Synthesis of 5?-Androstane-17-spiro-?-lactones with a 3-Keto, 3-Hydroxy, 3-Spirocarbamate or 3-Spiromorpholinone as Inhibitors of 17?-Hydroxysteroid Dehydrogenases


Abstract
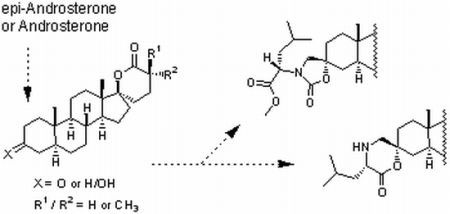
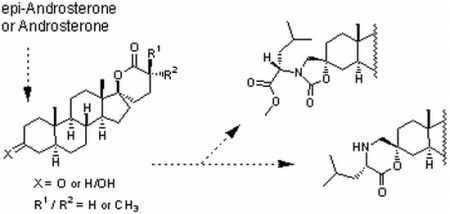
:
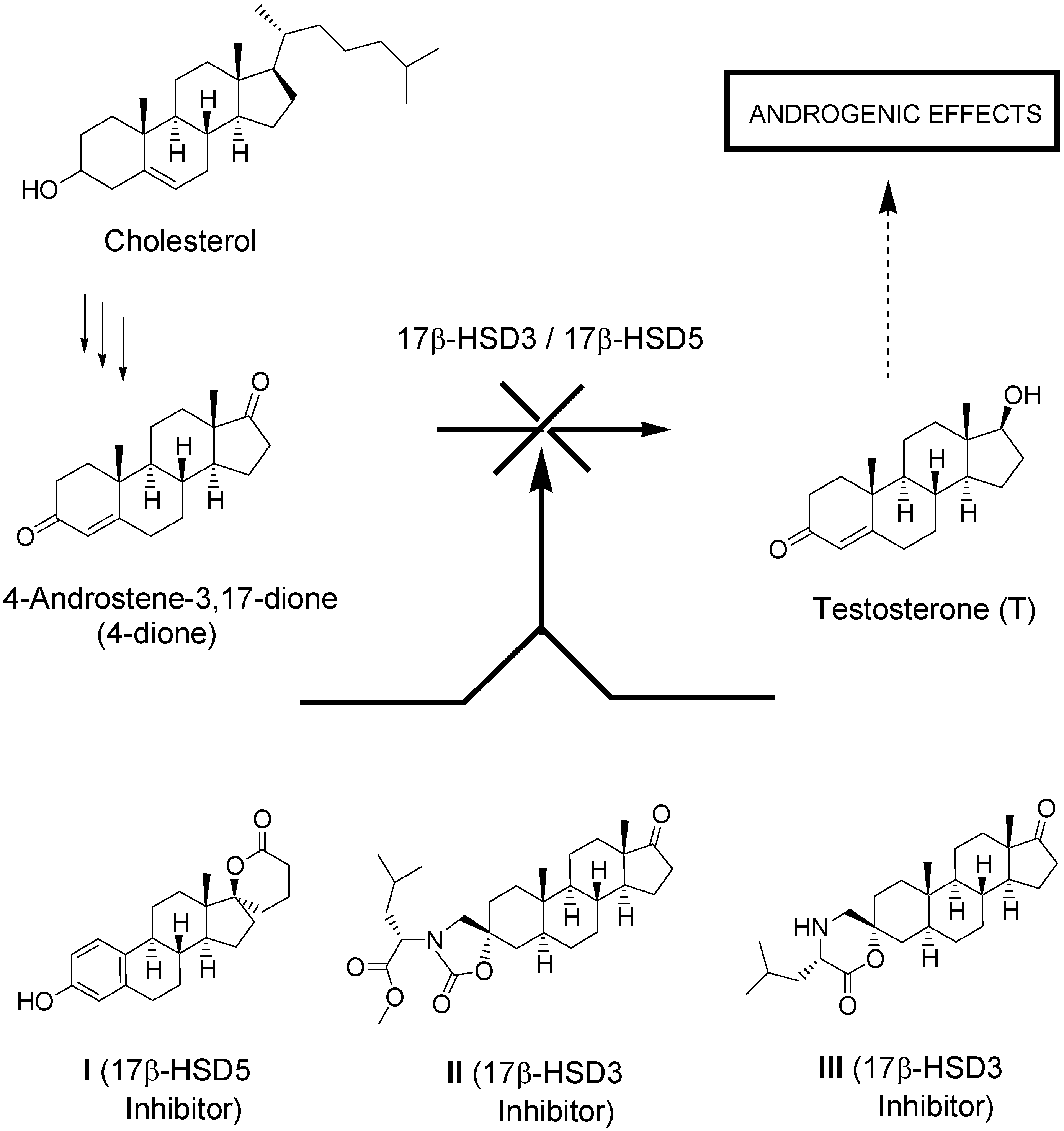
1. Introduction


2. Results and Discussion
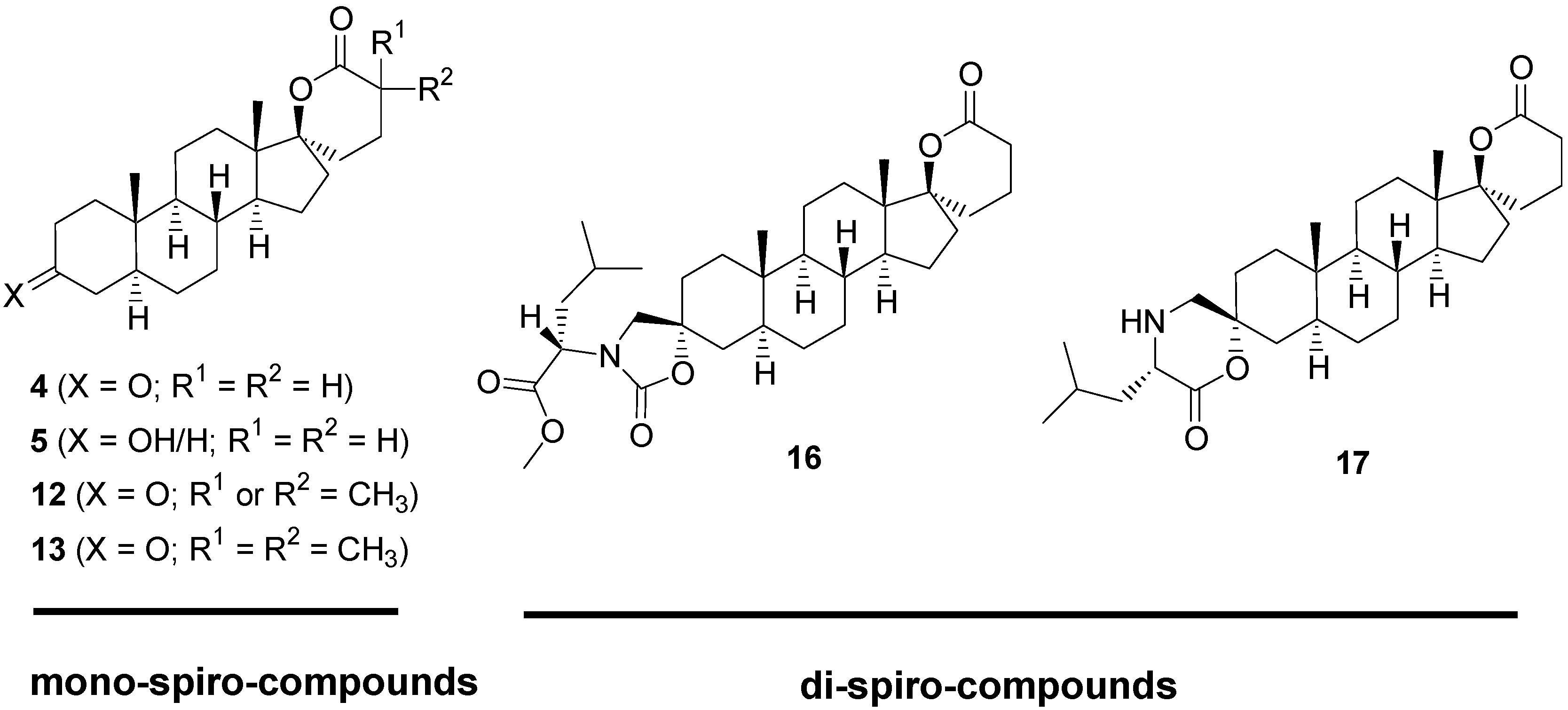
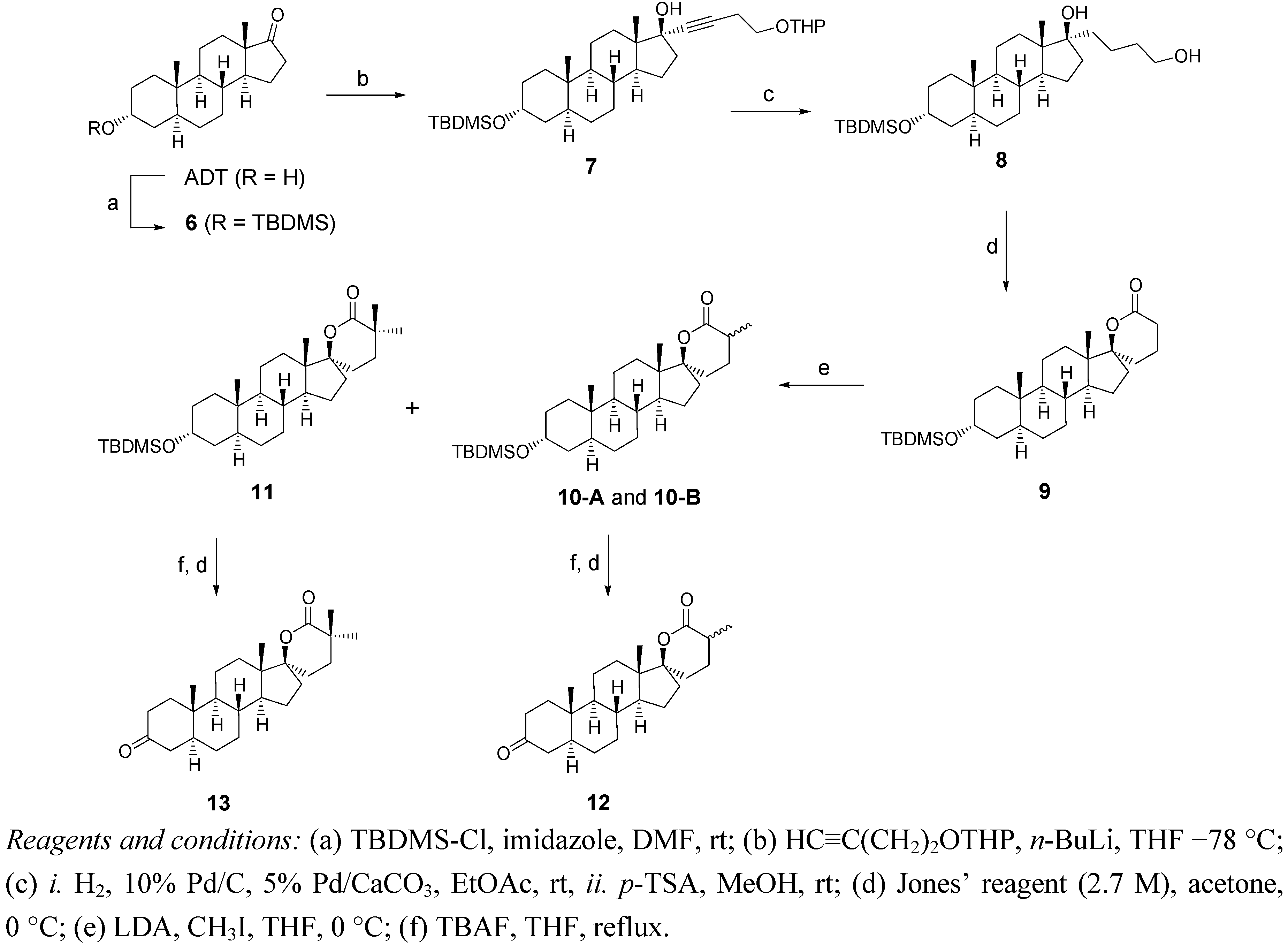
2.1. Synthesis of Spiro-δ-Lactones 4 and 5 (Scheme 1)

2.2. Synthesis of Methylated Spiro-δ-Lactones 12 and 13 (Scheme 2)
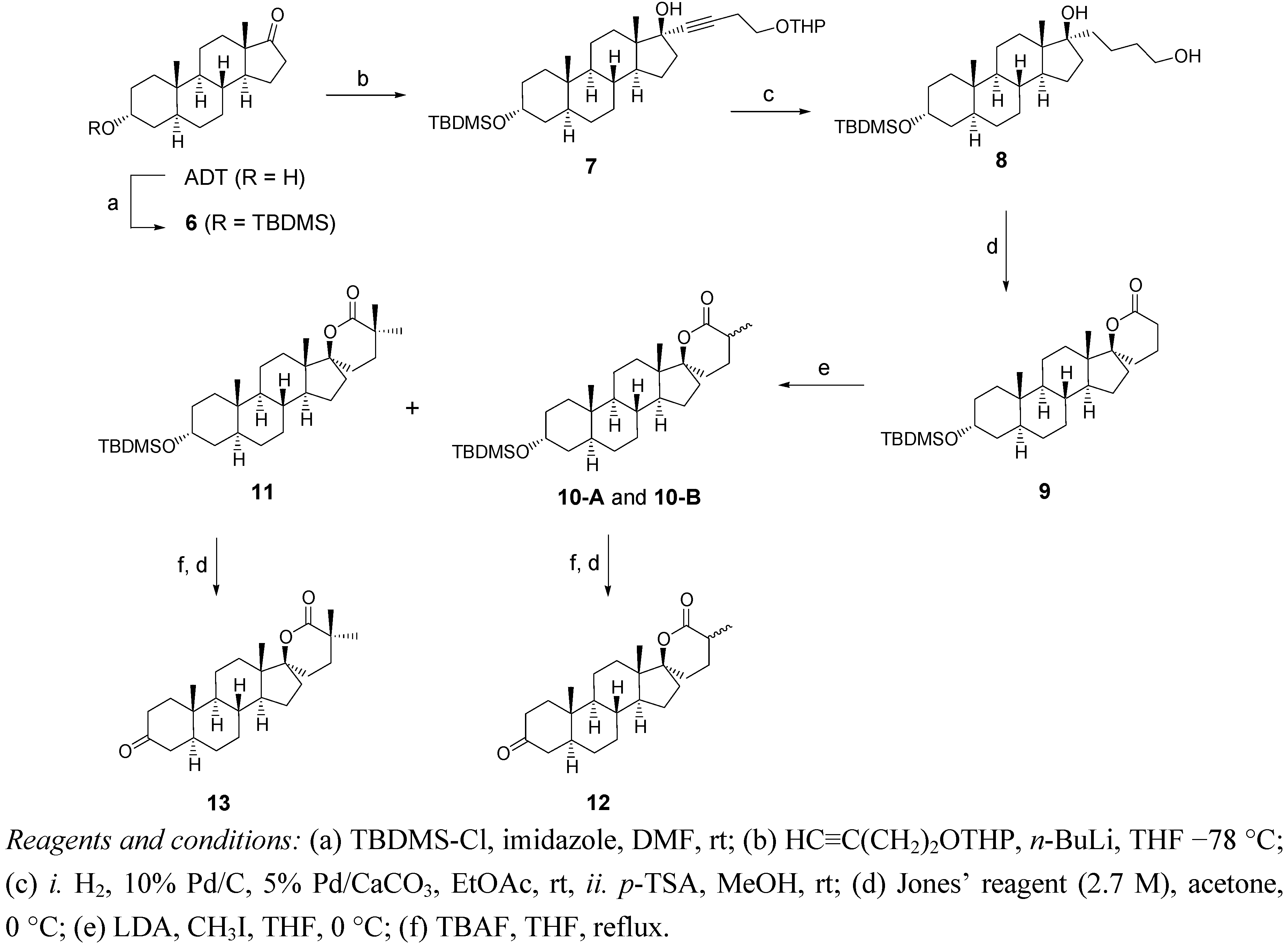
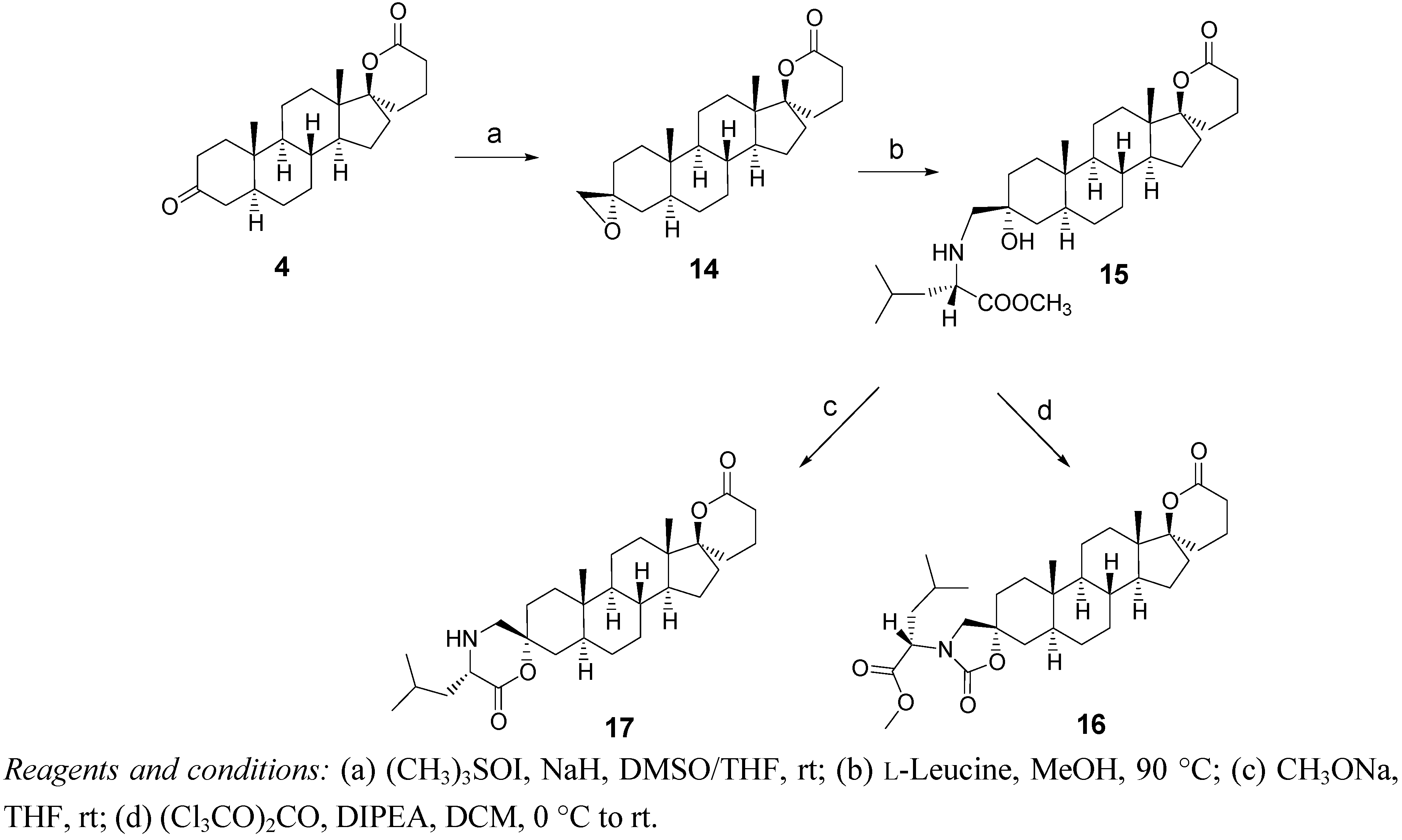
2.3. Synthesis of Spiro-Carbamate 16 and Spiro-Morpholinone 17 (Scheme 3)

2.4. Biological Evaluation of Monospiro Derivatives 4, 5, 12 and 13

| Compounds (characteristics) a | Inhibition of 17β-HSD5 at 0.3 μM (%) b | Inhibition of 17β-HSD5 at 3 μM (%) b | Inhibition of 17β-HSD3 at 0.1 μM (%) c | Inhibition of 17β-HSD3 at 1 μM (%) c | Inhibition of 17β-HSD3 at 10 μM (%) c |
|---|---|---|---|---|---|
| I (C18/17-lactone/3-OH | 92 | 95 | -- | -- | -- |
| II (C19/17-oxo/3-carbamate | -- | -- | 66.0 ± 1.7 | 88.3 ± 1.1 | 93.7 ± 0.8 |
| III (C19/17-oxo/3-morpholinone | -- | -- | 63.2 ± 2.6 | 81.0 ± 1.6 | 88.7 ± 4.0 |
| 4 (C19/17-lactone/3-oxo | 64 | 92 | 4.9 ± 4.8 | 15.0 ± 0.9 | 56.7 ± 2.2 |
| 5 (C19/17-lactone/3-OH | 56 | 91 | 0.6 ± 5.3 | 22.4 ± 2.8 | 45.7 ± 1.5 |
| 12 (C19/17-lactone; mono-CH3/3-oxo | 73 | 91 | 1.0 ± 3.3 | 22.2 ± 4.9 | 53.4 ± 5.0 |
| 13 (C19/17-lactone; bis-CH3/3-oxo | 54 | 91 | 1.0 ± 7.2 | 14.4 ± 3.3 | 58.0 ± 3 |
| 16 (C19/17-lactone/3-carbamate | -- | -- | 32.0 ± 3.3 | 60.4 ± 4.4 | 60.9 ± 0.7 |
| 17 (C19/17-lactone/3-morpholinone | -- | -- | 11.2 ± 2.6 | 51.0 ± 1.5 | 87.2 ± 0.5 |
| Compounds | Proliferative activity at 0.1 μM (%) a | Proliferative activity at 1 μM (%) a | Antiproliferative activity at 0.1 μM (%) b | Antiproliferative activity at 1 μM (%) b |
|---|---|---|---|---|
