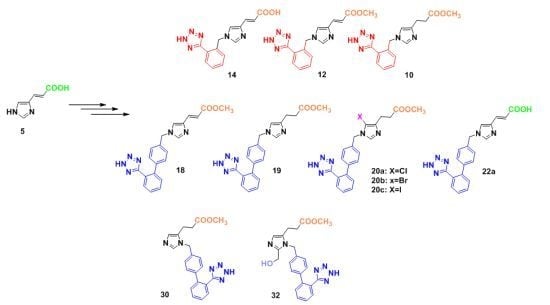
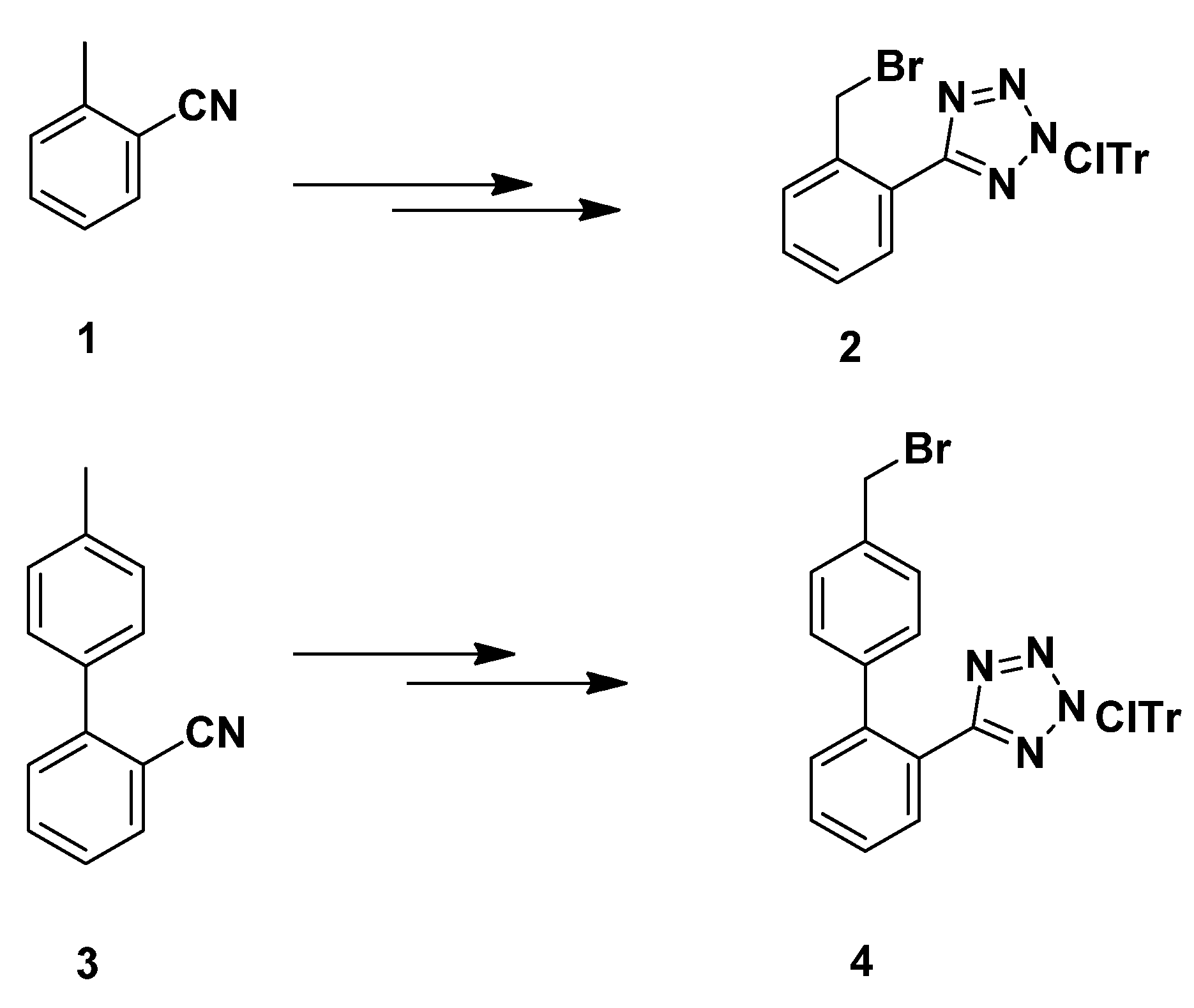
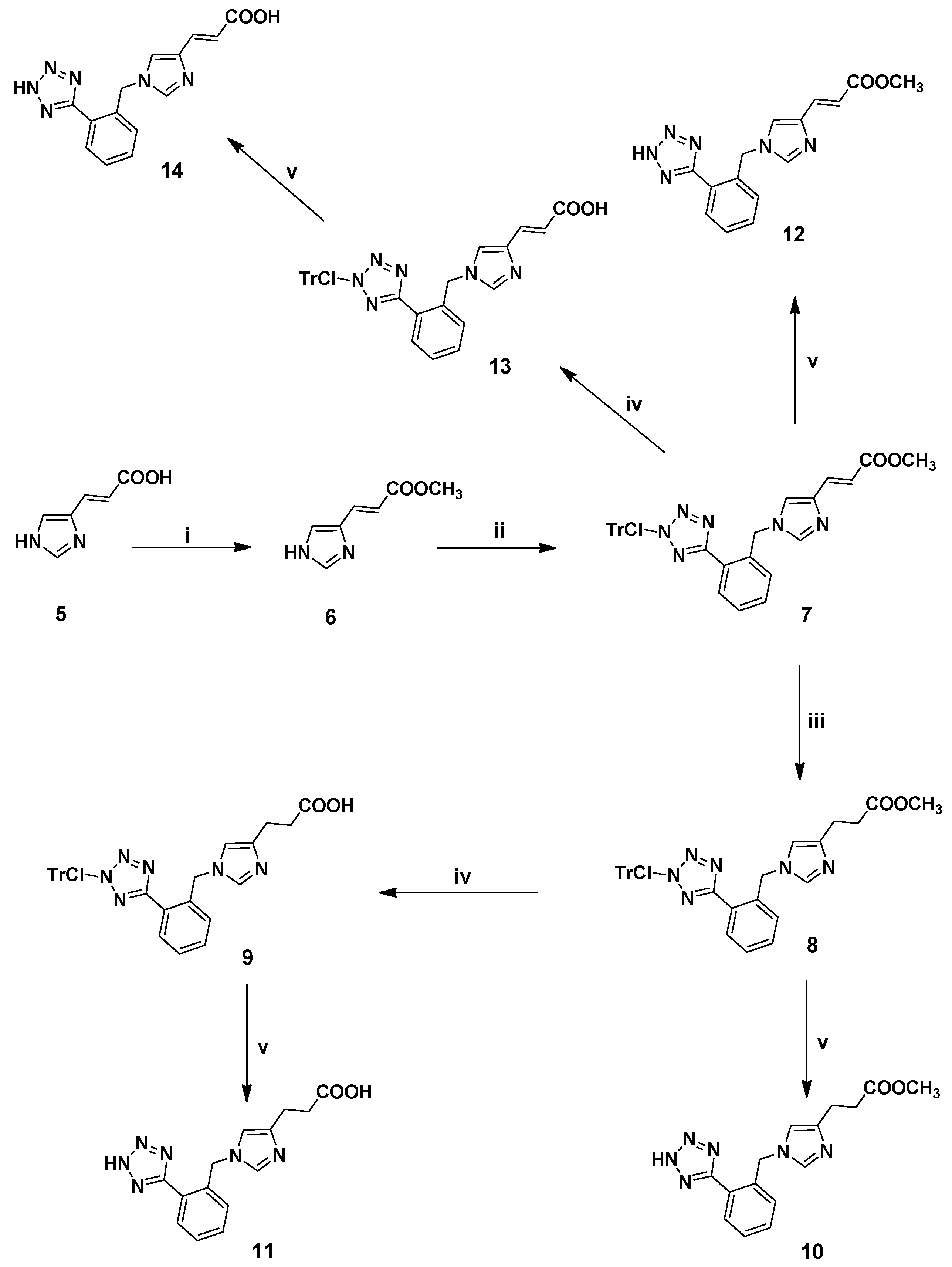
3.2.7. Synthesis of 1,4-Disubstituted Benzyl Analogues 10, 11, 12, 14
N-(2-Chlorotrityl)-5-(2-bromobenzyl)tetrazole (
2)
. Prepared from
1 according to the literature method [
8,
18,
24,
25]. Yield 78%; R
f = 0.46 (2:8 EtOAc:hexanes); ESI-MS (
m/z): 238.27 (M+H
+-ClTr), 277.78 (ClTr);
1H-NMR (CDCl
3):
δ 8.22–8.18 (m, 1H), 7.51–7.32 (m, 12H), 7.25–7.21 (m, 4H), 6.86 (d, 1H,
J = 7.6 Hz), 4.92 (s, 2H);
13C-NMR (CDCl
3):
δ 162.90, 141.28, 136.87, 131.64, 130.46, 129.96, 128.94, 128.42, 127.96, 127.84, 127.29, 126.43, 83.45, 32.42. Anal. Calcd for C
27H
20N
4ClBr (%): C: 62.87; H: 3.91; N: 10.86. Found (%): C: 62.99; H: 4.00; N: 10.54.
Methyl 3-(1H-imidazol-4-yl)acrylate (
