Nascimento, S.; Leger, J.; Guillon, J.; Sonnet, P. First enantioselective synthesis of 4-aminoalcohol quinoline derivatives through a regioselective SN2 epoxide opening mechanism. Tetrahedron Asymmetry 2011, 22, 138–148. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, Q.; Gudise, C.; Meng, L.; Wei, L.; Smitha, E.; Kong, Y. Synthesis and evaluation of naphthyridine compounds as antimalarial agents. Bioorg. Med. Chem. Lett. 2007, 17, 6101–6106. [Google Scholar] [CrossRef]
- Mello, H.; Echevarria, A.; Bernardino, A.M.; Canto-Cavalheiro, M.; Leon, L.L. Antileishmanial pyrazolopyridine derivatives: Synthesis and structure-activity relationship analysis. J. Med. Chem. 2004, 47, 5427–5432. [Google Scholar] [CrossRef]
- Dutra, K.D.B. Síntese e Avaliação Antimalárica de Novos Análogos Triazolopirimidínicos da Cloroquina. MSc. Thesis, Departamento de Química Orgânica, Universidade Federal Fluminense, Niterói, RJ, Brazil, 2004. [Google Scholar]
- Boechat, N.; Dutra, K.D.B.; Valverde, A.L.; Wardell, S.; Low, J.N.; Glidewell, C. Hydrogen-bonded chains in 5-methyl-2-trifluoromethyl-1,2,4-triazolo-[1,5-a]pyrimidin-7(4H)-one and hydrogen- bonded chains of rings in 5-amino-3-trifluoromethyl-1H-1,2,4-triazolo-5-methyl-2-trifluromethyl-1,2, 4-triazolo[1,5-a]pyrimidin-7(4H)-one (1/1) the co-crystal of a reaction product and one of its precursors. Acta Crystallogr. Sect. C-Cryst. Struct. Commun. 2004, 60, 733–736. [Google Scholar] [CrossRef]
- Gujjar, R.; Marwaha, A.; El Mazouni, F.; White, J.; White, K.L.; Creason, S.; Shackleford, D.M.; Baldwin, J.; Charman, W.N.; Buckner, F.S.; et al. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J. Med. Chem. 2009, 52, 1864–1872. [Google Scholar] [CrossRef]
- Phillips, M.A.; Gujjar, R.; Malmquist, N.A.; White, J.; El Mazouni, F.; Baldwin, J.; Rathod, P.K. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium Falciparum. J. Med. Chem. 2008, 51, 3649–3653. [Google Scholar] [CrossRef]
- Phillips, M.A.; Rathod, P.K. Plasmodium dihydroorotate dehydrogenase: A promising target for novel anti-malarial chemotherapy. Infect. Dis. Drug Targets 2010, 10, 226–239. [Google Scholar]
- Gujjar, R.; El Mazouni, F.; White, K.L.; White, J.; Creason, S.; Shackleford, D.M.; Deng, X.; Charman, W.N.; Bathurst, I.; Burrows, J.; et al. Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J. Med. Chem. 2011, 54, 3935–3949. [Google Scholar]
- Coteron, J.M.; Marco, M.; Esquivias, J.; Deng, X.; White, K.L.; White, J.; Koltun, M.; El Mazouni, F.; Kokkonda, S.; Katneni, K.; et al. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 2011, 54, 5540–5561. [Google Scholar]
- Zohdi, H.F. Reactions with 3-amino-5-(trifluoromethyl)-1,2,4-triazole: A simple route to fluorinated polysubstituted triazolo[1,5-a]pyrimidine and triazolo[5,1-c]triazine derivatives. J. Chem. Res. 1997, 392–393. [Google Scholar] [CrossRef]
- Erkin, A.V.; Krutikov, V.I. Formation, structure and heterocyclization of aminoguanidine and ethyl acetoacetate condensation products. Russ. J. Gen. Chem. 2009, 79, 1204–1209. [Google Scholar] [CrossRef]
- Deng, X.; Gujjar, R.; Mazouni, F.E.; Kaminsky, W.; Malmquist, N.A.; Goldsmith, E.J.; Rathod, P.K.; Phillips, M.A. Structural platicity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J. Biol. Chem. 2009, 284, 26999–27009. [Google Scholar]
- Tripos 1699 South Hanley Road, St. Louis, MO 63144–2319, USA.
