The Effect of Conformational Variability of Phosphotriesterase upon N-acyl-L-homoserine Lactone and Paraoxon Binding: Insights from Molecular Dynamics Studies


Abstract
:
1. Introduction

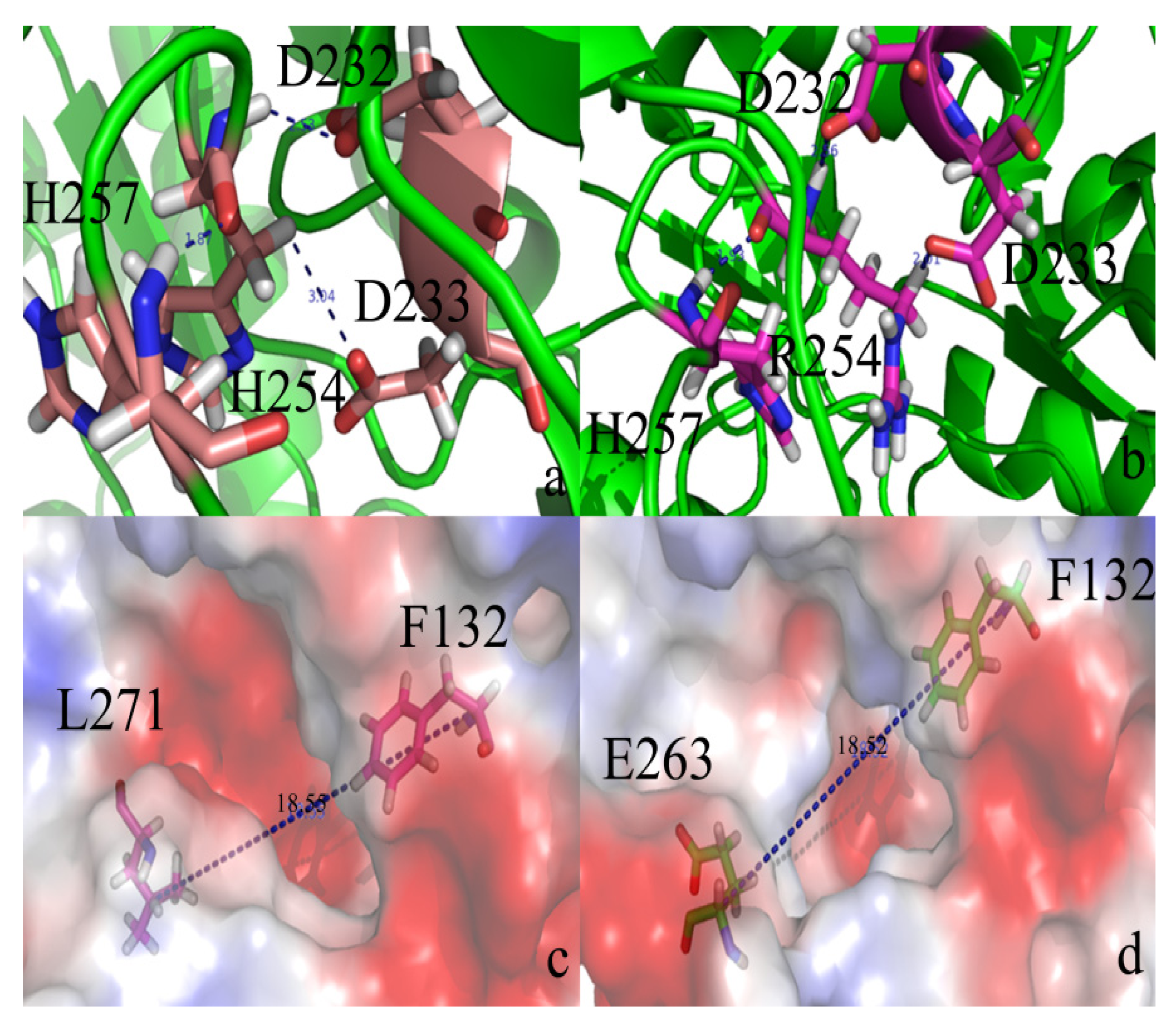
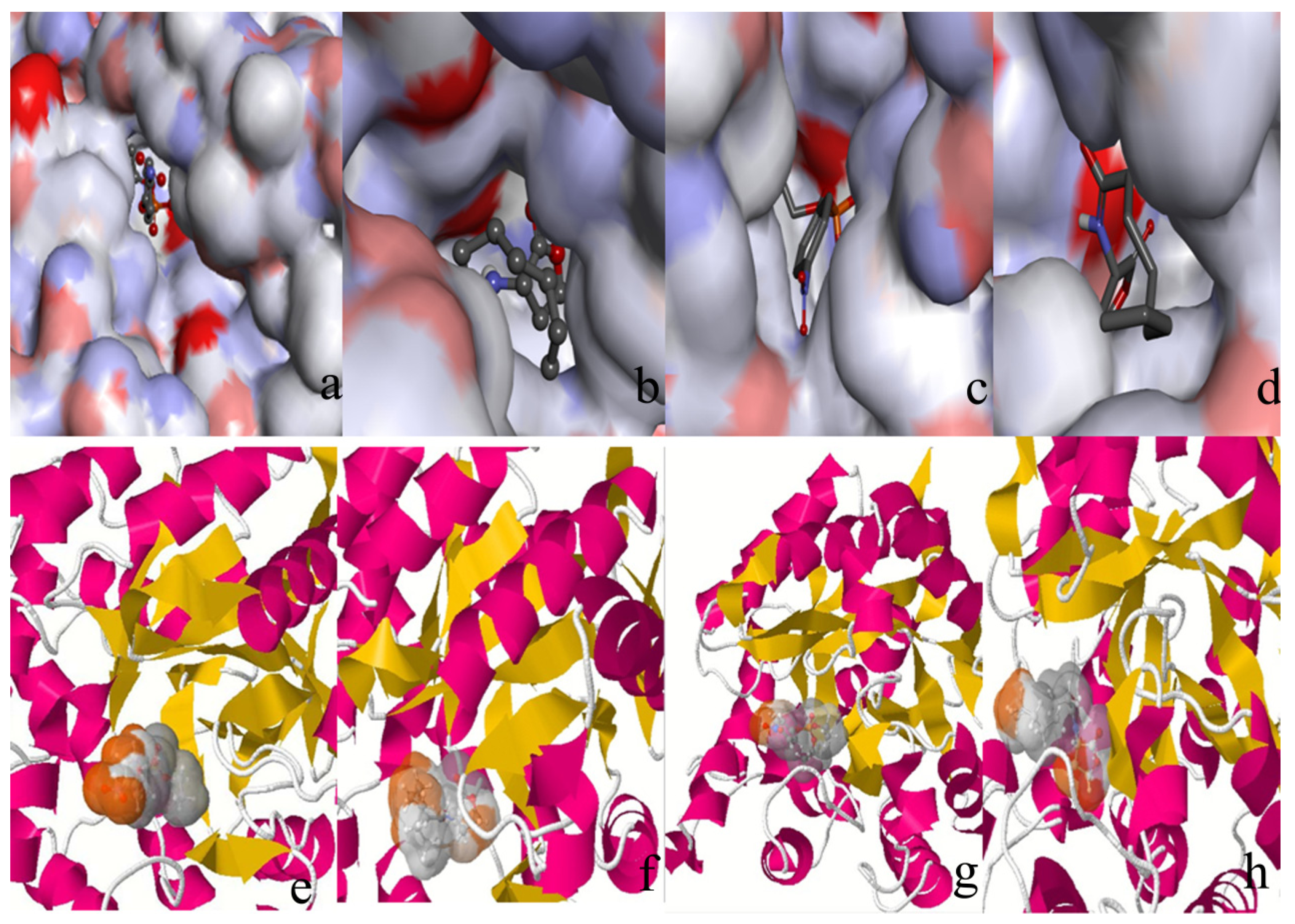
2. Results and Discussion


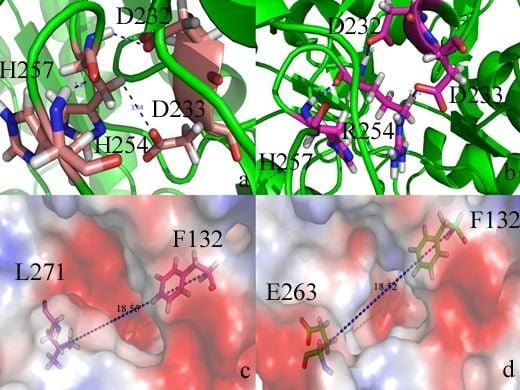
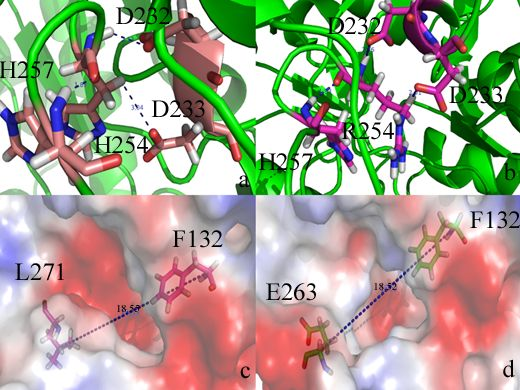{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | QMEAN Z-Score | Procheck | Verify_3D | Errat |
|---|---|---|---|---|
| Loop7 -2/H254R | 0.74 | 86.7% core 12.6% allow 0.7% gener 0.0% disall | 96.90% of the residues had an averaged score > 0.2 | 98.722 |
| PTE(PDB ID 1HZY) | 0.83 | 89.6% core 9.7% allow 0.7% gener 0.0% disall | 98.49% of the residues had an averaged score > 0.2 | 99.379 |
| Hydrogen bond Donor | Hydrogen bond acceptor |
|---|---|
| Asn165:NH | Leu262 :O |
| Ser267:HG | Asn265:OD1 |
| Ala268:HN | Asn265:OD1 |
| Ser269:HN | Asn265:O |
| Ala270:HN | Ala266:O |
| Leu271:HN | Ser267:O |
| Leu272:HN | Ala268:O |
| Gly273:HN | Ser269:O |

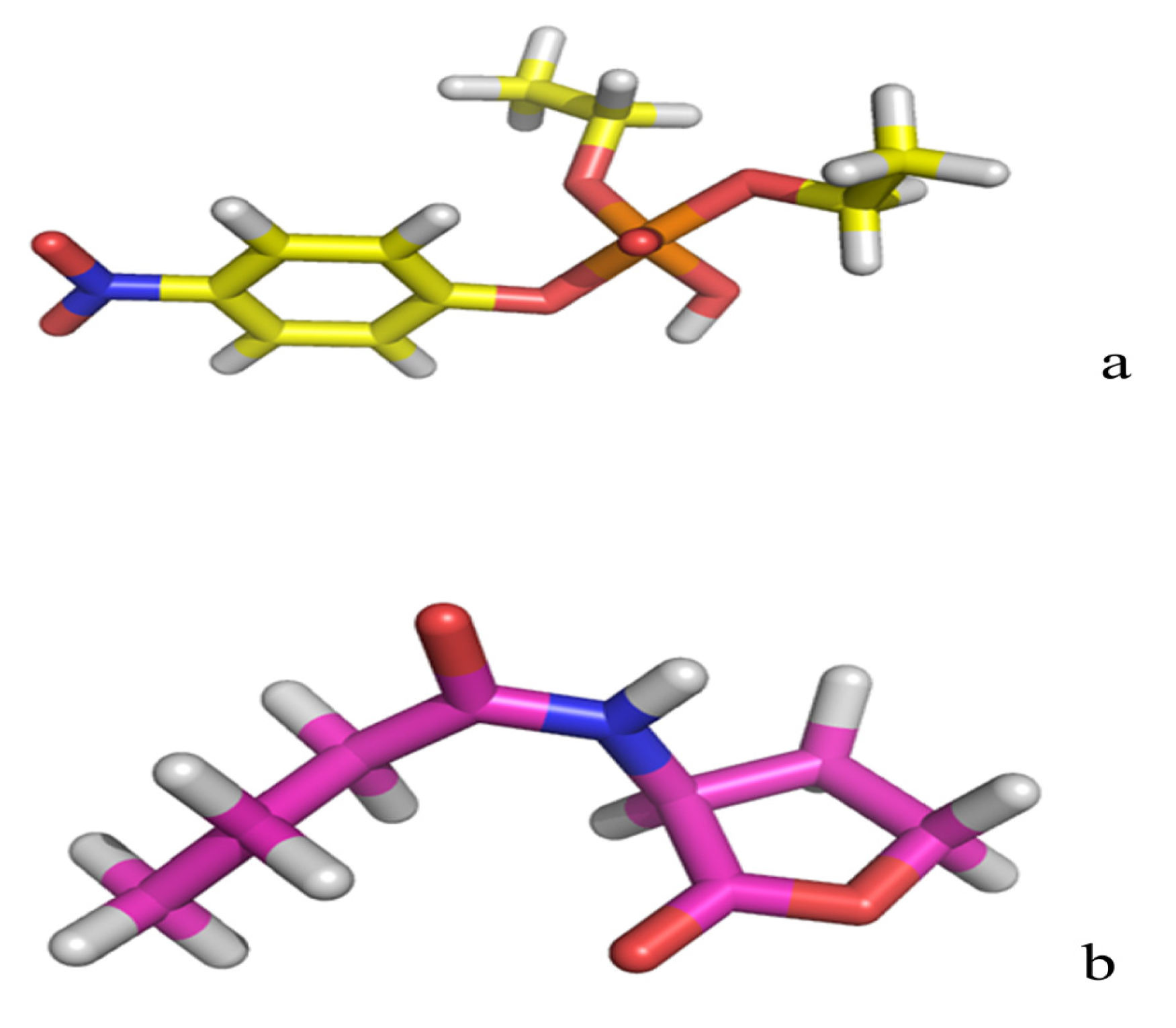
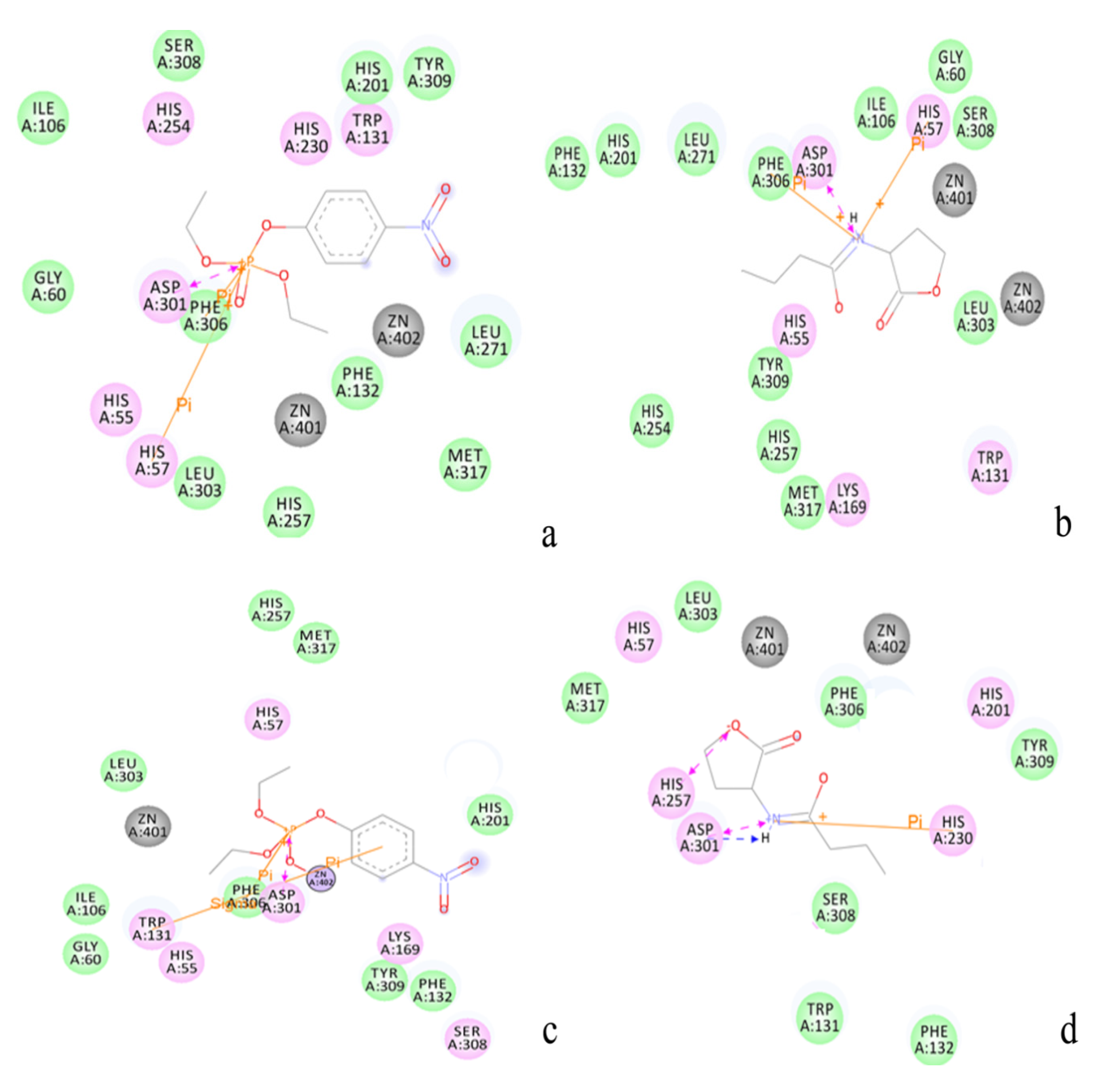
2.2. Docking Study

| Protein/substrate | Paraoxon (Kcal/mol) | C4-HSL (Kcal/mol) |
|---|---|---|
| PTE | −6.1 | −4.4 |
| loop7-2-H254R | −5.4 | −6.6 |


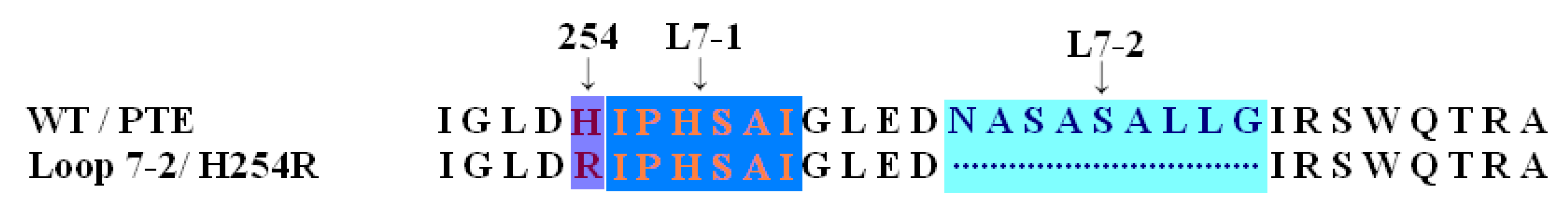
2.3. Protonation States of Residues His254 (Arg254) and Arg275
| Protein/substrate | His254 (R254) | Arg275 |
|---|---|---|
| PTE | 6.9 | 9.2 |
| loop7-2/H254R | 10.4 | 9.4 |
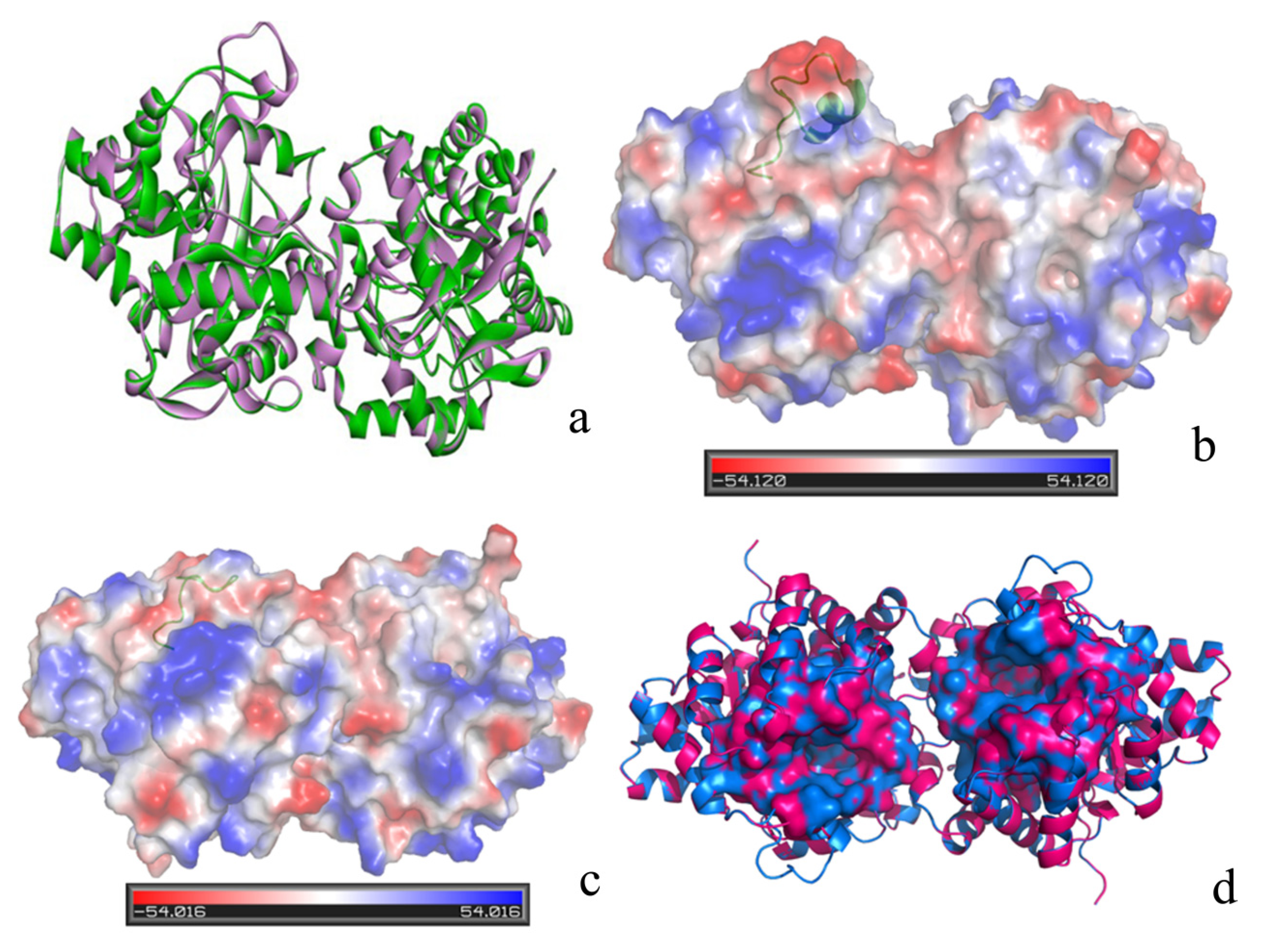
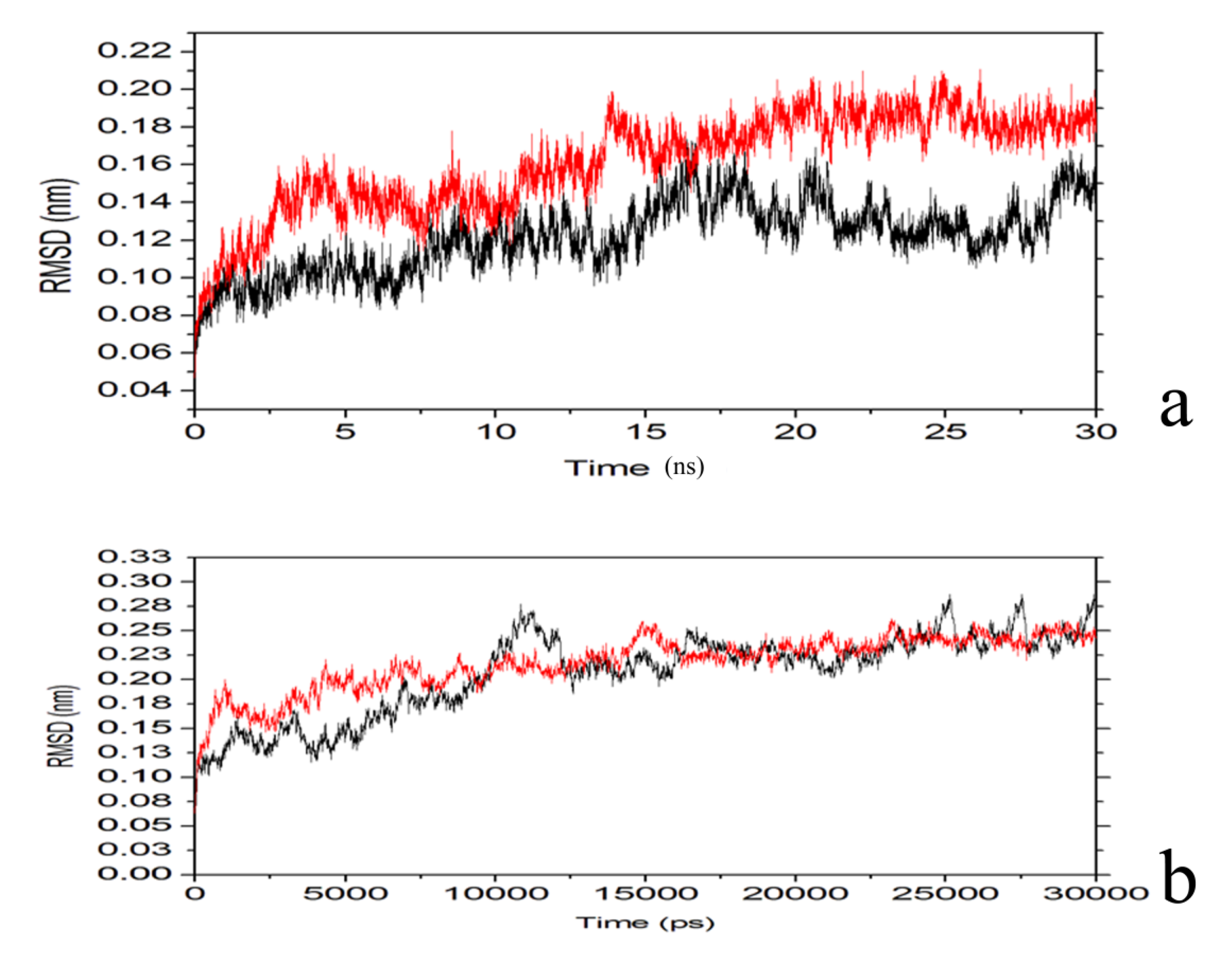
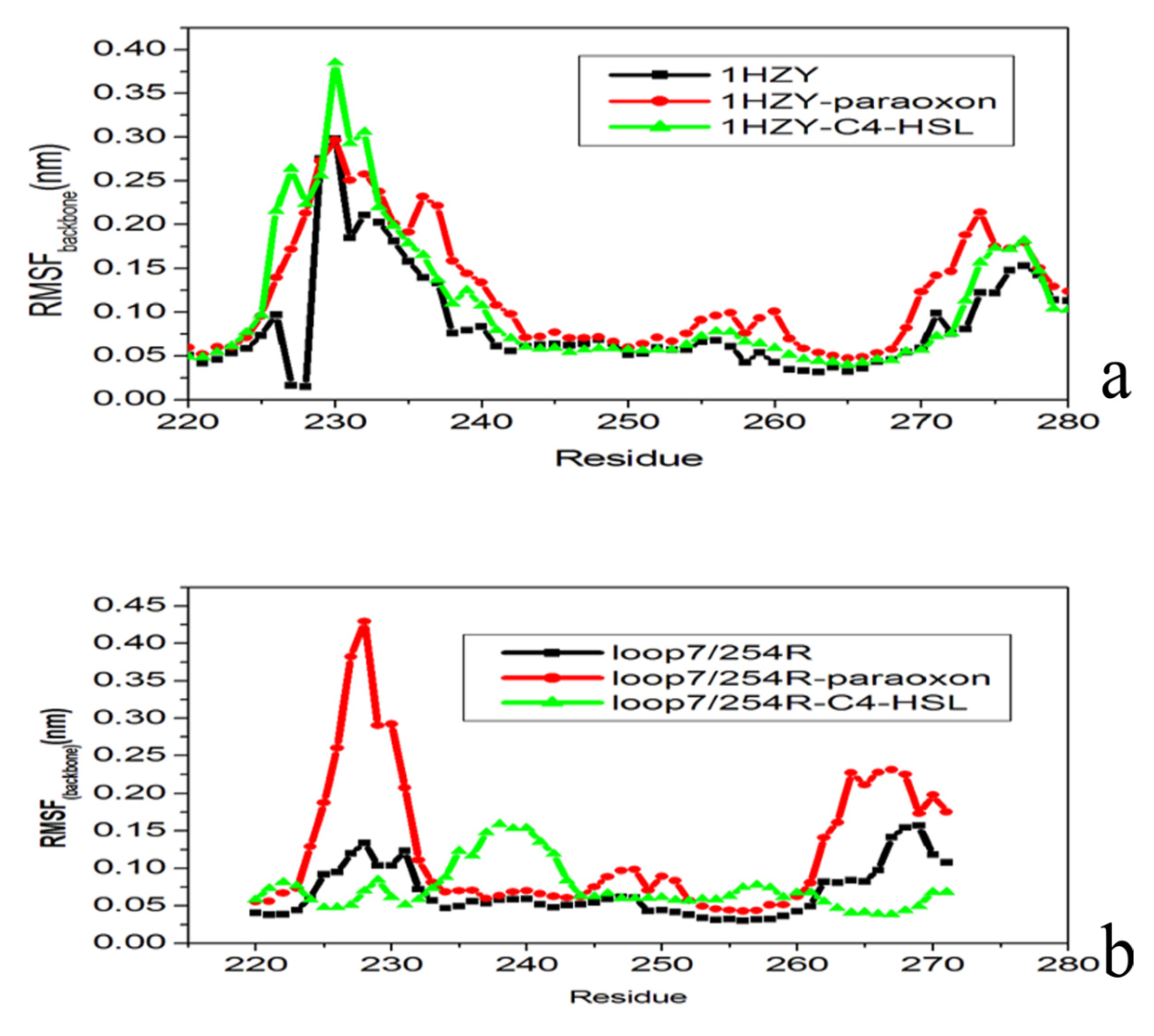