6)
. Prepared from
5 according to the literature method [
20,
26]. Yield 95%; M.p. 92–94 °C; R
f = 0.42 (9:1 CHCl
3:MeOH); ESI-MS (
m/z): 153.22 (M+H
+);
1Η-ΝΜR (CD
3OD)
: δ 7.78 (s, 1H), 7.61 (d, 1Η,
J = 16.0 Hz), 7.43 (s, 1H), 6.44 (d, 1H,
J = 16.0 Hz), 3.78 (s, 3H);
13C-NMR (CDCl
3):
δ 168.14, 137.43, 135.25, 134.35, 124.0, 114.35, 50.61. Anal. Calcd for C
7H
8N
2O
2 (%): C: 55.26; H: 5.30; N: 18.41. Found (%): C: 55.21; H: 5.39; N: 18.37. All data were consistent with literature [
26].
(E)-Methyl 1-[[1-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]phenyl-2-yl]methyl]imidazole-4-acrylate (7). General procedure 1 was employed for the preparation of 7 using 2 as alkylating agent. Yield 74%; Rf = 0.38 (8:2 EtOAc:hexanes); ESI-MS (m/z): 587.27 (M+H+), 309.37 (M+H+-ClTr), 277.88 (ClTr); 1Η-ΝΜR (CDCl3): δ 8.28 (dd, 1H, J = 2.0, 7.6 Hz), 7.53–7.10 (m, 19H), 6.76 (s, 1H), 6.48 (d, 1H, J = 15.6 Hz), 5.38 (s, 2H), 3.77 (s, 3H). Anal. Calcd for C34H27N6O2Cl (%): C: 74.02; H: 4.30; N: 13.28. Found (%): C: 73.96; H: 4.22; N: 13.32.
Methyl 1-[[1-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]phenyl-2-yl]methyl]imidazole-4-propanoate (8). General procedure 2 was employed for the preparation of 8. Yield 90%; Rf = 0.45 (9:1 CHCl3:MeOH); ESI-MS (m/z): 589.17 (M+H+), 311.45 (M+H+-ClTr), 277.38 (ClTr); 1Η-ΝΜR (CDCl3): δ 8.18–8.15 (m, 1H), 7.42–7.26 (m, 9H), 7.07–6.99 (m, 9H), 6.37 (s, 1H), 5.27 (s, 2H), 3.57 (s, 3H), 2.76 (t, 2H, J = 7.2 Hz), 2.56 (t, 2H, J = 7.2 Hz). Anal. Calcd for C34H29N6O2Cl (%): C: 69.32; H: 4.96; N: 14.27. Found (%): C: 69.45; H: 4.88; N: 14.32.