| 4 | 12 ± 2 | 0 ± 5 | 23 ± 1 | 100 ± 1 |
| 5 | 19 ± 4 | 0 ± 5 | 10 ± 1 | 61 ± 3 |
| 12 | 9 ± 5 | 0 ± 3 | 0 ± 5 | 58 ± 2 |
| 13 | 0 ± 9 | 0 ± 5 | 1 ± 2 | 49 ± 7 |
| OH-Flu c | 0 ± 6 | 0 ± 2 | 69 ± 3 | 100 ± 3 |
| Cpds | Androgen receptor (%) a | Estrogen receptor-α (%) a | Glucocorticoid receptor (%) a | Progestin receptor (%) a | ||||
| 10 nM | 1 µM | 10 nM | 1 µM | 10 nM | 1 µM | 10 nM | 1 µM | |
| 4 | 4 ± 1 | 9 ± 2 | 0 ± 1 | 1 ± 1 | 0 ± 3 | 0 ± 1 | 4 ± 2 | 0 ± 2 |
| 5 | 7 ± 1 | 5 ± 1 | 0 ± 1 | 0 ± 1 | 0 ± 1 | 1 ± 2 | 4 ± 1 | 1 ± 2 |
| 12 | 1 ± 2 | 4 ± 2 | 0 ± 2 | 1 ± 3 | 0 ± 3 | 0 ± 1 | 0 ± 2 | 0 ± 1 |
| 13 | 0 ± 2 | 0 ± 2 | 0 ± 2 | 0 ± 3 | 0 ± 2 | 1 ± 3 | 0 ± 3 | 1 ± 3 |
| DHT b | 70 ± 1 | 100 ± 1 | 2 ± 2 | 4 ± 1 | 2 ± 2 | 6 ±2 | 3 ± 2 | 40 ± 2 |
| E2 b | 0 ± 2 | 34 ± 1 | 75 ± 1 | 100 ± 1 | 5 ± 2 | 12 ± 2 | 6 ± 3 | 25 ± 2 |
| DEX b | 0 ± 1 | 2 ± 1 | 0 ± 3 | 0 ± 1 | 66 ± 2 | 99 ± 1 | 0 ± 3 | 1 ± 2 |
| R5050 b | 1 ± 4 | 28 ± 2 | 5 ± 2 | 4 ± 1 | 9 ± 2 | 85 ± 2 | 65 ± 2 | 99 ± 2 |
2.5. Biological Evaluation of Dispiro Derivatives 16 and 17
3. Experimental
3.1. General
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
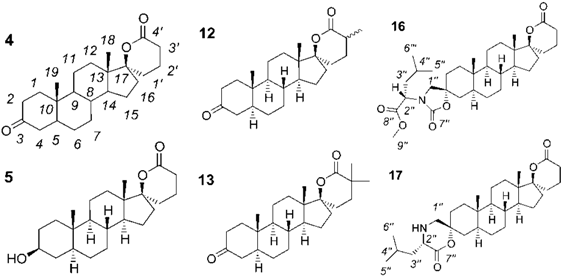
| Cpds | 4 | 5 | 12 | 13 | 16 | 17 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C1 | 38.55 | 36.98 | 38.60 | 38.58 | 33.86 | 32.98 | |||||
| C2 | 38.07 | 31.38 | 38.12 | 38.11 | 32.75 | 31.48 | |||||
| C3 | 211.67 | 71.07 | 211.79 | 211.84 | 79.74 | 82.37 | |||||
| C4 | 44.59 | 38.03 | 44.63 | 44.62 | 39.48 | 38.11 | |||||
| C5 | 46.71 | 44.84 | 46.75 | 46.74 | 40.86 | 39.29 | |||||
| C6 | 28.72 | 28.47 | 28.77 | 28.75 | 28.01 | 28.00 | |||||
| C7 | 31.40 | 31.72 | 31.44 | 31.41 | 31.47 | 31.38 | |||||
| C8 | 35.75 | 35.82 | 35.81 | 35.81 | 35.84 | 35.85 | |||||
| C9 | 53.61 | 54.12 | 53.67 | 53.63 | 53.61 | 53.50 | |||||
| C10 | 35.75 | 35.49 | 35.81 | 35.81 | 35.31 | 36.02 | |||||
| C11 | 20.90 | 20.65 | 20.91 (20.98) | 20.97 | 20.43 | 20.46 | |||||
