First-Principles Elucidation of the Surface Chemistry of the C2Hx (x = 0–6) Adsorbate Series on Fe(100)

Abstract
:1. Introduction
2. Results and Discussion
2.1. Acetylene, Ethylene, Ethane and Intermediates: Stable Adsorption States


2.2. Elementary Reaction Steps Connecting Reaction Intermediates and Molecules: Co-Adsorption Configurations and Minimum-Energy Paths
2.3. Surface Reaction Mechanisms


{kind=link}
{kind=link}
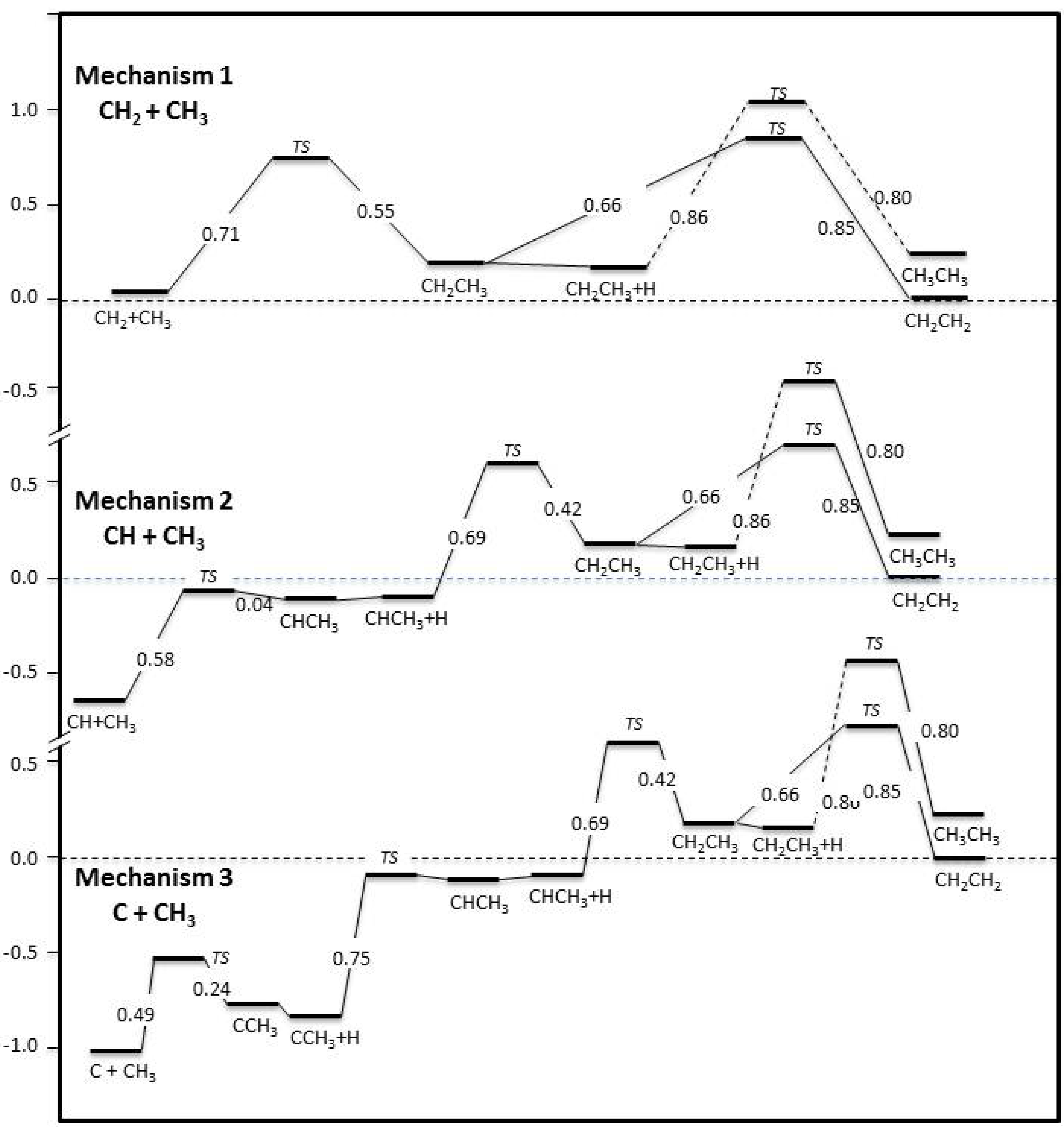{kind=link}
{kind=link}

2.4. Chemical Potential Profiles for Acetylene, Ethylene and Ethane Formation

2.5. Selection of Three Viable Reaction Mechanisms

2.6. Co-Existence of Many Surface Species: An Interconversion Scheme

3. Computational Method
4. Conclusions
Acknowledgments
References
- Chorkendorff, I.; Niemantsverdriet, J.W. Introduction to Catalysis. In Concepts of Modern Kinetics and Catalysis; Wiley-VCH: Weinheim, Germany, 2003; Volume 1, pp. 1–21. [Google Scholar]
- Claeys, M.; van Steen, E. Basic Studies. In Vol. 152: Fischer-Tropsch Technology; Steynberg, A.P., Dry, M.E., Eds.; Elsevier B.V: Amsterdam, The Netherlands, 2004; Volume 1, pp. 601–680. [Google Scholar]
- Van der Laan, G.P.; Beenackers, A.A.C.M. Kinetics and selectivity of the Fischer-Tropsch synthesis: A literature review. Catal. Rev.-Sci. Eng. 1999, 41, 255–318. [Google Scholar] [CrossRef]
- Schulz, H. Short history and present trends of Fischer-Tropsch synthesis. Appl. Catal. A-Gen. 1999, 186, 3–12. [Google Scholar] [CrossRef]
- Schulz, H.; Claeys, M. Reactions of α-olefins of different chain length added during Fischer-Tropsch synthesis on a cobalt catalyst in a slurry reactor. Appl. Catal. A-Gen. 1999, 186, 91–107. [Google Scholar] [CrossRef]
- Nijs, H.H.; Jacobs, P.A. New evidence for the mechanism of the Fischer-Tropsch synthesis of hydrocarbons. J. Catal. 1980, 66, 401–411. [Google Scholar] [CrossRef]
- Henrici-Olive, G.; Olive, S. Mechanism of the Fischer-Tropsch synthesis: Origin of oxygenates. J. Mol. Catal. 1984, 24, 7–13. [Google Scholar]
- Joyner, R.W. The role of surface science studies in elucidating the mechanism of the Fischer-Tropsch hydrocarbon synthesis. Vacuum 1988, 38, 309–315. [Google Scholar] [CrossRef]
- Adesina, A.A.; Hudgins, R.R.; Silveston, P.L. Effect of ethene addition during the Fischer-Tropsch reaction. Appl. Catal. 1990, 62, 295–308. [Google Scholar] [CrossRef]
- Carter, M.K. A molecular mechanism for Fischer–Tropsch catalysis. J. Mol. Catal. A-Chem. 2001, 172, 193–206. [Google Scholar] [CrossRef]