1-[[1-[[N-(2-Chlorotrityl)]-1H-tetrazol-5-yl]phenyl-2-yl]methyl]imidazole-4-propanoic acid (9). General procedure 4 was employed for the preparation of 9. Yield 93%; Rf = 0.31 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 576.19 (M+H+), 299.63 (M+H+-ClTr), 277.77 (ClTr); 1Η-ΝΜR (CDCl3): δ 8.31 (d, 1Η, J = 7.2 Hz), 7.54–7.11 (m, 17H), 6.85 (d, 1H, J = 7.6 Hz), 6.42 (s, 1H), 5.40 (s, 2H), 2.73 (bs, 2H), 2.65 (bs, 2H). Anal. Calcd for C33H27N6O2 (%): C: 68.92; H: 4.73; N: 14.61. Found (%): C: 68.92; H: 4.73; N: 14.61.
Methyl 1-[[1-(1H-tetrazol-5-yl)phenyl-2-yl]methyl]imidazole-4-propanoate (10). General procedure 6 was employed for the preparation of 10. Yield 91%; Rf = 0.48 (8:2 CHCl3:MeOH); ESI-MS (m/z): 313.34 (M+H+); 1Η-ΝΜR (CD3OD): δ 8.80 (s, 1H), 7.96-7.93 (m, 1H), 7.64–7.60 (m, 3H), 7.33 (s, 1Η), 5.75 (s, 2H), 3.65 (s, 3H), 2.94 (t, 2H, J = 7.2 Hz), 2.69 (t, 2H, J = 7.2 Hz). Anal. Calcd for C15H16N6O2∙CF3COOH (%): C: 47.89; H: 4.02; N: 19.71. Found (%): C: 47.78; H: 3.95; N: 19.87.
1-[[1-(1H-Tetrazol-5-yl)phenyl-2-yl]methyl]imidazole-4-propanoic acid (11). General procedure 6 was employed for the preparation of 11. Yield 88%; Rf = 0.37 (4:1:1 n-butanol:acetic acid:H2O); ESI-MS (m/z): 297.24 (M+H+); 1Η-ΝΜR (CD3OD): δ 8.83 (s, 1H), 7.97–7.93 (m, 1H), 7.65–7.59 (m, 3H), 7.36 (s, 1H), 5.76 (s, 2H), 2.93 (t, 2H, J = 7.2 Hz), 2.66 (t, 2H, J = 7.2 Hz). Anal. Calcd for C14H14N6O2∙CF3COOH (%): C: 46.11; H: 3.67; N: 20.38. Found (%): C: 46.02; H: 3.57; N: 20.48.
(E)-Methyl 1-[[1-(1H-tetrazol-5-yl)phenyl-2-yl]methyl]imidazole-4-acrylate (12). General procedure 6 was employed for the preparation of 12. Yield 95%; Rf = 0.48 (8:2 CHCl3:MeOH); ESI-MS (m/z): 311.24 (M+H+); 1Η-ΝΜR (DMSO-d6): δ 8.46 (s, 1H), 7.90 (s, 1H), 7.79 (s, 1H), 7.62 (bs, 2H), 7.58 (m, 4Η), 7.50 (d, 1H, J = 15.6 Hz), 7.34 (s, 1H), 6.46 (d, 1H, J = 15.6 Hz), 5.68 (s, 2H), 3.78 (s, 3H). Anal. Calcd for C15H14N6O2∙CF3COOH (%): C: 48.12; H: 3.56; N: 19.81. Found (%): C: 48.22; H: 3.47; N: 19.71.
(E)-1-[[1-[[N-(2-Chlorotrityl)]-1H-tetrazol-5-yl]phenyl-2-yl]methyl]imidazole-4-acrylic acid (13). General procedure 4 was employed for the preparation of 13. Yield 95%; Rf = 0.30 (9:1 CHCl3:MeOH); ESI-MS (m/z): 574.15 (M+H+), 297.54 (M+H+-ClTr), 277.78 (ClTr); 1Η-ΝΜR (CDCl3): δ 8.28 (d, 2Η, J = 5.6 Hz), 7.52–7.23 (m, 12H), 7.10 (d, 6H, J = 5.6 Hz), 6.74 (bs, 1H), 6.44 (d, 1H, J = 15.6 Hz), 5.36 (s, 2H). Anal. Calcd for C33H25N6O2Cl (%): C: 69.17; H: 4.40; N: 14.67. Found (%): C: 69.11; H: 4.29; N: 14.72.