| C12 | 31.86 | 31.92 | 31.93 | 31.92 | 31.93 | 31.96 | |||||
| C13 | 46.99 | 46.95 | 46.96 | 47.01 | 46.99 | 46.99 | |||||
| C14 | 49.64 | 49.77 | 49.45 (49.61) | 49.49 | 49.77 | 49.83 | |||||
| C15 | 23.77 | 23.73 | 23.94 (23.72) | 23.61 | 23.76 | 23.78 | |||||
| C16 | 33.93 | 33.90 | 33.99 (33.50) | 34.77 | 33.95 | 33.92 | |||||
| C17 | 93.21 | 93.34 | 92.78 (93.65) | 93.70 | 93.40 | 93.42 | |||||
| C18 | 14.40 | 14.37 | 14.52 | 14.56 | 14.43 | 14.45 | |||||
| C19 | 11.43 | 12.24 | 11.47 | 11.45 | 11.40 | 11.36 | |||||
| C1' | 27.86 | 27.82 | 27.21 (28.58) | 25.51 | 27.91 | 27.89 | |||||
| C2' | 15.83 | 15.78 | 24.41 (24.21) | 31.55 | 15.84 | 15.84 | |||||
| C3' | 29.40 | 29.37 | 34.64 (36.16) | 37.76 | 29.47 | 29.47 | |||||
| C4' | 171.98 | 172.13 | 174.90 (175.89) | 177.90 | 172.22 | 172.25 | |||||
| C5’ | --- | --- | 17.35 (17.26) | 27.65 | --- | --- | |||||
| C6' | --- | --- | --- | 27.75 | --- | --- | |||||
| C1'' | --- | --- | --- | --- | 52.89 | 52.41 | |||||
| C2'' | --- | --- | --- | --- | 53.61 | 55.58 | |||||
| C3'' | --- | --- | --- | --- | 37.69 | 41.41 | |||||
| C4'' | --- | --- | --- | --- | 24.93 | 24.46 | |||||
| C5''/C6'' | --- | --- | --- | --- | 21.06/23.14 | 20.97/23.40 | |||||
| C7'' | --- | --- | --- | --- | 157.99 | 171.99 | |||||
| C8'' | --- | --- | --- | --- | 171.99 | --- | |||||
| C9'' | --- | --- | --- | --- | 52.22 | --- | |||||
3.2. Synthesis of δ-Lactones 4 and 5 (Scheme 1)
3.3. Synthesis of α-Methylated δ-lactones 12 and 13 (Scheme 2)
Methylation of Lactone 9
Hydrolysis of the Silylated Ethers of 10A, 10B and 11
3.4. Synthesis of the Dispiro Compounds 16 and 17 (Scheme 3)
3.5. Inhibition of 17β-HSD5
3.6. Inhibition of 17β-HSD3 (Microsomal Fraction of Rat Testes)
3.7. Proliferative and Antiproliferative Shionogi (AR+) Cell Assay
3.8. Steroids Receptor Binding Assays
4. Conclusions
Acknowledgments