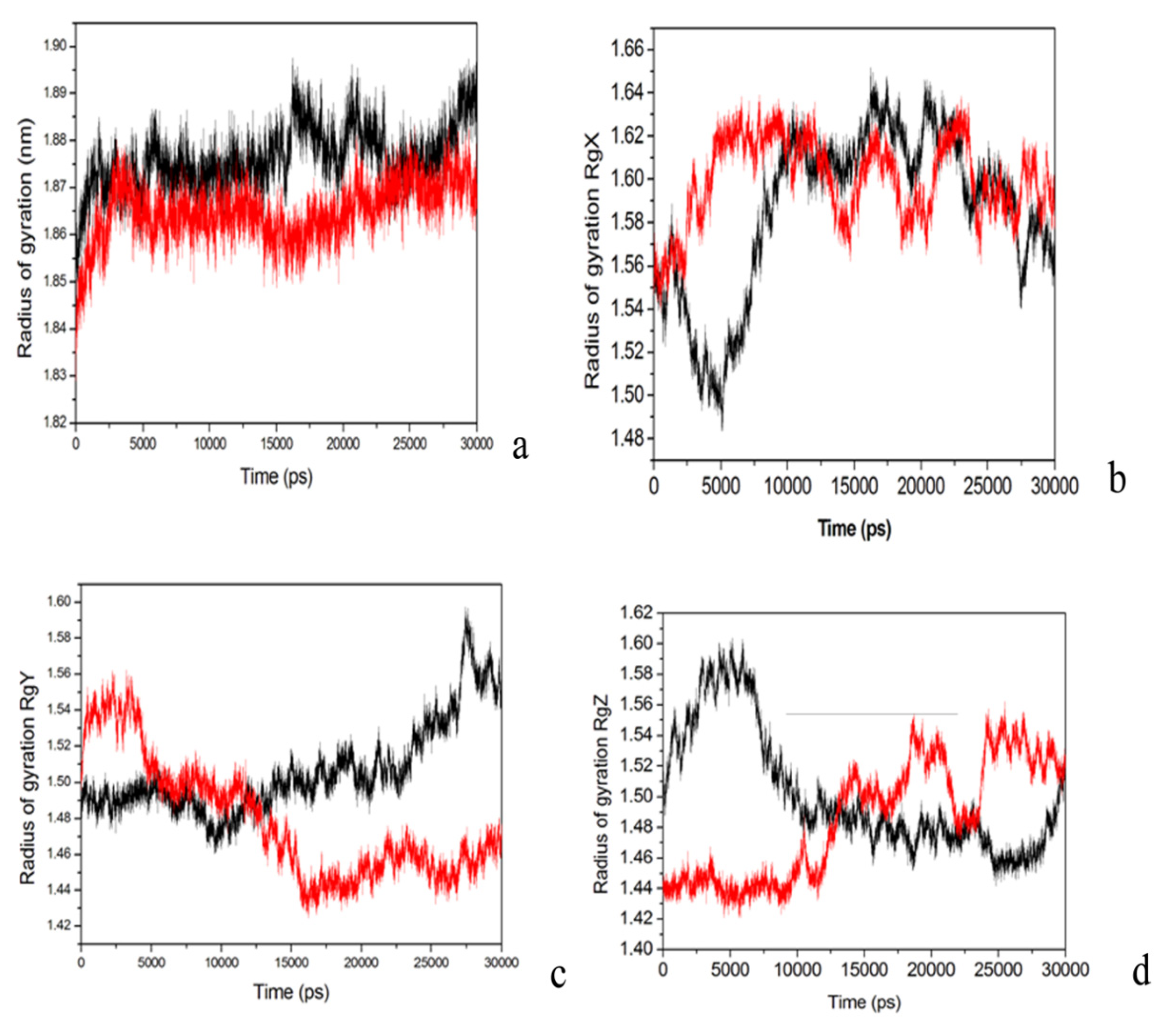
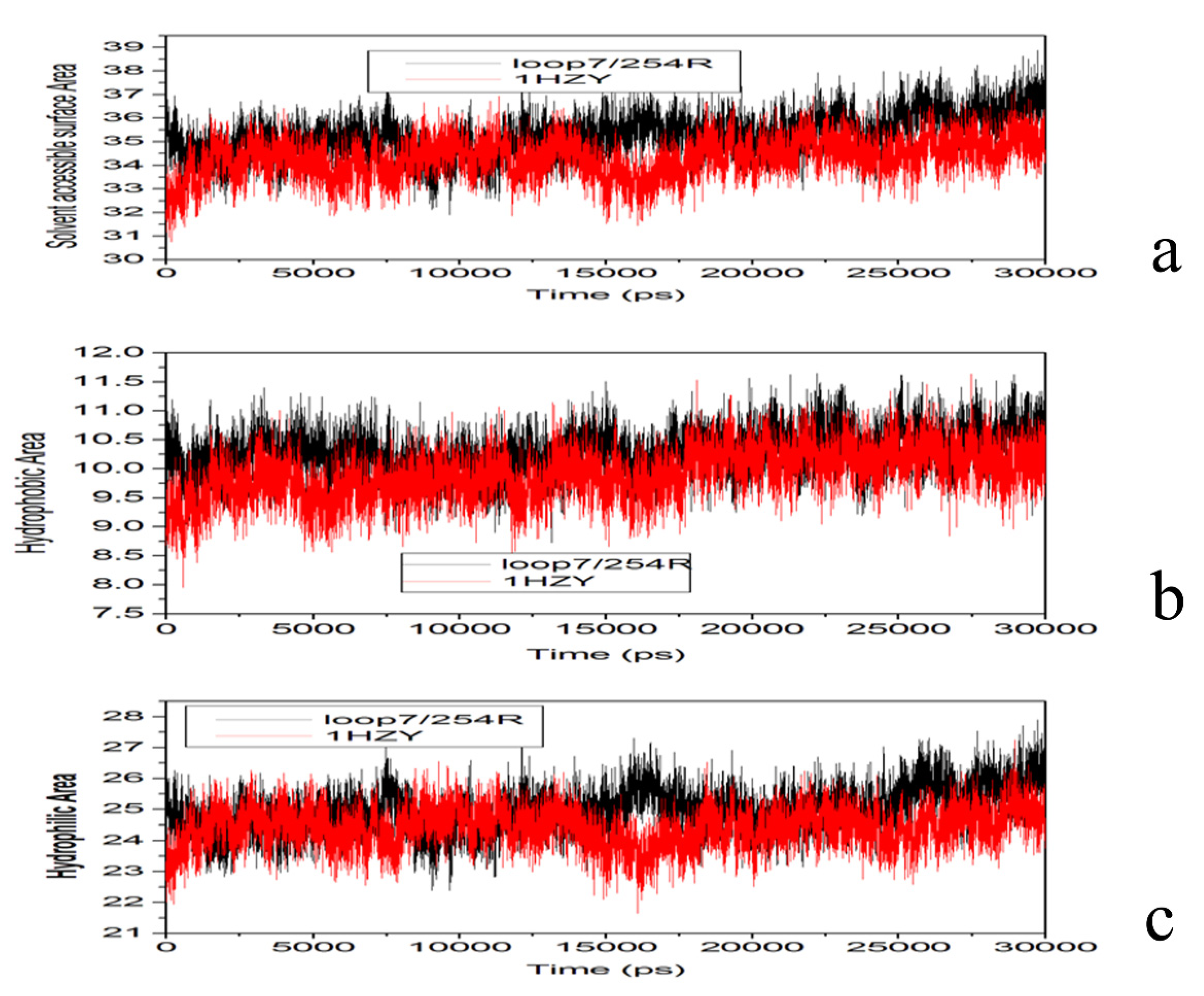
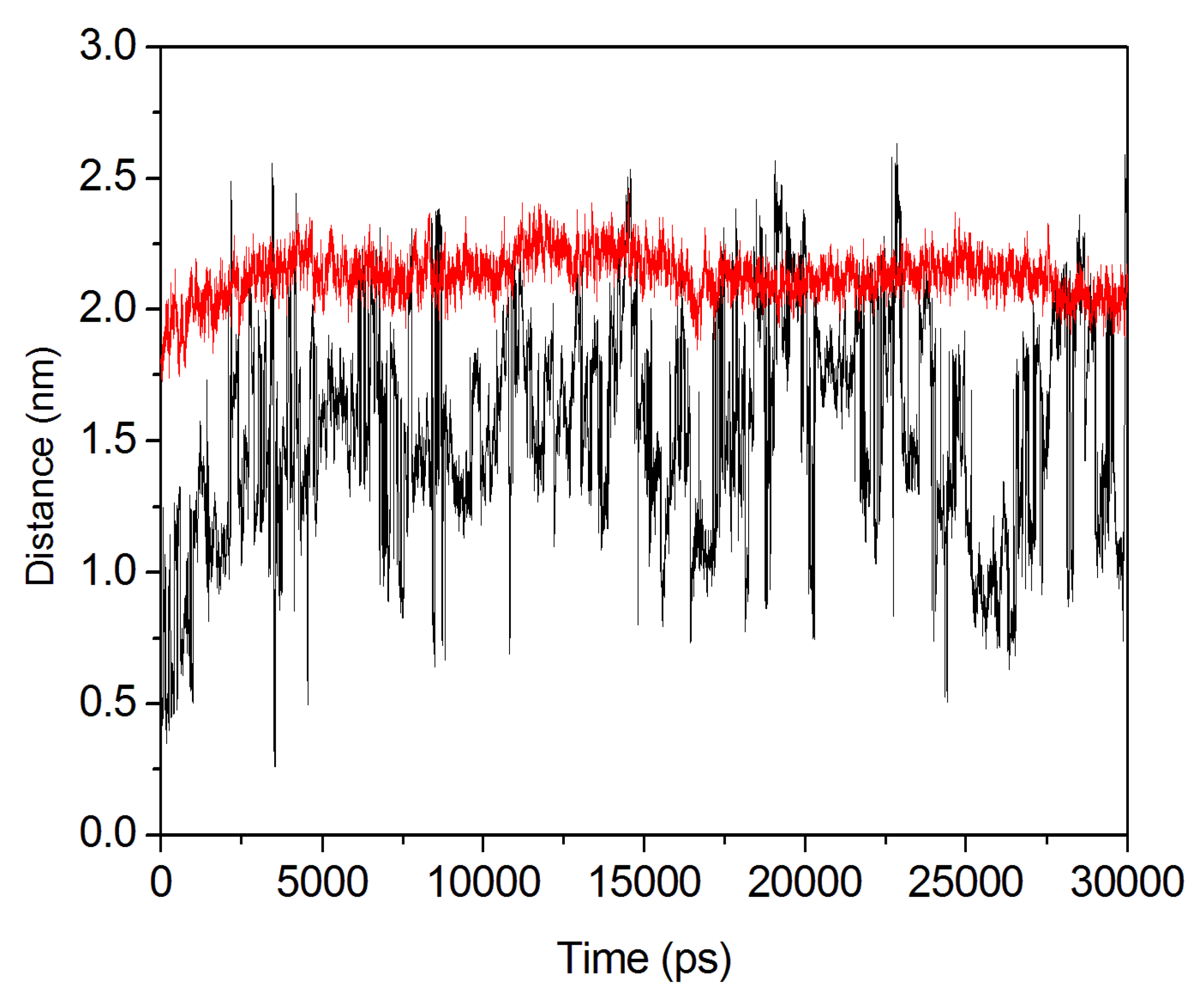
2.4. Conformational Dynamics of the PTE and the Variant upon Substrate Binding





3. Experimental
3.1. Computational Methodology
3.1.1. Protein Preparation
3.1.2. Molecular Dynamics Simulation
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Tsai, P.C.; Bigley, A.; Li, Y.C.; Ghanem, E.; Gadieu, E.; Kasten, S.A.; Reeves, T.E.; Gerasoli, D.C.; Raushel, F.M. Stereoselective hydrolysis of organophosphate nerve agents by the bacterial phosphotriesterase. Biochemistry 2010, 49, 7978–7987. [Google Scholar] [CrossRef]
- Hiblot, J.; Gotthard, G.; Champion, C.; Chabrierea, E.; Elias, M. Crystallization and preliminary X-ray diffraction analysis of the lactonase VmoLac from Vulcanisaeta moutnovskia. Acta Crystallogr. F 2013, F69, 1235–1238. [Google Scholar]
- Benschop, H.P.; de Jong, L.P.A. Nerve agent stereoisomers: Analysis, isolation, and toxicology. Acc. Chem. Res. 1988, 21, 368–374. [Google Scholar] [CrossRef]
- Tsai, P.C.; Fox, N.; Bigley, A.N.; Harvey, S.P.; Barondeau, D.P.; Raushel, F.M. Enzymes for the homeland defense: Optimizing phosphotriesterasefor the hydrolysis of organophosphate nerve agents. Biochemistry 2012, 51, 6463–6475. [Google Scholar] [CrossRef]
- Ordentlich, A.; Barak, D.; Sod-Moriah, G.; Kaplan, D.; Mizrahi, D.; Segall, Y.; Kronman, C.; Karton, Y.; Lazar, A.; Marcus, D.; et al. Stereoselectivity toward VX is determined by interactions with residues of the acyl pocket as well as of the peripheral anionic site of AChE. Biochemistry 2004, 43, 11255–11265. [Google Scholar] [CrossRef]