(E)-1-[[1-(1H-Tetrazol-5-yl)phenyl-2-yl]methyl]imidazole-4-acrylic acid (14). General procedure 6 was employed for the preparation of 14. Yield 95%; Rf = 0.50 (4:1:1 n-butanol:acetic acid:H2O); ESI-MS (m/z): 297.30 (M+H+); 1Η-ΝΜR (CD3OD): δ 8.85 (s, 1H), 7.98–7.95 (m, 1H), 7.84 (s, 1H), 7.72–7.64 (m, 3H), 7.48 (d, 1H, J = 16.0 Hz), 6.49 (d, 1H, J = 16.0 Hz), 5.80 (s, 2H). Anal. Calcd for C14H12N6O2∙CF3COOH (%): C: 48.12; H: 3.56; N: 19.81. Found (%): C: 48.22; H: 3.47; N: 19.71.
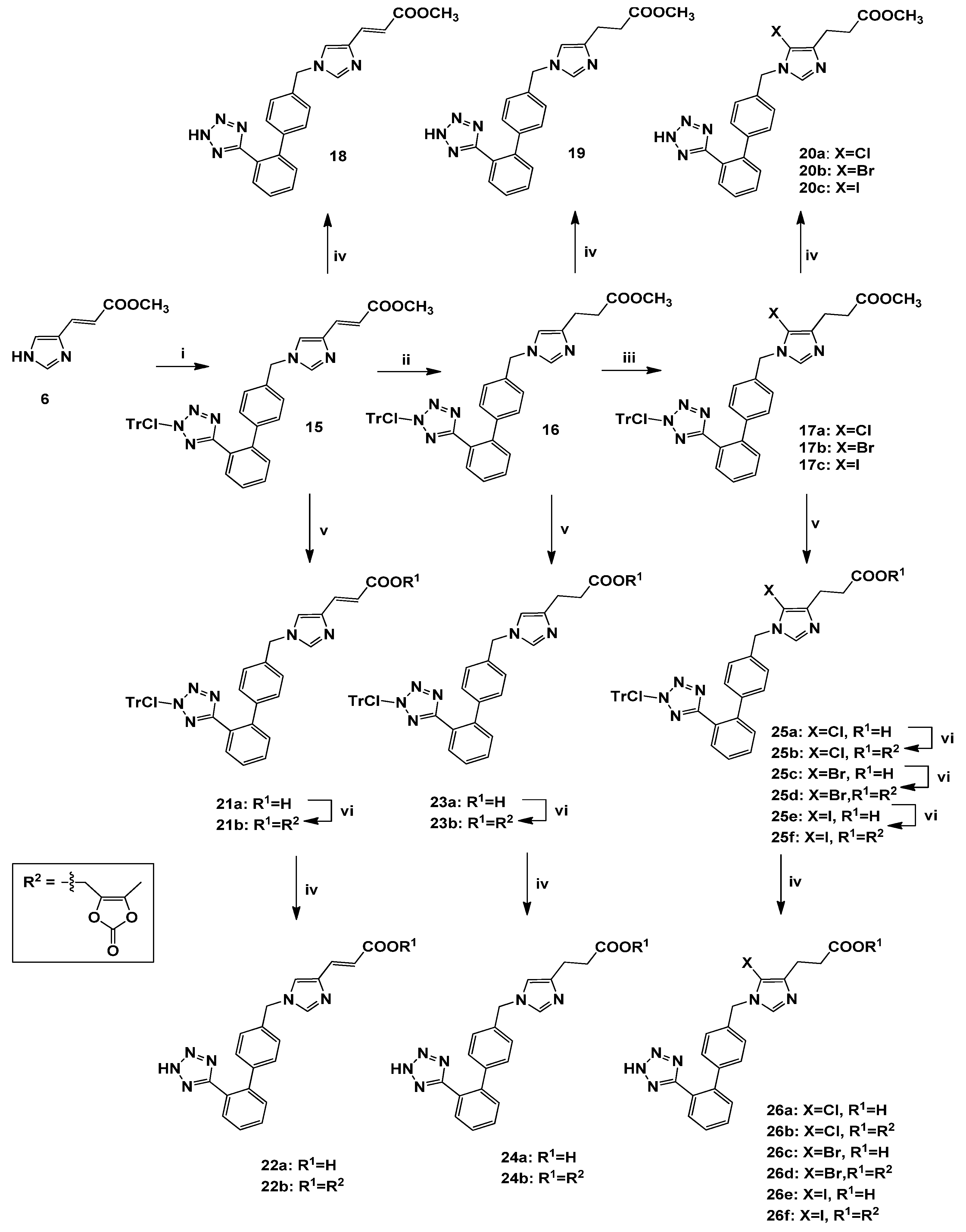
3.2.8. Synthesis of 1,4-Disubstituted biphenylmethyl analogues 18, 19, 20a–c, 22a–b, 24a–b, 26a–f
Ν-(2-Chlorotrityl)-5-[4′-(bromomethyl)biphenyl-2-yl]tetrazole (
4). Prepared from
3 according to the literature method [
8,
19,
25,
26]. Yield 80%; M.p. 155–157 °C; R
f = 0.33 (1.5:8.5 EtOAc:hexanes); ESI-MS (
m/z): 315.48 (M+H
+-ClTr), 277.22 (ClTr);
1Η-ΝΜR (CDCl
3):
δ 8.02–7.89 (m, 1H), 7.60–6.87 (m, 20H), 6.79 (dd, 1H,
J = 1.5, 8.0 Hz), 4.43 (s, 2H);
13C-NMR (CDCl
3):
δ 163.95, 141.63, 141.47, 140.64, 139.49, 136.34, 132.13, 131.75, 130.79, 130.53, 130.15, 129.69, 128.87, 128.66, 127.97, 127.91, 126.68, 81.96, 33.45. Anal. Calcd for C
33H
24Ν
4ClBr (%): C: 66.96; H: 4.09; N: 9.47. Found (%): C: 66.88; H: 4.15; N: 9.42.