- Sample Availability: Samples of the final compounds 4, 5, 12, 13, 16 and 17 are available from the authors.
References
- Huggins, C.; Hodges, C.V. Studies of prostate cancer. I Effect of castration, estrogen and androgen injections on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941, 1, 293–307. [Google Scholar]
- Gittes, R.F. Carcinoma of the prostate. N. Engl. J. Med. 1991, 24, 236–245. [Google Scholar] [CrossRef]
- Labrie, F. Intracrinology. Mol. Cell. Endocrinol. 1991, 78, C113–C118. [Google Scholar] [CrossRef]
- Labrie, F.; Bélanger, A.; Simard, J.; Luu-The, V.; Labrie, C. DHEA and peripheral androgen and estrogen formation: Intracrinology. NYAcad. Sci. 1995, 774, 16–28. [Google Scholar] [CrossRef]
- Poirier, D. 17β-Hydroxysteroid dehydrogenase inhibitors: A patent review. Exp. Opin. Ther. Patents 2010, 20, 1123–1145. [Google Scholar] [CrossRef]
- Mohler, M.L.; Narayanan, R.; He, Y.; Miller, D.D.; Dalton, J.T. Hydroxysteroid dehydrogenase (17β-HSD3, 17β-HSD5, and 3α-HSD3) inhibitors: Extragonadal regulation of intracellular sex steroid hormone levels. Recent Pat.Endocr. Metab. Immune Drug Discov. 2007, 1, 103–118. [Google Scholar] [CrossRef]
- Day, J.M.; Tutill, H.J.; Purohit, A.; Reed, M.J. Design and validation of specific inhibitors of 17β-hydroxysteroid dehydrogenases for therapeutic application in breast and prostate cancer, and in endometriosis. Endocr. Relat. Cancer 2008, 15, 665–692. [Google Scholar] [CrossRef]
- Penning, T.M. Human hydroxysteroid dehydrogenases and pre-receptor regulation: Insights into inhibitor design and evaluation. J. Steroid Biochem. Mol. Biol. 2011, 125, 46–56. [Google Scholar] [CrossRef]
- Marchais-Oberwinkler, S.; Henn, C.; Moller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; et al. 7β-Hydroxysteroid dehydrogenases (17β-HSDs) as therapeutic targets: Protein structures, functions, and recent progress in inhibitor development. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. [Google Scholar] [CrossRef]
