Cytotoxic and Antioxidant Compoundsfrom the Stem Bark of Goniothalamus tapisoides Mat Salleh

 ,
,
Abstract
:1. Introduction
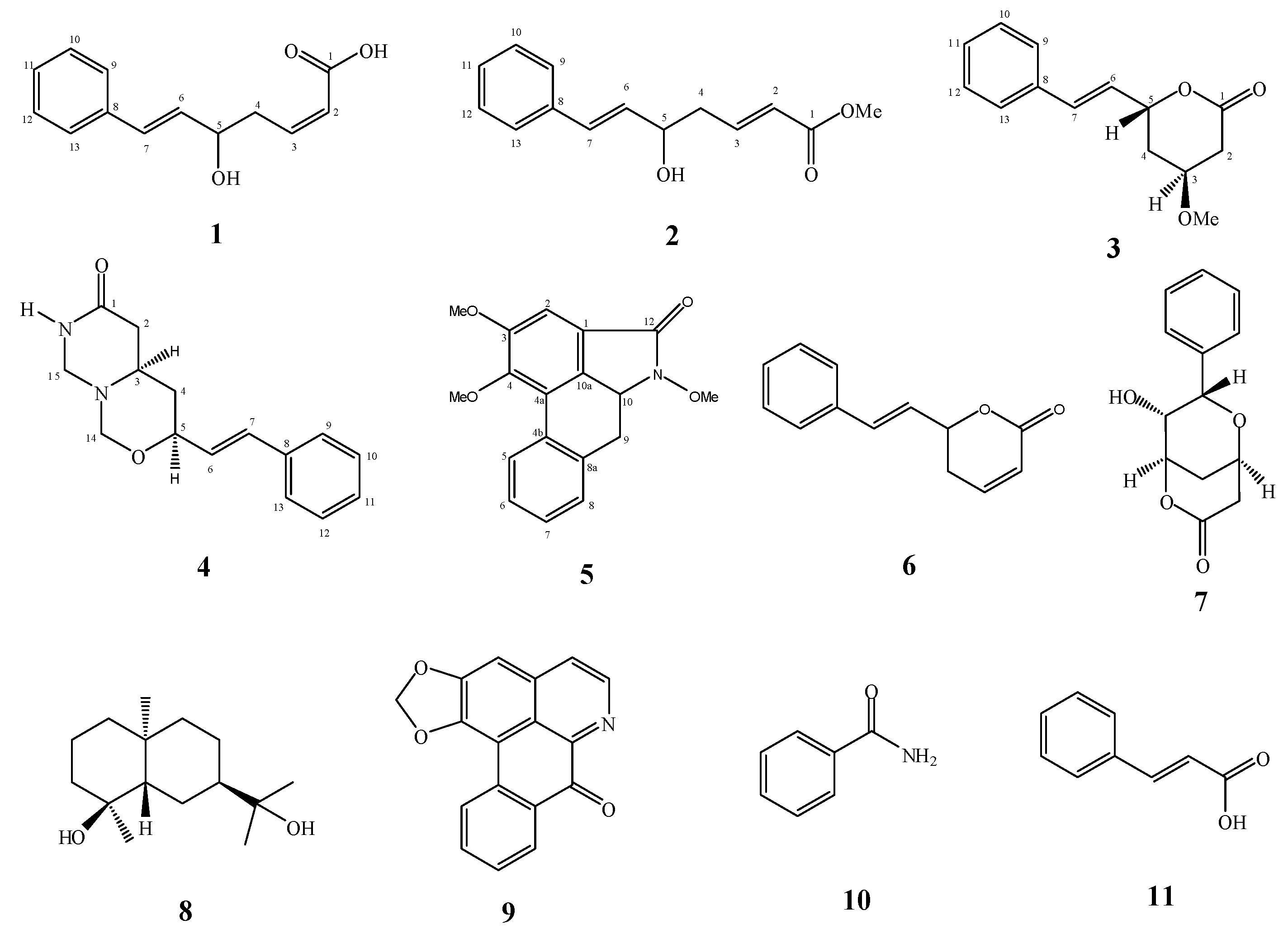
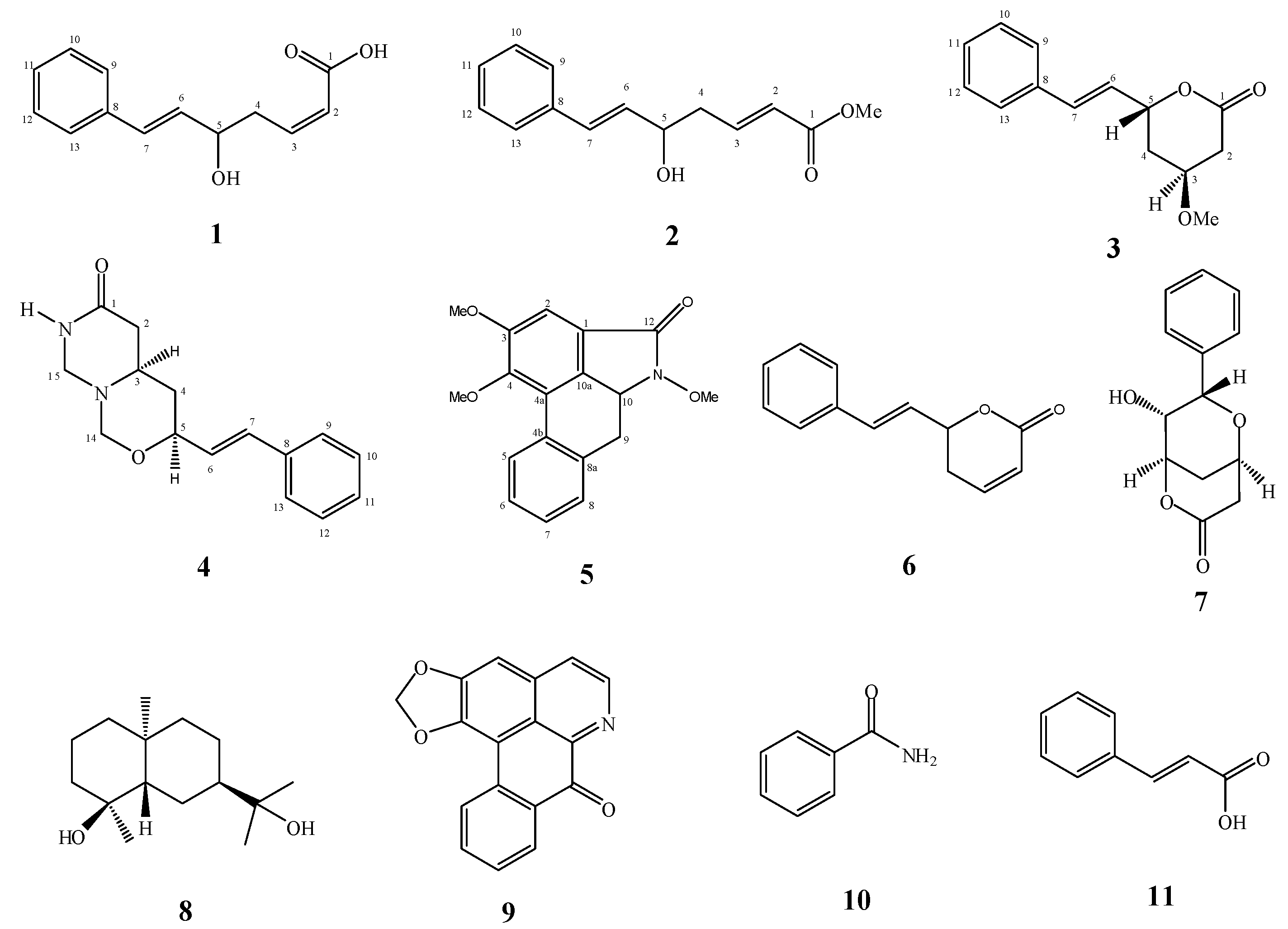
2. Results and Discussion
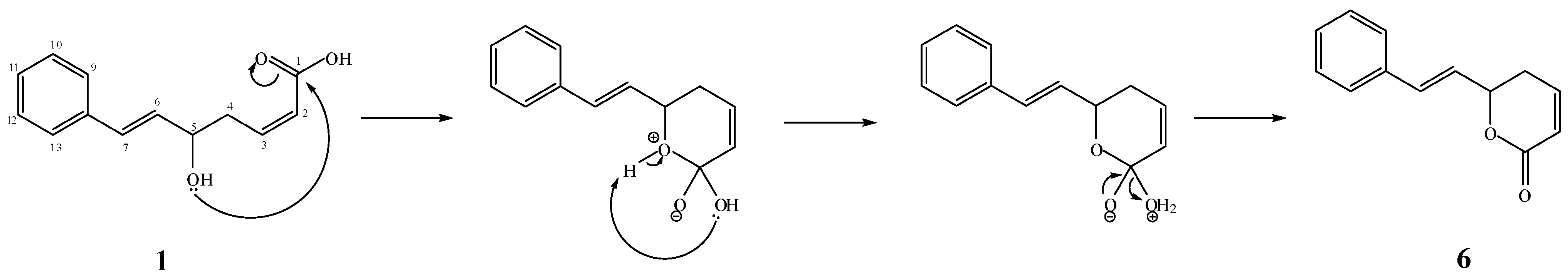
2.1. Isolation and Chemistry
 +2.5° (c 0.0239, MeOH). A molecular formula of C13H14O3 was deduced from the ESI-TOF-MS spectrum, that showed a strong fragmentation peak at m/z 199.1020 [M−H−H2O]− (calc. 199.0759) corresponding to the loss of a water molecule, thus indicating the presence of a hydroxyl group. This is in agreement with the 13C-NMR and HSQC spectra which confirmed the presence of thirteen carbons. The UV spectrum (λmax 206 and 251 nm) suggested the presence of a phenyl group [11]. The IR spectrum showed absorptions of hydroxyl (νmax 3344 cm−1) and carbonyl (νmax 1668 cm−1) functionalities.