(E)-Methyl 1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-acrylate (15). General procedure 1 was employed for the preparation of 15 using 4 as alkylating agent. Yield 73%; Rf = 0.50 (EtOAc); ESI-MS (m/z): 664.19 (M+H+), 387.29 (M+H+-ClTr), 277.38 (ClTr); 1Η-ΝΜR (CDCl3): δ 8.28 (dd, 1H, J = 1.8, 7.6 Hz), 7.53–7.12 (m, 22H), 6.84 (m, 2H), 6.54 (d, 1H, J = 15.6 Hz), 4.95 (s, 2H), 3.77 (s, 3H); 13C-NMR (CDCl3): δ 168.20, 137.43, 162.87, 140.48, 139.14, 138.89, 138.22, 136.33, 134.05, 132.22, 131.84, 130.99, 130.54, 129.29, 128.86, 128.30, 126.97, 126.31, 122.18, 115.53, 82.66, 51.63, 49.53. Anal. Calcd for C40H31N6O2Cl (%): C: 72.44; H: 4.71; N: 12.67. Found (%): C: 72.38; H: 4.77; N: 12.62.
Methyl 1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-propanoate (16). General procedure 2 was employed for the preparation of 16. Yield 88%; Rf = 0.55 (9:1 CHCl3:MeOH); ESI-MS (m/z): 666.19 (M+H+), 388.39 (M+H+-ClTr), 277.77 (ClTr); 1Η-ΝΜR (CDCl3): δ 7.98 (dd, 1H, J = 2.0, 7.6 Hz), 7.53–7.12 (m, 14H), 6.89–6.83 (m, 7H), 6.72 (dd, 1H, J = 1.2, 8.0 Hz), 6.47 (s, 1H), 4.87 (s, 2H), 3.65 (s, 3H), 2.84 (t, 2H, J = 7.6 Hz), 2.64 (t, 2H, J = 7.6 Hz). Anal. Calcd for C40H33N6O2Cl (%): C: 72.23; H: 5.00; N: 12.63. Found (%): C: 72.18; H: 5.09; N: 12.57.