- Ndlovu, S.B.; Phala, N.S.; Hearshaw-Timme, M.; Beagly, P.; Moss, J.R.; Claeys, M.; van Steen, E. Some evidence refuting the alkenyl mechanism for chain growth in iron-based Fischer–Tropsch synthesis. Catal. Tod. 2002, 71, 343–349. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Fan, L.T.; Shafie, S.; Bertok, B.; Friedler, F. Generation of light hydrocarbons through Fischer–Tropsch synthesis: Identification of potentially dominant catalytic pathways via the graph–theoretic method and energetic analysis. Comput. Chem. Eng. 2009, 33, 1182–1186. [Google Scholar] [CrossRef]
- Biloen, P.; Sachtler, W.M.H. Mechanism of hydrocarbon synthesis over Fischer-Tropsch catalysts. Adv. Catal. 1981, 30, 165–216. [Google Scholar] [CrossRef]
- Gaube, J.; Klein, H.F. Studies on the reaction mechanism of the Fischer–Tropsch synthesis on iron and cobalt. J. Mol. Catal. A-Chem. 2008, 283, 60–68. [Google Scholar]
- Gaube, J.; Klein, H.F. Further support for the two-mechanisms hypothesis of Fischer–Tropsch synthesis. Appl. Catal. A-Gen. 2010, 374, 120–125. [Google Scholar] [CrossRef]
- Schulz, H.; Riedel, T.; Schaub, G. Fischer–Tropsch principles of co-hydrogenation on iron catalysts. Top. Catal. 2005, 32, 117–124. [Google Scholar] [CrossRef]
- Maitlis, P.M.; Zanotti, V. The role of electrophilic species in the Fischer–Tropsch reaction. Chem. Commun. 2009, 1619–1634. [Google Scholar] [CrossRef]
- Maitlis, P.M.; Quyoum, R.; Long, H.C.; Turner, M.L. Towards a chemical understanding of the Fischer–Tropsch reaction: alkene formation. Appl. Catal. A-Gen. 1999, 186, 363–374. [Google Scholar] [CrossRef]
- Dry, M.E. Practical and theoretical aspects of the catalytic Fischer-Tropsch process. Appl. Catal. A-Gen. 1996, 138, 319–344. [Google Scholar] [CrossRef]
- Dry, M.E. Catalytic aspects of industrial Fischer-Tropsch synthesis. J. Mol. Catal. 1982, 17, 133–144. [Google Scholar]
- Davis, B.H. Fischer–Tropsch synthesis: Current mechanism and futuristic needs. Fuel Process. Technol. 2001, 71, 157–166. [Google Scholar] [CrossRef]
- Davis, B.H. Fischer–Tropsch Synthesis: Reaction mechanisms for iron catalysts. Catal. Tod. 2009, 141, 25–33. [Google Scholar] [CrossRef]
- Van der Laan, G.P.; Beenackers, A.A.C.M. Intrinsic kinetics of the gas–solid Fischer–Tropsch and water gas shift reactions over a precipitated iron catalyst. Appl. Catal. A-Gen. 2000, 193, 39. [Google Scholar] [CrossRef]