- Gomes, D.E.; Lins, R.D.; Pascutti, P.G.; Lei, C.; Soares, T.A. Conformational variability of organophosphorous hydrolase upon soman and paraoxon binding. J. Phys. Chem. B 2011, 115, 15389–15398. [Google Scholar] [CrossRef]
- Porzio, E.; di Gennaro, S.; Palma, A.; Manco, G. Mn2+ modulates the kinetic properties of an archaeal member of the PLL family. Chem. Biol. Interact. 2013, 203, 251–256. [Google Scholar] [CrossRef]
- Bigley, A.N.; Xu, C.; Henderson, T.J.; Harvey, S.P.; Raushel, F.M. Enzymatic neutralization of the chemical warfare agent VX: Evolution of phosphotriesterase for phosphorothiolate hydrolysis. J. Am. Chem. Soc. 2013, 135, 10426–10432. [Google Scholar] [CrossRef]
- Bird, S.B.; Sutherland, T.D.; Gresham, C.; Oakeshott, J.; Scott, C.; Eddleston, M. OpdA, a bacterial organophosphorus hydrolase, prevents lethality in rats after poisoning with highly toxic organophosphorus pesticides. Toxicology 2008, 247, 88–92. [Google Scholar] [CrossRef]
- Horne, I.; Sutherland, T.D.; Harcourt, R.L.; Russell, R.J.; Oakeshott, J.G. Identification of an opd (organophosphate degradation) gene in an Agrobacterium isolate. Appl. Environ. Microbiol. 2002, 68, 3371–3376. [Google Scholar] [CrossRef]
- Jackson, C.J.; Scott, C.; Carville, A.; Mansfield, K.; Ollis, D.L.; Bird, S.B. Pharmacokinetics of OpdA, an organophosphorus hydrolase, in the African green monkey. Biochem. Pharmacol. 2010, 80, 1079–1086. [Google Scholar]
