Bortezomib Congeners Induce Apoptosis of Hepatocellular Carcinoma via CIP2A Inhibition

Abstract
:1. Introduction

2. Results and Discussion
2.1. Chemistry

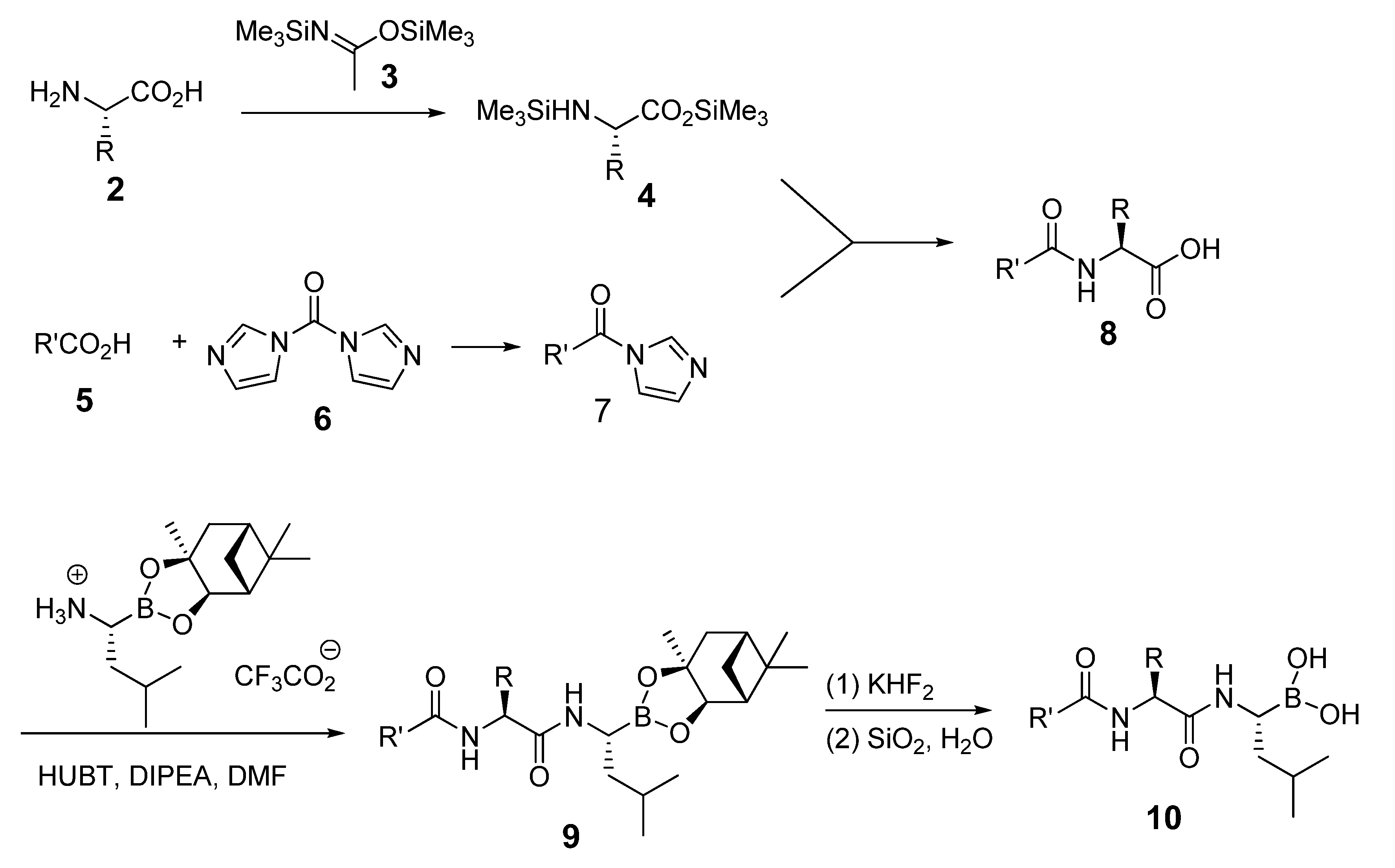
The Synthetic Route to Bortezomib Derivatives
2.2. Biological Evaluations
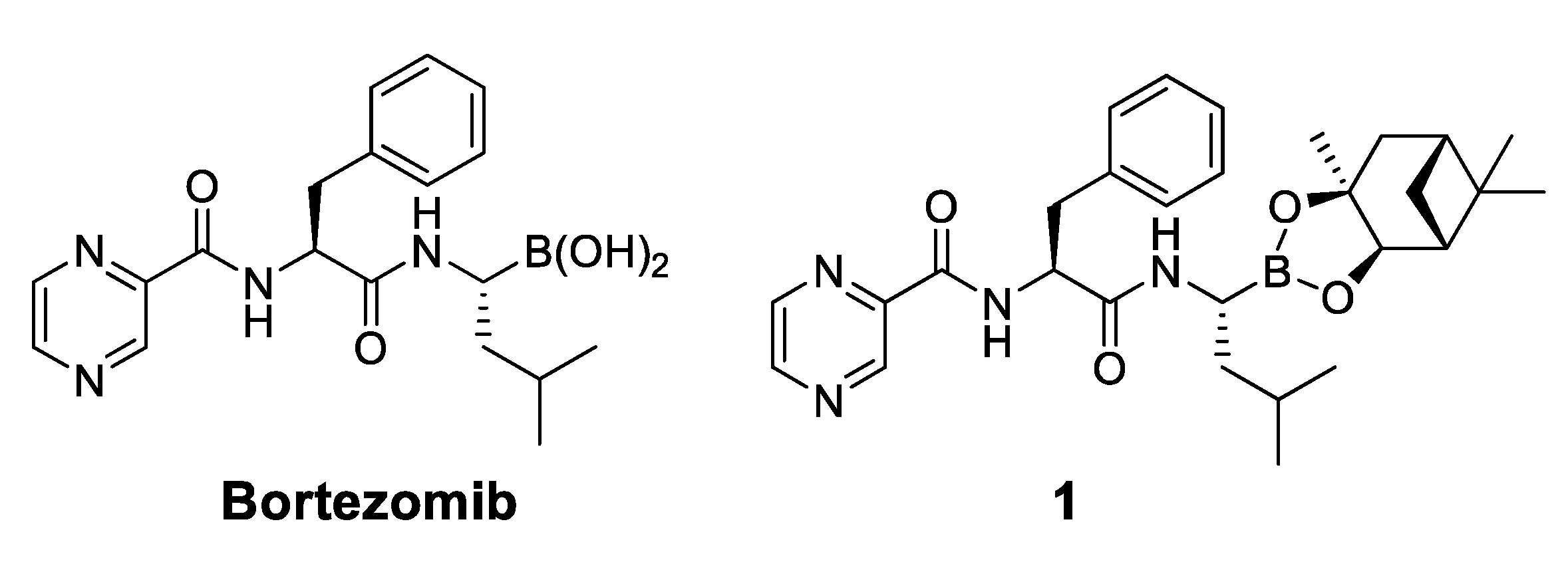
2.2.1. Development of a Bortezomib Derivative with a Bulky Pinanediol Group

2.2.2. Structure-Activity Relationship between Boronic Acid and the Pyrazinoic Ring, and Cell Growth Inhibition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Cpd | R | IC 50 (nM) in Sk-hep1 Cells |
|---|---|---|
| Bortezomib |  | 105 |
| 1 |  | 55 |
| 11 |  | >1000 |
| 12 |  | >1000 |
| 13 |  | >1000 |
| 14 | 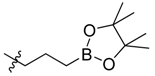 | >1000 |

| Cpd | R1 | R2 | IC 50 (nM) in Sk-hep1 Cells |
|---|---|---|---|
| 15 | 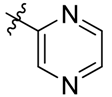 | H | >1000 |
| 16 |  |  | 58.7 |
| 17 | CH3 | | 432.6 |

| Cpd | R1 | R2 | IC 50 (nM) in Sk-hep1 Cells |
|---|---|---|---|
| 18 | | H | >1000 |
| 19 | | | 177.8 |
| 20 | CH3 | | 314.6 |
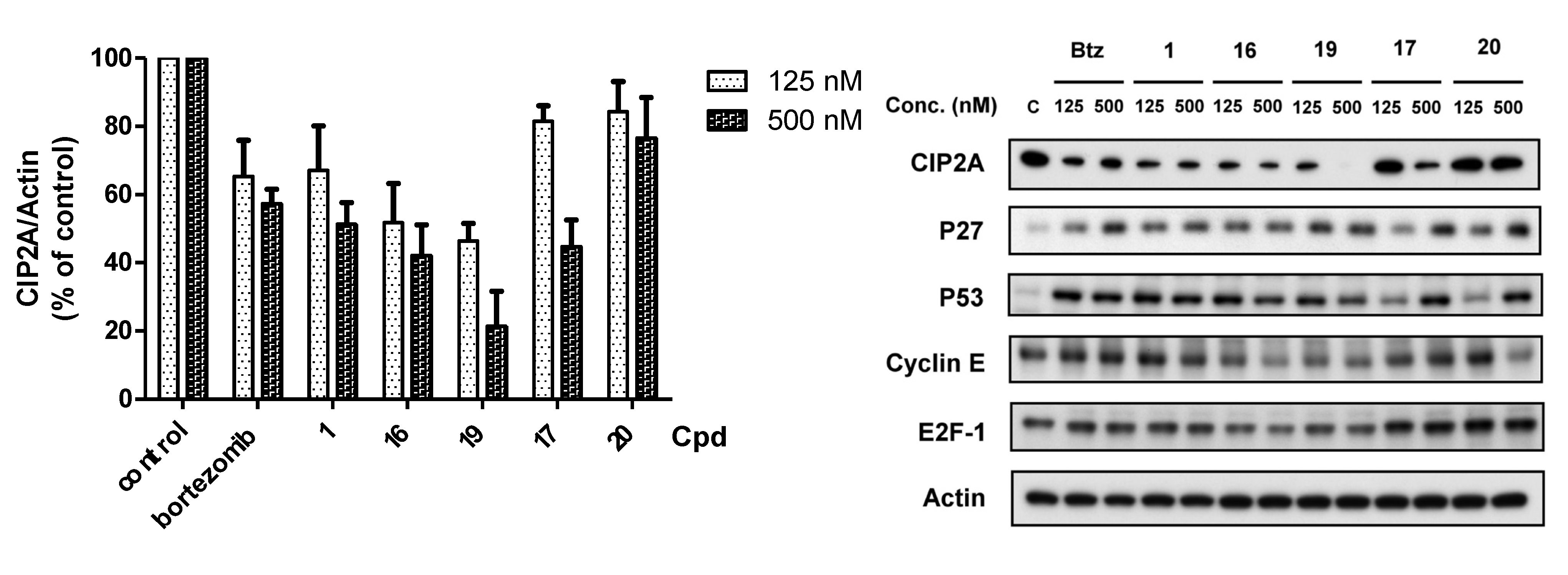
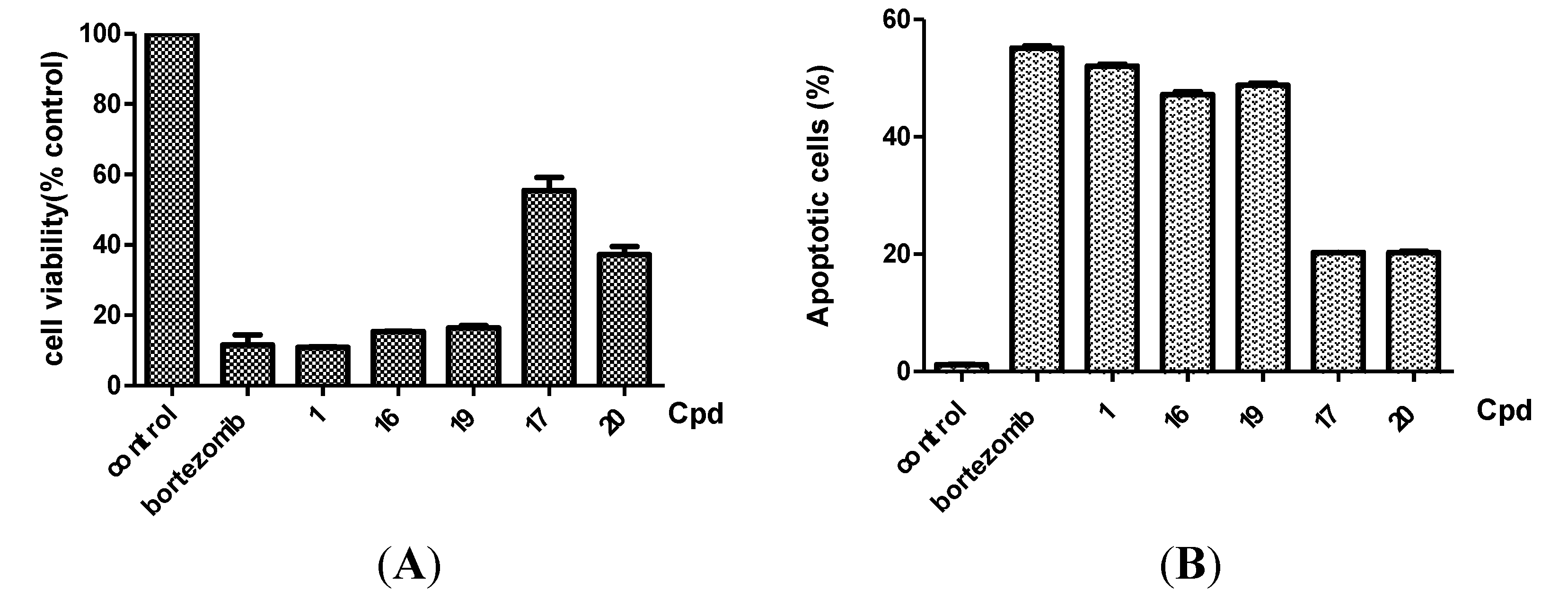
2.2.3. Target Validation of Bortezomib Derivatives