+2.5° (c 0.0239, MeOH). A molecular formula of C13H14O3 was deduced from the ESI-TOF-MS spectrum, that showed a strong fragmentation peak at m/z 199.1020 [M−H−H2O]− (calc. 199.0759) corresponding to the loss of a water molecule, thus indicating the presence of a hydroxyl group. This is in agreement with the 13C-NMR and HSQC spectra which confirmed the presence of thirteen carbons. The UV spectrum (λmax 206 and 251 nm) suggested the presence of a phenyl group [11]. The IR spectrum showed absorptions of hydroxyl (νmax 3344 cm−1) and carbonyl (νmax 1668 cm−1) functionalities. 
{kind=link}
{kind=link}
{kind=link}
| Atom | 1 | 2 | ||||
|---|---|---|---|---|---|---|
| No. | δ 13C | δ 1H | HMBC | δ 13C | δ 1H | HMBC |
| 1 | 169.6 | - | 166.8 | - | ||
| 2 | 125.3 | 5.96 (1H, d, J = 11.5) | C-1, C-3, C-4 | 123.9 | 5.94 (1H, d, J = 15.6) | C-1, C-4 |
| 3 | 140.6 | 6.12 (1H, dt, J = 8.6, 11.5) | C-1, C-4, C-5 | 144.7 | 7.00 (1H, m) | C-1, C-4 |
| 4α | 36.6 | 2.76 (1H, m) | C-2, C-3, C-5, C-6 | 40.2 | 2.54 (2H, m) | C-2, C-3, C-5, C-6 |
| 4β | 2.81 (1H, m) | |||||
| 5 | 71.5 | 4.41 (1H, m) | C-3, C-4, C-7 | 71.6 | 4.44 (1H, dd, J = 6.6, 13.3) | C-3, C-7 |
| 6 | 131.9 | 6.20 (1H, dd, J = 16.0, 6.7) | C-4, C-5, C-8 | 130.9 | 6.22 (1H, dd, J = 16.0, 6.6) | C-5, C-8 |
| 7 | 129.9 | 6.59 (1H, d, J = 16.0) | C-5, C-8, C-9, C-13 | 131.3 | 6.61 (1H, d, J = 16.0) | C-5, C-9, C-13 |
| 8 | 136.7 | - | 136.3 | - | ||
| 9,13 | 126.5 | 7.19–7.34 (m) | 126.5 | 7.24–7.38 (m) | ||
| 10,12 | 128.6 | 7.19–7.34 (m) | C-8 | 128.6 | 7.24–7.38 (m) | C-8 |
| 11 | 127.6 | 7.19–7.34 (m) | 128.0 | 7.24–7.38 (m) | ||
| 1-OH | - | 6.27 (OH, br s) | C-2 | - | - | |
| 5-OH | - | 6.57 (OH, br s) | C-5 | - | - | |
| 1-OMe | - | - | 51.6 | 3.72 (3H, s) | C-1 | |
 +2.4° (c 0.0082, CH2Cl2). Its molecular formula, C14H16O3, was deduced from the ESI-TOF-MS (m/z 231.1201, [M−H]−; calc. 231.1021). The IR spectrum showed a strong absorptionfora conjugated carbonyl group of an ester at 1718 cm−1, while the UV absorption bands at 207 and 251 nmindicated the presence of a phenyl group [11].
+2.4° (c 0.0082, CH2Cl2). Its molecular formula, C14H16O3, was deduced from the ESI-TOF-MS (m/z 231.1201, [M−H]−; calc. 231.1021). The IR spectrum showed a strong absorptionfora conjugated carbonyl group of an ester at 1718 cm−1, while the UV absorption bands at 207 and 251 nmindicated the presence of a phenyl group [11]. −4.9° (c 0.0122, CH2Cl2). The ESI-TOF-MS spectrum gave a prominent peak at m/z 231.1077, [M−H]−; (calc. 231.1021), corresponding to the molecular formula C14H16O3. The IR spectrum showed a C=O stretching absorption bands at 1731 cm−1 and C-O stretching ones at 1241 and 1090 cm−1. The UV absorptions at 206 and 251 nmsuggested the existence of a phenyl group [11].