- Labrie, F.; Luu-The, V.; Lin, S.-X.; Labrie, C.; Simard, J.; Breton, R.; Bélanger, A. The key role of 17β-hydroxysteroid dehydrogenase in sex steroid biology. Steroids 1997, 62, 148–158. [Google Scholar] [CrossRef]
- Bydal, P.; Luu-The, V.; Labrie, F.; Poirier, D. Steroidal lactones as inhibitors of 17β-hydroxysteroid dehydrogenase type 5: Chemical synthesis, enzyme inhibitory activity, and assessment of estrogenic and androgenic activities. Eur. J. Med. Chem. 2009, 44, 632–644. [Google Scholar]
- Maltais, R.; Fournier, M.A.; Poirier, D. Development of 3-substituted-androsterone derivatives as potent inhibitors of 17β-hydroxysteroid dehydrogenase type 3. Bioorg. Med. Chem. 2011, 19, 4652–4668. [Google Scholar] [CrossRef]
- Tchédam Ngatcha, B.; Luu-The, V.; Labrie, F.; Poirier, D. Androsterone 3α-ether-3β-substituted and androsterone 3β-substituted derivatives as inhibitors of type 3 17β-hydroxysteroid dehydrogenase: Chemical synthesis and structure-activity relationship. J. Med. Chem. 2005, 48, 5257–5268. [Google Scholar]
- Maltais, R.; Luu-The, V.; Poirier, D. Synthesis and optimization of a new family of type 3 17β-hydroxysteroid dehydrogenase inhibitors by parallel liquid-phase chemistry. J. Med. Chem. 2002, 45, 640–653. [Google Scholar] [CrossRef]
- Maltais, R.; Luu-The, V.; Poirier, D. Parallel solid-phase synthesis of 3β-peptido-3α-hydroxy-5α-androstan-17-one derivatives for inhibition of type 3 17β-hydroxysteroid dehydrogenase. Bioorg. Med. Chem. 2001, 9, 3101–3111. [Google Scholar] [CrossRef]
- Tchédam Ngatcha, B.; Luu-The, V.; Poirier, D. Androsterone derivatives substituted at position 16: Chemical synthesis, inhibition of type 3 17β-hydroxysteroid dehydrogenase, binding affinity for steroid receptors and proliferative/antiproliferative activity on Shionogi (AR+) cells. J. Enzyme Inhib. Med. Chem. 2002, 17, 155–165. [Google Scholar] [CrossRef]