| Cpd | Bortezomib | 1 | 16 | 19 | 17 | 20 | |
|---|---|---|---|---|---|---|---|
| Cells MTT IC50 (nM) | |||||||
| Sk-hep-1 | 105.0 | 54.9 | 58.7 | 177.8 | 432.6 | 314.5 | |
| Huh 7 | 273.5 | 489.4 | 118.3 | 158.5 | 416.5 | 448.2 | |
| Hep 3B | 20.7 | 12.8 | 5.8 | 34.3 | 93.0 | 31.1 | |


2.3. Discussion
3. Experimental
3.1. Materials
3.2. Chemical Synthesis
3.2.1. General Procedure for the Preparation of Acylated Amino Acids
3.2.2. General Procedure for the Preparation of Dipeptide Boronates
3.2.3. General Procedure for the Hydrolysis of Boronic Esters
3.3. Biological Assay
3.3.1. Cell Culture
3.3.2. Proteasome Activity Assay
3.3.3. Western Blot
3.3.4. Cell Viability Analysis
3.3.5. Apoptosis Analysis
4. Conclusions
Supplementary Material
Acknowledgments
Conflicts of Interest
References
- Junttila, M.R.; Puustinen, P.; Niemela, M.; Ahola, R.; Arnold, H.; Bottzauw, T.; Ala-aho, R.; Nielsen, C.; Ivaska, J.; Taya, Y.; et al. CIP2A inhibits PP2A in human malignancies. Cell 2007, 130, 51–62. [Google Scholar] [CrossRef]
- Sablina, A.A.; Hahn, W.C. SV40 small T antigen and PP2A phosphatase in cell transformation. Cancer Metastasis Rev. 2008, 27, 137–146. [Google Scholar] [CrossRef]