- Russell, R.J.; Scott, C.; Jackson, C.J.; Pandey, R.; Pandey, G.; Taylor, M.C.; Coppin, C.W.; Liu, J.W.; Oakeshott, J.G. The evolution of new enzyme function: Lessons from xenobiotic metabolizing bacteria versus insecticide-resistant insects. Evol. Appl. 2011, 4, 225–248. [Google Scholar] [CrossRef]
- Scott, C.; Pandey, G.; Hartley, C.J.; Jackson, C.J.; Cheesman, M.J.; Taylor, M.C.; Pandey, R.; Khurana, J.L.; Teese, M.; Coppin, C.W.; et al. The enzymatic basis for pesticide bioremediation. Indian J. Microbiol. 2008, 48, 65–79. [Google Scholar] [CrossRef]
- Hiblot, J.; Gotthard, G.; Elias, M.; Chabriere, E. Differential active site loop conformations mediate promiscuous activities in the lactonase SsoPox. PLoS One 2013, 8, e75272. [Google Scholar]
- Dawson, R.M.; Pantelidis, S.; Rose, H.R.; Kotsonis, S.E. Degradationof nerve agents by an organophosphate-degrading agent (OpdA). J. Hazard Mater. 2008, 157, 308–314. [Google Scholar] [CrossRef]
- Chen-Goodspeed, M.; Sogorb, M.A.; Wu, F.; Hong, S.B.; Raushel, F.M. Structural determinants of the substrate and stereochemical specificity of phosphotriesterase. Biochemistry 2001, 40, 1325–1331. [Google Scholar]
- Goldsmith, M.; Ashani, Y.; Simo, Y.; Ben-David, M.; Leader, H.; Silman, I.; Sussman, J.L.; Tawfik, D.S. Evolved Stereoselective hydrolases for broad-spectrum G-type nerve agent detoxification. Chem. Biol. 2012, 19, 456–466. [Google Scholar] [CrossRef]
- Benning, M.M.; Kuo, J.M.; Raushel, F.M.; Holden, H.M. Three-dimensional structure of the binuclear metal center of phosphotriesterase. Biochemistry 1995, 34, 7973–7938. [Google Scholar] [CrossRef]
- Vanhooke, J.L.; Benning, M.M.; Raushel, F.M.; Holden, H.M. Three-dimensional structure of the zinc-containing phosphotriesterase with bound substrate analog diethyl 4-methylbenzylphosphonate. Biochemistry 1996, 35, 6020–6025. [Google Scholar]