- Djigoué, G.B.; Simard, M.; Kenmogne, L.C.; Poirier, D. Two androsterone derivatives as inhibitors of androgen biosynthesis. Acta Cryst. 2012, c68, o231–o234. [Google Scholar]
- Salman, M.; Stotter, P.L.; Chamness, G.C. 125I-ligand for progesterone receptor: 17 Alpha-(6'-iodohex-1'-ynyl)-19-nortestosterone. J. Steroid Biochem. 1989, 33, 25–31. [Google Scholar]
- Sam, K.-M.; Labrie, F.; Poirier, D. N-Butyl-N-methyl-11-(3′-hydroxy-21′,17′-carbolactone-19'-nor-17'α-pregna-1',3',5'(10')-trien-7'α-yl)-undecanamide: An inhibitor of type 2 17β-hydroxysteroid dehydrogenase that does not have estrogenic or androgenic activity. Eur. J. Med. Chem. 2000, 35, 217–225. [Google Scholar] [CrossRef]
- Bydal, P.; Auger, S.; Poirier, D. Inhibition of type 2 17β-hydroxysteroid dehydrogenase by estradiol derivatives bearing a lactone on the D-ring: Structure-activity relationships. Steroids 2004, 69, 325–342. [Google Scholar] [CrossRef]
- Rouillard, F.; Roy, J.; Poirier, D. Chemical synthesis of (S)-spiro(estradiol-17,2'-[1,4]oxazinan)-6'-one derivatives bearing two levels of molecular diversity. Eur. J. Org. Chem. 2008, 2008, 2446–2453. [Google Scholar]
- Neri, R.; Florance, K.; Koziol, P.; van Cleave, S. A biological profile of a nonsteroidal antiandrogen, SCH1352 (4'-nitro-3'-trifluoromethyl-iso-butyranilide). Endocrinology 1972, 91, 427–437. [Google Scholar] [CrossRef]
- Poyet, P.; Labrie, F. Comparison of the antiandrogenic/androgenic activities of flutamide, cyproterone acetate and megestrol acetate. Mol. Cell. Endocrinol. 1985, 42, 283–288. [Google Scholar] [CrossRef]
- Claridge, T.D.W. High-Resolution NMR Techniques in Organic Chemistry; Elsevier Science LTD.: Oxford, UK, 1999. [Google Scholar]
- Tchédam Ngatcha, B.; Trottier, M.C.; Poirier, D. 13C Nuclear magnetic resonance spectroscopy data of a variety of androsterone and epi-androsterone derivatives substituted at position 3beta or/and 3 alpha. Curr. Top. Steroids Res. 2011, 8, 35–45. [Google Scholar]