- Wang, Y.N.; Ma, W.P.; Lu, Y.J.; Yang, J.; Xu, Y.Y.; Xiang, H.W.; Li, Y.W.; Zhao, Y.L.; Zhang, B.J. Kinetics modelling of Fischer–Tropsch synthesis over an industrial Fe–Cu–K catalyst. Fuel 2003, 82, 195–213. [Google Scholar] [CrossRef]
- Ciobîca, I.M.; Kramer, G.J.; Ge, Q.; Neurock, M.; van Santen, R.A. Mechanisms for chain growth in Fischer–Tropsch synthesis over Ru(0001). J. Catal. 2002, 212, 136–144. [Google Scholar] [CrossRef]
- Ciobîca, I.M. The Molecular Basis of the Fischer Tropsch Reaction. Ph.D. thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2002. [Google Scholar]
- Van Santen, R.A.; Ciobîca, I.M.; van Steen, E.; Ghouri, M.M. Mechanistic issues in Fischer-Tropsch catalysis. Adv. Catal. 2011, 54, 127–187. [Google Scholar] [CrossRef]
- Cheng, J.; Gong, X.Q.; Hu, P.; Lok, C.M.; Ellis, P.; French, S. A quantitative determination of reaction mechanisms from density functional theory calculations: Fischer–Tropsch synthesis on flat and stepped cobalt surfaces. J. Catal. 2008, 254, 285–295. [Google Scholar] [CrossRef]
- Cheng, J.; Hu, P.; Ellis, P.; French, S.; Kelly, G.; Lok, C.M. Chain growth mechanism in Fischer−Tropsch synthesis: A DFT study of C−C coupling over Ru, Fe, Rh, and Re surfaces. J. Phys. Chem. C 2008, 112, 6082–6086. [Google Scholar] [CrossRef]
- Cheng, J.; Hu, P.; Ellis, P.; French, S.; Kelly, G.; Lok, C.M. A DFT study of the chain growth probability in Fischer–Tropsch synthesis. J. Catal. 2008, 257, 221–228. [Google Scholar] [CrossRef]
- Liu, Z.P.; Hu, P. A new insight into Fischer-Tropsch synthesis. J. Am. Chem. Soc. 2002, 124, 11568–11569. [Google Scholar] [CrossRef]
- Zhuo, M.; Tan, K.F.; Borgna, A.; Saeys, M. Density functional theory study of the CO insertion mechanism for Fischer−Tropsch synthesis over Co catalysts. J. Phys. Chem. C 2009, 113, 8357–8365. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Sun, K.J.; Ma, X.F.; Liu, J.X.; Sun, D.P.; Su, H.Y.; Li, W.X. Carbon chain growth by formyl insertion on rhodium and cobalt catalysts in syngas conversion. Angew. Chem. Int. Ed. 2011, 50, 5335–5338. [Google Scholar]
- Shetty, S.G.; Ciobîca, I.M.; Hensen, E.J. M.; van Santen, R.A. Site regeneration in the Fischer–Tropsch synthesis reaction: a synchronized CO dissociation and C–C coupling pathway. Chem. Commun. 2011, 47, 9822–9824. [Google Scholar]
- Cao, D.-B.; Li, Y.-W.; Wang, J.; Jiao, H. Chain growth mechanism of Fischer–Tropsch synthesis on Fe5C2(001). J. Mol. Catal. A-Chem. 2011, 346, 55–69. [Google Scholar] [CrossRef]