- Khanna, A.; Bockelman, C.; Hemmes, A.; Junttila, M.R.; Wiksten, J.P.; Lundin, M.; Junnila, S.; Murphy, D.J.; Evan, G.I.; Haglund, C.; et al. MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J. Natl. Cancer Inst. 2009, 101, 793–805. [Google Scholar] [CrossRef]
- Chen, K.F.; Liu, C.Y.; Lin, Y.C.; Yu, H.C.; Liu, T.H.; Hou, D.R.; Chen, P.J.; Cheng, A.L. CIP2A mediates effects of bortezomib on phospho-Akt and apoptosis in hepatocellular carcinoma cells. Oncogene 2010, 29, 6257–3266. [Google Scholar] [CrossRef]
- Chen, K.F.; Yeh, P.Y.; Hsu, C.; Hsu, C.H.; Lu, Y.S.; Hsieh, H.P.; Chen, P.J.; Cheng, A.L. Bortezomib overcomes tumor necrosis factor-related apoptosis-inducing ligand resistance in hepatocellular carcinoma cells in part through the inhibition of the phosphatidylinositol 3-kinase/Akt pathway. J. Biol. Chem. 2009, 284, 11121–11133. [Google Scholar]
- Tseng, L.M.; Liu, C.Y.; Chang, K.C.; Chu, P.Y.; Shiau, C.W.; Chen, K.F. CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast Cancer Res. 2012, 14, R68. [Google Scholar] [CrossRef]
- Ma, L.; Wen, Z.S.; Liu, Z.; Hu, Z.; Ma, J.; Chen, X.Q.; Liu, Y.Q.; Pu, J.X.; Xiao, W.L.; Sun, H.D.; et al. Overexpression and small molecule-triggered downregulation of CIP2A in lung cancer. PLoS One 2011, 6, e20159. [Google Scholar] [CrossRef]