Methyl 5-chloro-1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-propanoate (17a). General procedure 3 was employed for the preparation of 17a using NCS in MeCN. Yield 85%; Rf = 0.49 (EtOAc); ESI-MS (m/z): 700.59 (M+H+), 422.86 (M+H+-ClTr), 277.77 (ClTr); 1Η-ΝΜR (CDCl3): δ 7.97 (dd, 1Η, J = 1.2, 7.2 Hz), 7.52–7.14 (m, 15H), 6.90–6.84 (m, 6H), 6.72 (d, 1H, J = 8.0 Hz), 4.89 (s, 2H), 3.68 (s, 3H), 2.87 (t, 2H, J = 7.6 Hz), 2.70 (t, 2H, J = 7.6 Hz). C40H32Cl2N6O2. Anal. Calcd for C40H32N6O2Cl2 (%): C: 68.67; H: 4.61; N: 12.01. Found (%): C: 67.92; H: 4.78; N: 11.84.
Methyl 5-bromo-1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-propanoate (17b). General procedure 3 was employed for the preparation of 17b using NBS in DMF. Yield 68%; Rf = 0.47 (EtOAc); ESI-MS (m/z): 745.54 (M+H+), 467.80 (M+H+-ClTr), 277.80 (ClTr); 1Η-ΝΜR (CDCl3): δ 7.84 (dd, 1H, J = 1.2, 7.2 Hz ), 7.46-7.24 (m, 8H), 7.18-6.79 (m, 13H), 6.68 (dd, 1H, J = 1.6, 8.0 Hz), 4.88 (s, 2H), 3.56 (s, 3H), 2.73 (t, 2H, J = 7.6 Hz), 2.58 (t, 2H, J = 7.6 Hz). Anal. Calcd for C40H32N6O2ClBr (%): C: 64.57; H: 4.33; N: 11.29. Found (%): C: 64.68; H: 4.40; N: 11.23.
Methyl 5-iodo-1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-propanoate (17c). General procedure 3 was employed for the preparation of 17c using NIS in DMF. Yield 70%; Rf = 0.46 (EtOAc); ESI-MS (m/z): 792.48 (M+H+), 514.44 (M+H+-ClTr), 277.76 (ClTr); 1Η-ΝΜR (CD3OD): δ 7.58 (d, 1H, J = 7.6 Hz), 7.49–7.23 (m, 13H), 7.16–7.00 (m, 8H), 6.83 (d, 1H, J = 8.0 Hz), 5.15 (s, 2H), 3.57 (s, 3H), 2.78 (t, 2H, J = 7.6 Hz), 2.57 (t, 2H, J = 7.6 Hz). Anal. Calcd for C40H32N6O2ClI (%): C: 60.73; H: 4.08; N: 10.62. Found (%): C: 60.66; H: 4.14; N: 10.56.
(E)-Methyl 1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-acrylate (18). General procedure 6 was employed for the preparation of 18. Yield 94%; Rf = 0.51 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 387.15 (M+H+); 1Η ΝΜR (400 MHz, DMSO-d6): δ 8.49 (s, 1H), 7.87 (s, 1H), 7.68–7.54 (m, 4H), 7.53 (d, 1H, J = 15.6 Hz), 7.27 (d, 2H, J = 7.6 Hz), 7.13 (d, 2H, J = 8.0 Hz), 6.48 (d, 1H, J = 15.6 Hz), 5.32 (s, 2H), 3.71 (s, 3H). Anal. Calcd for C21H18N6O2∙CF3COOH (%): C: 55.20; H: 3.83; N: 16.79. Found (%): C: 55.11; H: 3.72; N: 16.84.