- Cheng, J.; Hu, P.; Ellis, P.; French, S.; Kelly, G.; Lok, C.M. Density functional theory study of Iron and Cobalt carbides for Fischer-Tropsch synthesis. J. Phys. Chem. C 2010, 114, 1085–1093. [Google Scholar] [CrossRef]
- Lo, J.M.H.; Ziegler, T. Theoretical studies of the formation and reactivity of C2 hydrocarbon species on the Fe (100) surface. J. Phys. Chem. C 2007, 111, 13149–13162. [Google Scholar] [CrossRef]
- Lee, G.D.; Han, S.W.; Yu, J.J.; Ihm, J. Catalytic decomposition of acetylene on Fe (001): A first-principles study. Phys. Rev. B 2002, 66, 081403. [Google Scholar] [CrossRef]
- Anderson, A.B.; Mehandru, S.P. Acetylene adsorption to Fe (100), (110), and (111) surfaces; structures and reactions. Surf. Sci. 1984, 136, 398–418. [Google Scholar] [CrossRef]
- Mehandru, S.P.; Anderson, A.B. Dependence of carbon-carbon and carbon-hydrogen bond activation on d band position: acetylene on platinum(111) and iron(100). An electrochemical model. J. Am. Chem. Soc. 1985, 107, 844–849. [Google Scholar] [CrossRef]
- Cheng, J.; Hu, P.; Ellis, P.; French, S.; Kelly, G.; Lok, C.M. A DFT study of the transition metal promotion effect on ethylene chemisorption on Co (0001). Surf. Sci. 2009, 603, 2752–2758. [Google Scholar] [CrossRef]
- Xu, L.S.; Ma, Y.S.; Wu, Z.F.; Chen, B.H.; Yuan, Q.; Huang, W.X. A photoemission study of ethylene decomposition on a Co(0001) surface: Formation of different types of carbon species. J. Phys. Chem. C 2012, 116, 4167–4174. [Google Scholar]
- Swart, J.C.W.; Ciobîca, I.M.; van Santen, R.A.; van Steen, E. Intermediates in the formation of graphitic carbon on a flat FCC-Co (111) surface. J. Phys. Chem. C 2008, 112, 12899–12904. [Google Scholar]
- Swart, J.C.W.; van Steen, E.; Ciobîca, I.M.; van Santen, R.A. Interaction of graphene with FCC-Co(111). Phys. Chem. Chem. Phys. 2009, 11, 803–807. [Google Scholar] [CrossRef]
- Ciobîca, I.M.; van Santen, R.A.; van Berge, P.J.; de Loosdrecht, J.V. Adsorbate induced reconstruction of cobalt surfaces. Surf. Sci. 2008, 602, 17–27. [Google Scholar] [CrossRef]
- van Helden, P.; Ciobîca, I.M. A DFT study of carbon in the subsurface layer of cobalt surfaces. ChemPhysChem. 2011, 12, 2925–2928. [Google Scholar] [CrossRef]
- Bernardo, C.G.P.M.; Gomes, J.A.N.F. The adsorption of ethylene on the (100) surfaces of platinum, Palladium and nickel: A DFT study. J. Mol. Struct.(Theochem) 2001, 542, 263–271. [Google Scholar] [CrossRef]
- Whitten, J.L.; Yang, H. Theoretical studies of surface reactions on metals: I. Ethyl to ethylene conversion on Ni(100). II. Photodissociation of methane on platinum. Catal. Tod. 1999, 50, 603–612. [Google Scholar]