- Blunt, J.W.; Stothers, S.B. 13C-NMR spectra of steroids-a survey and commentary. Org. Magn. Reson. 1977, 9, 439–464. [Google Scholar] [CrossRef]
- Dionne, P.; Poirier, D. 13C nuclear magnetic resonance study of 17alpha-substituted estradiols. Steroids 1995, 60, 830–836. [Google Scholar] [CrossRef]
- Maltais, R.; Mercier, C.; Labrie, F.; Poirier, D. Solid-phase synthesis of model libraries of 3α,17β-dihydroxy-16α-(aminoethyl-N-substituted)-5α-androstanes for the development of steroidal therapeutic agents. Mol. Divers. 2005, 9, 67–79. [Google Scholar] [CrossRef]
- Dufort, I.; Rheault, P.; Huand, X.F.; Soucy, P.; Luu-The, V. Characteristics of a highly labile human type 5 17β-hydroxysteroid dehydrogenase. Endocrinology 1999, 140, 568–574. [Google Scholar] [CrossRef]
- Hu, G.; Zhou, H.Y.; Li, X.W.; Chen, B.B.; Xiao, Y.C.; Lian, Q.Q.; Kim, H.H.; Zheng, Z.Q.; Hardy, D.O.; Ge, R.S. The (+)- and (−)-gossypols potently inhibit both 3beta-hydroxysteroid dehydrogenase and 17beta-hydroxysteroid dehydrogenase 3 in human and rat testes. J. Steroid Biochem. Mol. Biol. 2009, 115, 14–19. [Google Scholar] [CrossRef]
- Blomquist, C.H.; Bonenfant, M.; McGinley, D.M.; Posalaky, Z.; Lakatua, D.J.; Tuli-Puri, S.; Bealka, D.G.; Tremblay, Y. Androgenic and estrogenic 17beta-hydroxysteroid dehydrogenase/17-ketosteroid reductase in human ovarian epithelial tumors: Evidence for the type 1, 2 and 5 isoforms. J. Steroid Biochem. Mol. Biol. 2002, 81, 343–351. [Google Scholar] [CrossRef]
- Moutaouakkil, M.; Prost, O.; Dahan, N.; Adessi, G.L. Estrone and dehydroepiandrosterone sulfatase activities in guinea-pig uterus and liver: Estrogenic effect of estrone sulphate. J. Steroid Biochem. 1984, 21, 321–328. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal.Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Luo, S.; Martel, C.; Sourla, A.; Gauthier, S.; Merand, Y.; Bélanger, A.; Labrie, C. Comparative potencies effects of 28-day treatment with the new anti-estrogen EM-800 and tamoxifen on estrogen sensitive parameters in intact mice. Int. J. Cancer 1997, 73, 381–391. [Google Scholar] [CrossRef]