- Thoden, J.B.; Phillips, G.N.J.; Neal, T.M.; Raushel, F.M.; Holden, H.M. Molecular structure of dihydroorotase: A paradigm for catalysis through the use of a binuclear metal center. Biochemistry 2001, 40, 6989–6997. [Google Scholar]
- Lewis, V.E.; Donarski, W.J.; Wild, J.R.; Raushel, F.M. Mechanism and stereochemical course at phosphorus of the reaction catalyzed by a bacterial phosphotriesterase. Biochemistry 2001, 27, 1591–1597. [Google Scholar]
- Aubert, S.D.; Li, Y.C.; Raushel, F.M. Mechanism for the hydrolysis of organophosphates by the bacterial phosphotriesterase. Biochemistry 2004, 43, 5707–5715. [Google Scholar] [CrossRef]
- Samples, C.R.; Raushel, F.M.; DeRose, V.J. Activation of the binuclear metal center through formation of phosphotriesterase-inhibitor complexes. Biochemistry 2007, 46, 3435–3442. [Google Scholar] [CrossRef]
- Afriat-Jurnou, L.; Jackson, C.J.; Tawfik, D.S. Reconstructing a missing link in the evolution of a recently diverged phosphotriesterase by active-site loop remodeling. Biochemistry 2012, 51, 6047–6055. [Google Scholar]
- Elias, M.; Tawfik, D.S. Divergence and convergence in enzyme evolution: Parallel evolution of paraoxonases from quorumquenching lactonases. J. Biol. Chem. 2011, 287, 11–20. [Google Scholar] [CrossRef]
- Xiang, D.F.; Kolb, P.; Fedorov, A.A.; Meier, M.M.; Fedorov, L.V.; Nguyen, T.T.; Sterner, R.; Almo, S.C.; Shoichet, B.K.; Raushel, F.M. Functional annotation and three-dimensional structure of Dr0930 from Deinococcus radiodurans, a close relative of phosphotriesterase in the amidohydrolase superfamily. Biochemistry 2009, 48, 2237–2247. [Google Scholar] [CrossRef]