- Molegro ApS. C. F. Moellers Alle 8, Building 1110, DK-8000 Aarhus C, Denmark.
- Oduola, A.M.; Weatherly, N.F.; Bowdre, J.H.; Desjardins, R.E. Plasmodium falciparum: Cloning by single-erythrocyte micromanipulation and heterogeneity in vitro. Exp. Parasitol. 1988, 66, 86–95. [Google Scholar] [CrossRef]
- Trager, W.; Jensen, J. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. [Google Scholar]
- Lambros, C.; Vanderberg, J. Synchronization of plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 1979, 65, 418–420. [Google Scholar] [CrossRef]
- Desjardins, R.; Canfield, C.; Haynes, J.; Chulay, J. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. [Google Scholar] [CrossRef]
- Noedl, H.; Wongsrichanalai, C.; Miller, R.; Myint, K.; Looareesuwan, S.; Sukthana, Y.; Wongchotigul, V.; Kollaritsch, H.; Wiedermann, G.; Wernsdorfer, W. Plasmodium falciparum: Effect of anti-malarial drugs on the production and secretion characteristics of histidine-rich protein II. Exp. Parasitol. 2002, 102, 157–163. [Google Scholar] [CrossRef]
- Carvalho, L.; Brandão, M.; Santos-Filho, D.; Lopes, J.; Krettli, A. Antimalarial activity of crude extracts from brazilian plants studied in vivo in plasmodium berghei-infected mice and in vitro against plasmodium falciparum in culture. Braz. J. Med. Biol. Res. 1991, 24, 1113–1123. [Google Scholar]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Madureira, A.M.; Martins, A.P.; Gomes, M.; Paiva, J.; Cunha, A.P.; Rosário, V. Antimalarial activity of medicinal plants used in traditional medicine in S. Tome and Principe Island. J. Ethnopharmacol. 2002, 81, 23–29. [Google Scholar] [CrossRef]
- Na-Bangchang, K.; Karbwang, J. Current status of malaria chemotherapy and the role of pharmacology in antimalarial drug research and development. Fundam. Clin. Pharmacol. 2009, 23, 387–409. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Boechat, N.; Pinheiro, L.C.S.; Silva, T.S.; Aguiar, A.C.C.; Carvalho, A.S.; Bastos, M.M.; Costa, C.C.P.; Pinheiro, S.; Pinto, A.C.; Mendonça, J.S.; et al. New Trifluoromethyl Triazolopyrimidines as Anti-Plasmodium falciparum Agents. Molecules 2012, 17, 8285-8302. https://doi.org/10.3390/molecules17078285
Boechat N, Pinheiro LCS, Silva TS, Aguiar ACC, Carvalho AS, Bastos MM, Costa CCP, Pinheiro S, Pinto AC, Mendonça JS, et al. New Trifluoromethyl Triazolopyrimidines as Anti-Plasmodium falciparum Agents. Molecules. 2012; 17(7):8285-8302. https://doi.org/10.3390/molecules17078285
Chicago/Turabian StyleBoechat, Núbia, Luiz C. S. Pinheiro, Thiago S. Silva, Anna C. C. Aguiar, Alcione S. Carvalho, Monica M. Bastos, Carolina C. P. Costa, Sergio Pinheiro, Angelo C. Pinto, Jorge S. Mendonça, and et al. 2012. "New Trifluoromethyl Triazolopyrimidines as Anti-Plasmodium falciparum Agents" Molecules 17, no. 7: 8285-8302. https://doi.org/10.3390/molecules17078285
APA StyleBoechat, N., Pinheiro, L. C. S., Silva, T. S., Aguiar, A. C. C., Carvalho, A. S., Bastos, M. M., Costa, C. C. P., Pinheiro, S., Pinto, A. C., Mendonça, J. S., Dutra, K. D. B., Valverde, A. L., Santos-Filho, O. A., Ceravolo, I. P., & Krettli, A. U. (2012). New Trifluoromethyl Triazolopyrimidines as Anti-Plasmodium falciparum Agents. Molecules, 17(7), 8285-8302. https://doi.org/10.3390/molecules17078285