−4.9° (c 0.0122, CH2Cl2). The ESI-TOF-MS spectrum gave a prominent peak at m/z 231.1077, [M−H]−; (calc. 231.1021), corresponding to the molecular formula C14H16O3. The IR spectrum showed a C=O stretching absorption bands at 1731 cm−1 and C-O stretching ones at 1241 and 1090 cm−1. The UV absorptions at 206 and 251 nmsuggested the existence of a phenyl group [11].| Atom no. | δ 13C | δ 1H | HMBC | Atom no. | δ 13C | δ 1H | HMBC |
|---|---|---|---|---|---|---|---|
| 1 | 169.7 | - | 7 | 132.5 | 6.68 (1H, d, J = 16.0) | C-5, C-9, C-13 | |
| 2 | 35.7 | 2.73 (2H, td, J = 4.3, 1.4) | C-1 | 8 | 136.0 | - | |
| 3 | 71.4 | 3.82 (1H, m) | 9,13 | 126.7 | 7.24–7.37 (m) | ||
| 4α | 33.7 | 1.87 (1H, ddd, J = 14.8, 11.0, 3.5) | 10,12 | 128.8 | 7.24–7.37 (m) | ||
| 4β | 2.18 (1H, m) | 11 | 128.3 | 7.24–7.37 (m) | |||
| 5 | 76.2 | 5.20 (1H, ddd, J = 11.0, 6.4, 3.5) | C-3 | 3-OMe | 56.3 | 3.36 (3H, s) | C-3 |
| 6 | 126.6 | 6.18 (1H, dd, J = 16.0, 6.4) | C-5, C-8 |
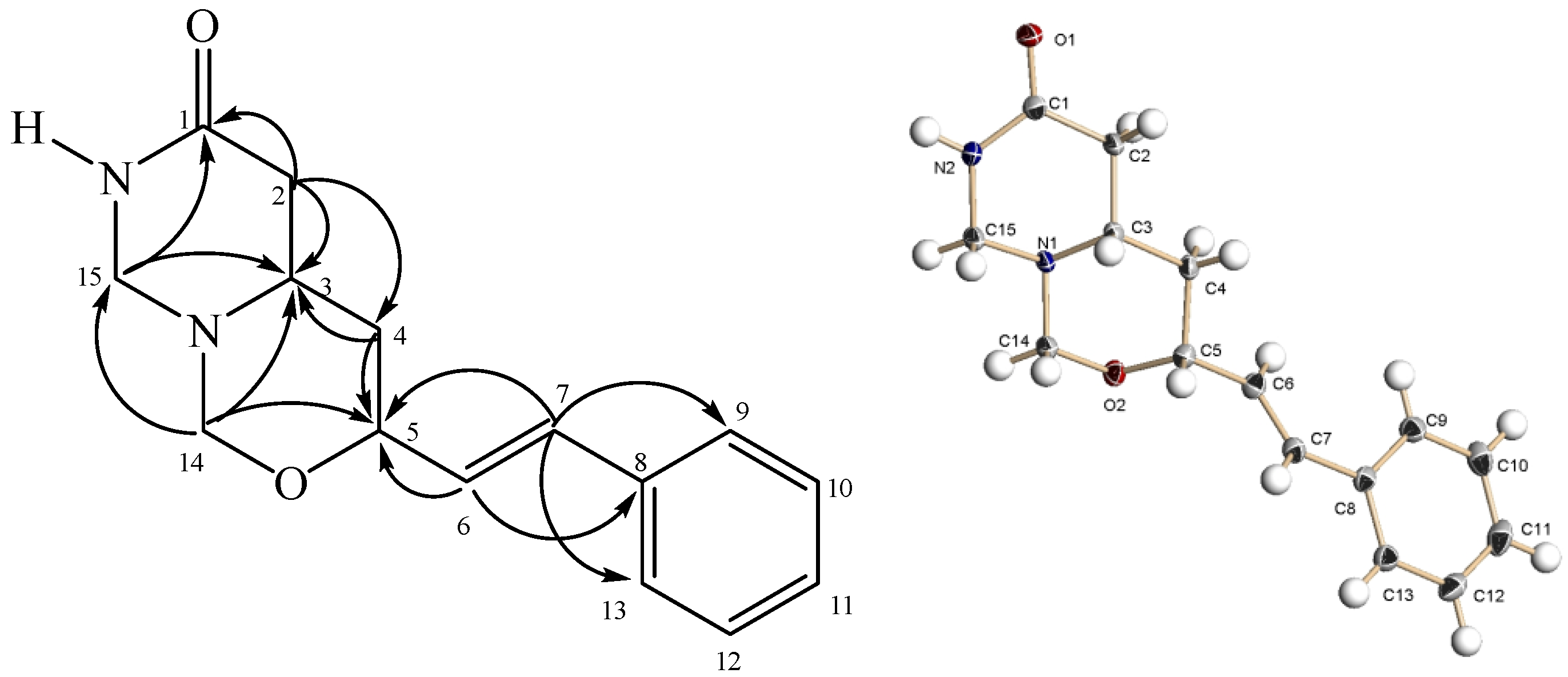
 +37.17° (c 0.0191, MeOH). Its molecular formula was determined to be C15H18O2N2 by ESI-TOF-MS (m/z 281.1278, [M+Na]+; calc. 281.1266). The IR spectrum showed absorption peaks of N-H stretching at 3413.15 cm−1 and a C=O group at 1662 cm−1 [11]. The UV spectrum revealed maxima at 206 and 252 nm, suggesting the presence of a phenyl group [11].