- Nencioni, A.; Grunebach, F.; Patrone, F.; Ballestrero, A.; Brossart, P. Proteasome inhibitors: Antitumor effects and beyond. Leukemia 2007, 21, 30–36. [Google Scholar] [CrossRef]
- Chen, K.F.; Yu, H.C.; Liu, C.Y.; Chen, H.J.; Chen, Y.C.; Hou, D.R.; Chen, P.J.; Cheng, A.L. Bortezomib sensitizes HCC cells to CS-1008, an antihuman death receptor 5 antibody, through the inhibition of CIP2A. Mol. Cancer Ther. 2011, 10, 892–901. [Google Scholar] [CrossRef]
- Chen, K.F.; Yu, H.C.; Liu, T.H.; Lee, S.S.; Chen, P.J.; Cheng, A.L. Synergistic interactions between sorafenib and bortezomib in hepatocellular carcinoma involve PP2A-dependent Akt inactivation. J. Hepatol. 2010, 52, 88–95. [Google Scholar]
- Zhu, Y.; Yao, S.; Xu, B.; Ge, Z.; Cui, J.; Cheng, T.; Li, R. Design, synthesis and biological evaluation of tripeptide boronic acid proteasome inhibitors. Bioorgan. Med. Chem. 2009, 17, 6851–6861. [Google Scholar] [CrossRef]