- Chow, J.Y.; Wu, L.; Yew, W.S. Directed evolution of a quorum-quenching lactonase from Mycobacterium avium subsp. paratuberculosis K-10 in the amidohydrolase superfamily. Biochemistry 2009, 48, 4344–4353. [Google Scholar] [CrossRef]
- Kiefer, F.; Arnold, K.; Künzli, M.; Bordoli, L.; Schwede, T. The SWISS-MODEL repository and associated resources. Nucleic Acids Res. 2009, 37, D387–D392. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W. PROCHECK—a program to check the stereochemical quality of protein structures. J. App. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 104, 435–447. [Google Scholar]
- Liang, J.; Woodward, C.; Edlsbrunner, H. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. 1998, 7, 1884–1897. [Google Scholar] [CrossRef]
- Tsai, P.C.; Fan, Y.; Kim, J.; Yang, L.; Almo, S.C.; Gao, Y.Q.; Raushel, F.M. Structural determinants for the stereoselective hydrolysis of chiral substrates by phosphotriesterase. Biochemistry 2010, 49, 7988–7997. [Google Scholar]
- Raushel, F.M.; Holden, H.M. Phosphotriesterase: An enzyme in search of its natural substrate. In Advances in Enzymology and Related Areas of Molecular Biology; Purich, D.L., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; Volume 74, pp. 51–93. [Google Scholar]
- Rochu, D.; Viguie, N.; Renault, F.; Crouzier, D.; Froment, M.T.; Masson, P. Contribution of the active-site metal cation to the catalytic activity and to the conformational stability of phosphotriesterase: temperature- and pH-dependence. Biochem. J. 2004, 380, 627–633. [Google Scholar] [CrossRef]