+37.17° (c 0.0191, MeOH). Its molecular formula was determined to be C15H18O2N2 by ESI-TOF-MS (m/z 281.1278, [M+Na]+; calc. 281.1266). The IR spectrum showed absorption peaks of N-H stretching at 3413.15 cm−1 and a C=O group at 1662 cm−1 [11]. The UV spectrum revealed maxima at 206 and 252 nm, suggesting the presence of a phenyl group [11]. | Atom no. | δ 13C | δ 1H | HMBC | Atom no. | δ 13C | δ 1H | HMBC |
|---|---|---|---|---|---|---|---|
| 1 | 168.9 | - | 9,13 | 126.6 | 7.19–7.34 (m) | ||
| 2α | 37.2 | 2.28 (1H, dd, J = 17.6, 7.8) | C-1, C-3, C-4 | 10,12 | 128.7 | 7.19–7.34 (m) | |
| 2β | 2.64 (1H, dd, J = 17.6, 5.5) | C-1 | 11 | 128.0 | 7.19–7.34 (m) | ||
| 3 | 54.1 | 2.98 (1H, m) | 14α | 82.9 | 4.13 (1H, d, J = 9.2) | C-15, C-3, C-5 | |
| 4 | 35.6 | 1.74 (2H, m) | C-3, C-5 | 14β | 4.64 (1H, d, J = 9.2) | C-3, C-5 | |
| 5 | 77.3 | 4.17 (1H, m) | 15α | 58.9 | 3.82 (1H, d, J = 8.7) | C-1, C-3 | |
| 6 | 128.3 | 6.20 (1H, dd, J = 16.0, 6.0) | C-5, C-8 | 15β | 4.34 (1H, dd, J = 8.7, 2.7) | ||
| 7 | 131.3 | 6.63 (1H, d, J = 16.0) | C-5, C-9, C-13 | N-H | - | 6.41 (NH, br s) | |
| 8 | 136.4 | - |

 −25.9° (c 0.0058, CH2Cl2). Its molecular formula was determined to be C18H17O4N by ESI-TOF-MS (m/z 334.1074, [M+Na]+; calc. 334.1055). The IR spectrum displayed a band at 1715 cm−1 representing the C=O group of the molecule. The UV spectrum (λmax 209, 251, 277 and 322 nm) indicated the presence of a basic aristolactam alkaloid structure [11]. The 1H-NMR spectrum (Table 4) showed signals for four adjacent aromatic protons at δ 8.35 (H-5, dd, J = 7.9, 1.4), δ 7.40 (H-6, m), δ 7.44 (H-7, m) and δ 7.35 (H-8, dd, J = 7.9, 1.4). Signals for three methyl groups at δ 3.96, δ 3.89 and δ 4.00 were assigned to the methoxyl groups at N-11, C-3 and C-4, respectively. The positions of the methoxyl groups were established from the NOESY and HMBC spectra. In the NOESY spectrum, the methyl protons of the methoxyl group at C-3 showed correlations with H-2. In the HMBC spectrum, the proton signal at δ 7.23 (H-2) correlated with the carbons signal at δ 59.7 (3-OCH3) and δ 56.0 (4-OCH3), thereby confirmed the assignments of the methoxyl groups.