- Ostrom, H.; Foehlisch, A.; Nyberg, M.; Weinelt, M.; Heske, C.; Pettersson, L.G.M.; Nilsson, A. Ethylene on Cu (110) and Ni (110): electronic structure and bonding derived from X-ray spectroscopy and theory. Surf. Sci. 2004, 559, 85–99. [Google Scholar] [CrossRef]
- Li, B.; Bao, S.N.; Zhuang, Y.Y.; Cao, P.L. The adsorption geometry of ethylene on the Ni (110) surface. Acta Phys. Sin. 2003, 52, 202–206. [Google Scholar]
- Li, B.; Bao, S.N.; Cao, P.L. Adsorption geometry of C2H4 and C2H on Ni (110) surface. Acta Phys. Sin. 2005, 54, 5784–5790. [Google Scholar]
- Gutdeutsch, U.; Birkenheuer, U.; Bertel, E.; Cramer, J.; Boettger, J.C.; Roesch, N. On the adsorption site of ethylene at the Ni (110) surface: A combined experimental and theoretical study involving the unoccupied band structure. Surf. Sci. 1996, 345, 331–346. [Google Scholar] [CrossRef]
- Vang, R.T.; Honkala, K.; Dahl, S.; Vestergaard, E.K.; Schnadt, J.; Laegsgaard, E.; Clausen, B.S.; Nørskov, J.K.; Besenbacher, F. Ethylene dissociation on flat and stepped Ni (111): A combined STM and DFT study. Surf. Sci. 2006, 600, 66–77. [Google Scholar] [CrossRef]
- Bromfield, T.C.; Curulla-Ferré, D.; Niemantsverdriet, J.W. A DFT study of the adsorption and dissociation of CO on Fe (100): Influence of surface coverage on the nature of accessible adsorption states. ChemPhysChem 2005, 6, 254–260. [Google Scholar]
- Scheijen, F.J.E.; Curulla-Ferré, D.; Niemantsverdriet, J.W. Adsorption and dissociation of CO on body-centered cubic transition metals and alloys: Effect of coverage and scaling relations. J. Phys. Chem. C 2009, 113, 11041–11049. [Google Scholar]
- Elahifard, M.R.; Perez Jigato, M.; Niemantsverdriet, J.W. Direct versus hydrogen-assisted CO dissociation on the Fe (100) Surface: a DFT study. ChemPhysChem 2012, 13, 89–91. [Google Scholar] [CrossRef]
- Govender, A.; Curulla-Ferré, D.; Niemantsverdriet, J.W. The surface chemistry of water on Fe (100): A density functional theory study. ChemPhysChem 2012, 13, 1583–1590. [Google Scholar] [CrossRef]
- Govender, A.; Curulla-Ferré, D.; Niemantsverdriet, J.W. A density functional theory study on the effect of zero-point energy corrections on the methanation profile on Fe (100). ChemPhysChem 2012, 13, 1591–1596. [Google Scholar] [CrossRef]