- Jackson, C.J.; Foo, J.L.; Kim, H.K.; Carr, P.D.; Liu, J.W.; Salem, G.; Ollis, D.L. In crystallo capture of a michaelis complex and product-binding modes of a bacterial phosphotriesterase. J. Mol. Biol. 2008, 375, 1189–1196. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Pei, B.Q.; Li, H.; Li, D.Y.; Fan, Y.B.; Wang, C.; Wu, S.Q. Creep bulging deformation of intervertebral disc under axial compression. Biomed. Mater. Eng. 2013, 23, S191–S198. [Google Scholar]
- Jenkins, A.B.; Batterham, M.; Samocha-Bonet, D.; Tonks, K.; Greenfield, J.R.; Campbell, L.V. Segregation of a latent high adiposity phenotype in families with a history of type 2 diabetes mellitus implicates rare obesity-susceptibility genetic variants with large effects in diabetes-related obesity. PLoS One 2013, 8, e70435. [Google Scholar]
- Huey, H.; Morris, G.M.; Olson, A.J.; Goodsell, D.H. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar]
- Oostenbrink, C.; Soares, T.A.; van der Vegt, N.F.A.; van Gunsteren, W.F. Validation of the 53A6 GROMOS force field. Eur. Biophys. J. 2005, 34, 273–284. [Google Scholar] [CrossRef]
- Soares, T.A.; Daura, X.; Oostenbrink, C.; Smith, L.J.; van Gunsteren, W.F. Validation of the GROMOS force-field parameter set 45A3 against nuclear magnetic resonance data of hen egg lysozyme. J. Biomol. NMR 2004, 30, 407–422. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. In Intermolecular Forces; Pullman, B., Ed.; Dordrecht Reidel: Boston, MA, USA, 1981; pp. 331–342. [Google Scholar]