−25.9° (c 0.0058, CH2Cl2). Its molecular formula was determined to be C18H17O4N by ESI-TOF-MS (m/z 334.1074, [M+Na]+; calc. 334.1055). The IR spectrum displayed a band at 1715 cm−1 representing the C=O group of the molecule. The UV spectrum (λmax 209, 251, 277 and 322 nm) indicated the presence of a basic aristolactam alkaloid structure [11]. The 1H-NMR spectrum (Table 4) showed signals for four adjacent aromatic protons at δ 8.35 (H-5, dd, J = 7.9, 1.4), δ 7.40 (H-6, m), δ 7.44 (H-7, m) and δ 7.35 (H-8, dd, J = 7.9, 1.4). Signals for three methyl groups at δ 3.96, δ 3.89 and δ 4.00 were assigned to the methoxyl groups at N-11, C-3 and C-4, respectively. The positions of the methoxyl groups were established from the NOESY and HMBC spectra. In the NOESY spectrum, the methyl protons of the methoxyl group at C-3 showed correlations with H-2. In the HMBC spectrum, the proton signal at δ 7.23 (H-2) correlated with the carbons signal at δ 59.7 (3-OCH3) and δ 56.0 (4-OCH3), thereby confirmed the assignments of the methoxyl groups.| Atom no. | δ 13C | δ1H | HMBC | Atom no. | δ 13C | δ1H | HMBC |
|---|---|---|---|---|---|---|---|
| 1 | 123.1 | - | 8a | 131.3 | - | ||
| 2 | 105.9 | 7.23 (1H, s) | C-3, C-4, C-10a, C-12 | 9α | 34.8 | 2.73 (1H, t, J = 13.8) | C-8a, C-10, C-10a |
| 3 | 150.5 | - | 9β | 3.49 (1H, dd, J = 13.8, 6.0) | C-10a | ||
| 4 | 155.7 | - | 10 | 57.3 | 4.60 (1H, dd, J = 13.8, 6.0) | C-10a | |
| 4a | 123.8 | - | 10a | 134.4 | - | ||
| 4b | 135.1 | - | 12 | 167.9 | - | ||
| 5 | 127.7 | 8.35 (1H, dd, J = 7.9, 1.4) | C-4a, C-4b | 3-OCH3 | 59.7 | 3.89 (3H, s) | C-3 |
| 6 | 127.9 | 7.40 (1H, dd, J = 7.9, 1.4) | C-7 | 4-OCH3 | 56.0 | 4.00 (3H, s) | C-4 |
| 7 | 130.0 | 7.44 (1H, dd, J = 7.9, 1.4) | C-6, C-8a | N-OCH3 | 63.7 | 3.96 (3H, s) | |
| 8 | 128.7 | 7.35 (1H, dd, J = 7.9, 1.4) | C-6, C-4b |
2.2. Bioactivity
| Extracts | Cell Viability (%) | ||
|---|---|---|---|
| A549 | MCF-7 | DU-145 | |
| Hexane | 2.9 | 6.2 | 9.4 |
| CH2Cl2 | 2.9 | 6.4 | 8.3 |
| Methanol | 87.2 | 94.8 | 82.3 |
| Cmpd. | Antioxidant activity | Cytotoxicity | |||||||
|---|---|---|---|---|---|---|---|---|---|
| A549 1 | DU-145 2 | SK-MEL-5 3 | BxPC-3 4 | Hep G2 5 | HT-29 6 | MCF-7 7 | MDA-MB-231 8 | ||
| 1 | 0.328 | >150 | >150 | >150 | >150 | >150 | >150 | >150 | >150 |
| 2 | 0.207 | >150 | >150 | >150 | >150 | >150 | >150 | >150 | >150 |
| 3 | 1.748 | >150 | >150 | >150 | >150 | >150 | >150 | >150 | >150 |
| 4 | 0.252 | >150 | >150 | >150 | >150 | >150 | >150 | >150 | >150 |
| 5 | 0.772 | >150 | >150 | >150 | >150 | >150 | >150 | >150 | >150 |
| 6 | 2.024 | 107.62 ± 4.67 | 71.79 ± 1.61 | 100.14 ± 11.84 | 130.48 ± 7.69 | 128.73 ± 1.81 | 64.17 ± 5.60 | 120.37 ± 11.11 | >150 |
| Cisplatin | - | 37.37 ± 3.00 | 15.18 ± 0.49 | 31.82 ± 0.23 | 20.10 ± 1.21 | 22.07 ± 0.64 | 77.24 ± 3.23 | 91.49 ± 6.54 | 276.53 ± 1.29 |
| Ascorbic acid | 0.075 | - | - | - | - | - | - | - | - |
3. Experimental
3.1. General
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Cytotoxicity Assay
3.5. Antioxidant Assay

4. Conclusions
Acknowledgments
Supplementary Materials
- Sample Availability: Samples of the compounds are available from the authors.