- Luo, S.; Martel, C.; Leblanc, G.; Candas, B.; Singh, S.M.; Labrie, C.; Simard, J.; Bélanger, A.; Labrie, F. Relative potencies of flutamide and casodex: Preclinical studies. Endocr.Relat. Cancer 1996, 3, 229–241. [Google Scholar] [CrossRef]
- Asselin, J.; Melançon, R.; Moachon, G.; Bélanger, A. Characteristics of binding to estrogen, androgen, progestin, and glucocorticoid receptors in 7,12-dimethylbenz(a)anthracene-induced mammary tumors and their hormonal control. Cancer Res. 1980, 40, 1612–1622. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Djigoué, G.B.; Ngatcha, B.T.; Roy, J.; Poirier, D. Synthesis of 5?-Androstane-17-spiro-?-lactones with a 3-Keto, 3-Hydroxy, 3-Spirocarbamate or 3-Spiromorpholinone as Inhibitors of 17?-Hydroxysteroid Dehydrogenases. Molecules 2013, 18, 914-933. https://doi.org/10.3390/molecules18010914
Djigoué GB, Ngatcha BT, Roy J, Poirier D. Synthesis of 5?-Androstane-17-spiro-?-lactones with a 3-Keto, 3-Hydroxy, 3-Spirocarbamate or 3-Spiromorpholinone as Inhibitors of 17?-Hydroxysteroid Dehydrogenases. Molecules. 2013; 18(1):914-933. https://doi.org/10.3390/molecules18010914
Chicago/Turabian StyleDjigoué, Guy Bertrand, Béatrice Tchédam Ngatcha, Jenny Roy, and Donald Poirier. 2013. "Synthesis of 5?-Androstane-17-spiro-?-lactones with a 3-Keto, 3-Hydroxy, 3-Spirocarbamate or 3-Spiromorpholinone as Inhibitors of 17?-Hydroxysteroid Dehydrogenases" Molecules 18, no. 1: 914-933. https://doi.org/10.3390/molecules18010914
APA StyleDjigoué, G. B., Ngatcha, B. T., Roy, J., & Poirier, D. (2013). Synthesis of 5?-Androstane-17-spiro-?-lactones with a 3-Keto, 3-Hydroxy, 3-Spirocarbamate or 3-Spiromorpholinone as Inhibitors of 17?-Hydroxysteroid Dehydrogenases. Molecules, 18(1), 914-933. https://doi.org/10.3390/molecules18010914