- Van Gunsteren, W.F.; Berendsen, H.J.C. A leap-frog algorithm for stochastic dynamics. Mol. Simul. 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Jackson, C.J.; Carr, P.D.; Kim, H.K.; Liu, J.W.; Herrald, P.; Mitić, N.; Schenk, G.; Smith, C.A.; Ollis, D.L. Anomalous scattering analysis of Agrobacterium radiobacter phosphotriesterase: The prominent role of iron in the heterobinuclear active site. Biochem. J. 2006, 397, 501–508. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhan, D.; Zhou, Z.; Guan, S.; Han, W. The Effect of Conformational Variability of Phosphotriesterase upon N-acyl-L-homoserine Lactone and Paraoxon Binding: Insights from Molecular Dynamics Studies. Molecules 2013, 18, 15501-15518. https://doi.org/10.3390/molecules181215501
Zhan D, Zhou Z, Guan S, Han W. The Effect of Conformational Variability of Phosphotriesterase upon N-acyl-L-homoserine Lactone and Paraoxon Binding: Insights from Molecular Dynamics Studies. Molecules. 2013; 18(12):15501-15518. https://doi.org/10.3390/molecules181215501
Chicago/Turabian StyleZhan, Dongling, Zhenhuan Zhou, Shanshan Guan, and Weiwei Han. 2013. "The Effect of Conformational Variability of Phosphotriesterase upon N-acyl-L-homoserine Lactone and Paraoxon Binding: Insights from Molecular Dynamics Studies" Molecules 18, no. 12: 15501-15518. https://doi.org/10.3390/molecules181215501
APA StyleZhan, D., Zhou, Z., Guan, S., & Han, W. (2013). The Effect of Conformational Variability of Phosphotriesterase upon N-acyl-L-homoserine Lactone and Paraoxon Binding: Insights from Molecular Dynamics Studies. Molecules, 18(12), 15501-15518. https://doi.org/10.3390/molecules181215501