References
- Wiart, C. Goniothalamus species: A source of drugs for the treatment of cancers and bacterial infections? Evid. Based Complement. Alternat. Med. 2007, 4, 299–311. [Google Scholar] [CrossRef]
- Ahmad, F.; Moharm, B.A.; Jantan, I. A comparative study of the constituents of the essential oils of Goniothalamus tapis Miq. and G. tapisoides Mat Salleh from Borneo. J. Essent. Oil Res. 2010, 22, 499–502. [Google Scholar] [CrossRef]
- Fátima, Â.; Modolo, L.V.; Conegero, L.S.; Pilli, R.A.; Ferreira, L.K.; Kohn, L.K.; Carvalho, J.E. Styryl lactones and their derivatives: Biological activities, mechanisms of action and potential leads for drug design. Curr. Med. Chem. 2006, 13, 3371–3384. [Google Scholar] [CrossRef]
- Inayat-Hussain, S.H.; Osman, A.B.; Din, L.B.; Ali, A.M.; Snowden, R.T.; MacFarlane, M.; Cain, K. Caspases-3 and -7 are activated in goniothalamin-induced apoptosis in human Jurkat T-cells. FEBS Lett. 1999, 456, 379–383. [Google Scholar] [CrossRef]
- Lee, A.T.C.; Azimahtol, H.L.P.; Tan, A.N. Strylpyrone Derivative (SPD) induces apoptosis in a capase-7-dependent manner in the human breast cancer cell line MCF-7. Cancer Cell Int. 2003, 3, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Inayat-Hussain, S.H.; Osman, A.B.; Din, L.B.; Ali, A.M.; Ross, D. Loss of mitochondrial transmembrane potential and caspase-9 activation during apoptosis induced by the novel styryl-lactone goniothalamin in HL-60 leukemia cells. Toxicol. In Vitro 2003, 17, 433–439. [Google Scholar] [CrossRef]
- Litaudon, M.; Guéritte, F.; Bousserouel, H.; Awang, K.; Nosjean, O.; Martin, M.T.; Tran Huu Dau, M.E.; Sévenet, T. A dimeric sesquiterpenoid from a Malaysian Meiogyne as a new inhibitor of Bcl-xL/BakBH3 domain peptide interaction. J. Nat. Prod. 2009, 72, 480–483. [Google Scholar] [CrossRef]
- Mohamad, K.; Hirasawa, Y.; Litaudon, M.; Awang, K.; Hadi, A.H.A.; Takeya, K.; Ekasari, W.; Morita, H. Ceramicines B–D, new antiplasmodial limonoids from Chisocheton ceramicus. Bioorg. Med. Chem. 2009, 17, 727–730. [Google Scholar] [CrossRef]
- Mohamad, K.; Hirasawa, Y.; Lim, C.S.; Awang, K.; Hadi, A.H.A.; Takeya, K.; Morita, H. Ceramicine A and walsogyne A, novel limonoids from two species of Meliaceae. Tetrahedron Lett. 2008, 49, 4276–4278. [Google Scholar] [CrossRef]
- Mukhtar, M.R.; Martin, M.T.; Domansky, M.; Pais, M.; Hadi, A.H.A.; Awang, K. Phoebegrandines A and B, proaporphine-tryptamine dimmers, from Phoebe grandis. Phytochemistry 1997, 45, 1543–1546. [Google Scholar] [CrossRef]
- Anderson, R.J.; Bendell, D.J.; Groundwater, P.W. Organic Spectroscopic Analysis; Royal Society of Chemistry: Cambridge, UK, 2004; pp. 13–80. [Google Scholar]
- Desai, S.J.; Charturvedi, R.N.; Badheka, L.P.; Mulchandani, N.P. Aristolactams and 4,5 dioxoaporphines from Indian PIPER species. Indian J. Chem. 1989, 28, 775–777. [Google Scholar]
- Fátima, Â.; Kohn, L.K.; Antônio, M.A.; Carvalho, J.E.; Pilli, R.A. (R)-Goniothalamin: Total syntheses and cytotoxic activity against cancer cell lines. Bioorg. Med. Chem. 2005, 13, 2927–2933. [Google Scholar] [CrossRef]
- Tai, B.H.; HuYen, V.T.; Huong, T.T.; Nhiem, N.X.; Choi, E.M. New prano–pyrone from Goniothalamus tamirensis Enchances the Proliferation and Differentiation of Osteoblastic MC3T3-E1 Cells. Chem. Pharm. Bull. 2010, 58, 521–525. [Google Scholar] [CrossRef]