- Codony-Servat, J.; Tapia, M.A.; Bosch, M.; Oliva, C.; Domingo-Domenech, J.; Mellado, B.; Rolfe, M.; Ross, J.S.; Gascon, P.; Rovira, A.; et al. Differential cellular and molecular effects of bortezomib, a proteasome inhibitor, in human breast cancer cells. Mol. Cancer Ther. 2006, 5, 665–675. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hou, D.-R.; Huang, A.-C.; Shiau, C.-W.; Wang, C.-Y.; Yu, H.-C.; Chen, K.-F. Bortezomib Congeners Induce Apoptosis of Hepatocellular Carcinoma via CIP2A Inhibition. Molecules 2013, 18, 15398-15411. https://doi.org/10.3390/molecules181215398
Hou D-R, Huang A-C, Shiau C-W, Wang C-Y, Yu H-C, Chen K-F. Bortezomib Congeners Induce Apoptosis of Hepatocellular Carcinoma via CIP2A Inhibition. Molecules. 2013; 18(12):15398-15411. https://doi.org/10.3390/molecules181215398
Chicago/Turabian StyleHou, Duen-Ren, Ann-Chi Huang, Chung-Wai Shiau, Chun-Yi Wang, Hui-Chuan Yu, and Kuen-Feng Chen. 2013. "Bortezomib Congeners Induce Apoptosis of Hepatocellular Carcinoma via CIP2A Inhibition" Molecules 18, no. 12: 15398-15411. https://doi.org/10.3390/molecules181215398
APA StyleHou, D.-R., Huang, A.-C., Shiau, C.-W., Wang, C.-Y., Yu, H.-C., & Chen, K.-F. (2013). Bortezomib Congeners Induce Apoptosis of Hepatocellular Carcinoma via CIP2A Inhibition. Molecules, 18(12), 15398-15411. https://doi.org/10.3390/molecules181215398