- Niemantsverdriet, J.W.; van der Kraan, A.M.; van Dijk, W.L.; van der Baan, H.S. Behavior of metallic iron catalysts during Fischer-Tropsch synthesis studied with Mössbauer spectroscopy, x-ray diffraction, carbon content determination, and reaction kinetic measuremen. J. Phys. Chem. 1980, 84, 3363–3370. [Google Scholar]
- de Smit, E.; Cinquini, F.; Beale, A.M.; Safonova, O.V.; van Beek, W.; Sautet, P.; Weckhuysen, B.M. Stability and Reactivity of ε-χ- θ Iron Carbide Catalyst Phases in Fischer-Tropsch Synthesis: Controlling μC. J. Am. Chem. Soc. 2010, 132, 14928–14941. [Google Scholar]
- Steynberg, P.J.; van den Berg, J.A.; van Rensburg, W.J. Bulk and surface analysis of Hägg Fe carbide (Fe5C2): A density functional theory study. J. Phys. Condens. Matter 2008, 20, 064238. [Google Scholar] [CrossRef]
- Gracia, J.M.; Prinsloo, F.F.; Niemantsverdriet, J.W. Mars-van Krevelen-like mechanism of CO hydrogenation on an iron carbide surface. Catal. Lett. 2009, 133, 257–261. [Google Scholar]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Govender, A. Towards a mechanism for the Fischer-Tropsch synthesis on Fe(100) using density functional theory. Ph.D. thesis, Eindhoven University of Technology, Eindhoven, The Netherlands, 2010. [Google Scholar]
- Bernasek, S.L. Reaction of small molecules at well-characterized iron surfaces. Ann. Rev. Phys. Chem. 1993, 44, 265–298. [Google Scholar] [CrossRef]
- Hung, W.H.; Bernasek, S.L. Adsorption and decomposition of ethylene and acetylene on Fe (100). Surf. Sci. 1995, 339, 272–290. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Govender, A.; Curulla-Ferré, D.; Pérez-Jigato, M.; Niemantsverdriet, H. First-Principles Elucidation of the Surface Chemistry of the C2Hx (x = 0–6) Adsorbate Series on Fe(100). Molecules 2013, 18, 3806-3824. https://doi.org/10.3390/molecules18043806
Govender A, Curulla-Ferré D, Pérez-Jigato M, Niemantsverdriet H. First-Principles Elucidation of the Surface Chemistry of the C2Hx (x = 0–6) Adsorbate Series on Fe(100). Molecules. 2013; 18(4):3806-3824. https://doi.org/10.3390/molecules18043806
Chicago/Turabian StyleGovender, Ashriti, Daniel Curulla-Ferré, Manuel Pérez-Jigato, and Hans Niemantsverdriet. 2013. "First-Principles Elucidation of the Surface Chemistry of the C2Hx (x = 0–6) Adsorbate Series on Fe(100)" Molecules 18, no. 4: 3806-3824. https://doi.org/10.3390/molecules18043806
APA StyleGovender, A., Curulla-Ferré, D., Pérez-Jigato, M., & Niemantsverdriet, H. (2013). First-Principles Elucidation of the Surface Chemistry of the C2Hx (x = 0–6) Adsorbate Series on Fe(100). Molecules, 18(4), 3806-3824. https://doi.org/10.3390/molecules18043806