- Zhu, W.M.; Zhao, Q.; Li, S.L.; Hao, X.J. Sesquiterpenoids from Hedychium yunnanense and Porana discifera, and the structural revision of two sesquiterpenoids from Laggera pterodonta. J. Asian Nat. Prod. Res. 2007, 9, 277–283. [Google Scholar] [CrossRef]
- Bick, I.R.C.; Douglas, G.K. Yellow alkaloids of Atherosperma moschatum. Tetrahedron Lett. 1964, 25, 1629–1633. [Google Scholar]
- Aires-de-Sousa, J.; Hemmer, M.C.; Gasteiger, J. Prediction of 1H-NMR chemical shifts using nueral networks. Anal. Chem. 2002, 74, 80–90. [Google Scholar] [CrossRef]
- Hanai, K.; Kuwae, A.; Takai, T.; Senda, H.; Kunimoto, K. A comparative vibrational and NMR study of cis-cinnamic acid polymorphs and trans-cinnamic acid. Spectrochim. Acta A 2001, 57, 513–519. [Google Scholar] [CrossRef]
- Wattanapiromsakul, C.; Wangsintaweekul, B.; Sangprapan, P.; Itharat, A.; Keawpradub, N. Goniothalamin, a cytotoxic compound, isolated from Goniothalamus macrophyllus (Blume) Hook. f. & Thomson var. macrophyllus. Songklanakarin J. Sci. Technol. 2005, 27, 479–487. [Google Scholar]
- Pihie, A.H.L.; Stanslas, J.; Bin, D.L. Non-steroid receptor-mediated antiproliferative activity of styrylpyrone derivative in human breast cancer cell lines. Anticancer Res. 1998, 18, 1739–1744. [Google Scholar]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C.M. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Miller, N.J.; Sampson, J.; Candeias, L.P.; Bramley, P.M.; Rice-Evans, C.A. Antioxidant activities ofcarotenes and xanthophylls. FEBS Lett. 1996, 384, 240–242. [Google Scholar] [CrossRef]
- Tian, B.; Xu, Z.; Sun, Z.; Lin, J.; Hua, Y. Evaluation of the antioxidant effects of carotenoids from Deinococcus radiodurans through targeted mutagenesis, chemiluminescence, and DNA damage analyses. Biochim. Biophys. Acta 2007, 1770, 902–911. [Google Scholar] [CrossRef]
- Prism, 5.0 software; GraphPad Software Inc.: La Jolla, CA, USA, 2009.
- Shimada, K.; Fujikawa, K.; Yahara, K.; Nakamura, T. Antioxidative properties of xanthin onautoxidation of soybean oil in cyclodextrin emulsion. J. Agric. Food Chem. 1992, 40, 945–948. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, R.P.T.; Bihud, V.; Bin Mohamad, K.; Leong, K.H.; Bin Mohamad, J.; Bin Ahmad, F.; Hazni, H.; Kasim, N.; Halim, S.N.A.; Awang, K. Cytotoxic and Antioxidant Compoundsfrom the Stem Bark of Goniothalamus tapisoides Mat Salleh. Molecules 2013, 18, 128-139. https://doi.org/10.3390/molecules18010128
Kim RPT, Bihud V, Bin Mohamad K, Leong KH, Bin Mohamad J, Bin Ahmad F, Hazni H, Kasim N, Halim SNA, Awang K. Cytotoxic and Antioxidant Compoundsfrom the Stem Bark of Goniothalamus tapisoides Mat Salleh. Molecules. 2013; 18(1):128-139. https://doi.org/10.3390/molecules18010128
Chicago/Turabian StyleKim, Rosalind Pei Theng, Vicky Bihud, Khalit Bin Mohamad, Kok Hoong Leong, Jamaludin Bin Mohamad, Fasihuddin Bin Ahmad, Hazrina Hazni, Noraini Kasim, Siti Nadiah Abdul Halim, and Khalijah Awang. 2013. "Cytotoxic and Antioxidant Compoundsfrom the Stem Bark of Goniothalamus tapisoides Mat Salleh" Molecules 18, no. 1: 128-139. https://doi.org/10.3390/molecules18010128
APA StyleKim, R. P. T., Bihud, V., Bin Mohamad, K., Leong, K. H., Bin Mohamad, J., Bin Ahmad, F., Hazni, H., Kasim, N., Halim, S. N. A., & Awang, K. (2013). Cytotoxic and Antioxidant Compoundsfrom the Stem Bark of Goniothalamus tapisoides Mat Salleh. Molecules, 18(1), 128-139. https://doi.org/10.3390/molecules18010128