Methyl 1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoate (19). General procedure 6 was employed for the preparation of 19. Yield 89%; Rf = 0.49 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 389.13 (M+H+); 1Η-ΝΜR (CD3OD): δ 7.91 (d, 1H, J = 7.5 Hz), 7.62-6.76 (m, 9H), 6.62 (s, 1H), 5.04 (s, 2H), 3.64 (s, 3H), 2.76 (t, 2H, J = 7.2 Hz), 2.56 (t, 2H, J = 7.2 Hz); 13C-NMR (CD3OD): δ 167.99, 163.90, 141.79, 141.32, 140.68, 139.38, 138.96, 137.53, 136.06, 135.08, 131.47, 130.72, 130.30, 129.66, 128.36, 127.80, 127.31, 126.52, 114.82, 50.85, 50.37, 35.75, 29.83. Anal. Calcd for C21H20N6O2∙CF3COOH (%): C: 54.98; H: 4.21; N: 16.73. Found (%): C: 54.86; H: 4.33; N: 16.61.
Methyl 5-chloro-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoate (20a). General procedure 6 was employed for the preparation of 20a. Yield 95%; Rf = 0.53 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 423.92 (M+H+); 1Η-ΝΜR (CD3OD): δ 8.30 (s, 1H), 7.71-7.60 (m, 2H), 7.23–7.18 (m, 4H), 5.32 (s, 2H), 3.66 (s, 3H), 2.92 (t, 2H, J = 7.2 Hz), 2.70 (t, 2H, J = 7.2 Hz). Anal. Calcd for C21H19N6O2Cl∙CF3COOH (%): C: 51.45; H: 3.75; N: 15.65. Found (%): C: 51.37; H: 3.83; N: 16.51.
Methyl 5-bromo-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoate (20b). General procedure 6 was employed for the preparation of 20b. Yield 93%; Rf = 0.52 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 468.39 (M+H+); 1Η-ΝΜR (CD3OD): δ 8.80 (s, 1H), 7.70–7.56 (m, 4H), 7.26–7.18 (m, 4H), 5.40 (s, 2H), 3.66 (s, 3H), 2.97 (t, 2H, J = 7.2 Hz), 2.73 (t, 2H, J = 7.2 Hz). Anal. Calcd for C21H19N6O2Br∙CF3COOH (%): C: 47.52; H: 3.47; N: 14.46. Found (%): C: 47.63; H: 3.36; N: 14.55.
Methyl 5-iodo-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoate (20c). General procedure 6 was employed for the preparation of 20c. Yield 95%; Rf = 0.53 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 515.38 (M+H+); 1Η-ΝΜR (CD3OD): δ 8.37 (s, 1H), 7.62–7.51 (m, 4H), 7.09 (s, 4H), 5.23 (s, 2H), 3.58 (s, 3H), 2.84 (t, 2H, J = 7.2 Hz), 2.60 (t, 2H, J = 7.2 Hz). Anal. Calcd for C21H19N6O2I∙CF3COOH (%): C: 43.96; H: 3.21; N: 13.37. Found (%): C: 43.87; H: 3.34; N: 13.44.
(E)-1-[[2′-[[N-(2-Chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-acrylic acid (21a). General procedure 4 was employed for the preparation of 21a. Yield 90%; Rf = 0.38 (9:1 CHCl3:MeOH); ESI-MS (m/z): 650.17 (M+H+), 372.80 (M+H+-ClTr), 277.76 (ClTr); 1Η-ΝΜR (DMSO-d6): δ 7.81 (d, 1H, J = 7.6 Hz), 7.62–7.49 (m, 4H), 7.45–7.30 (m, 9H), 7.29 (d, 1H, J = 15.6 Hz), 7.11–7.06 (m, 4H), 6.78 (d, 4H, J = 7.6 Hz), 6.68 (d, 1Η, J = 8.0 Hz), 6.44 (d, 1H, J = 15.6 Hz), 5.12 (s, 2H). Anal. Calcd for C39H29N6O2Cl (%): C: 72.16; H: 4.50; N: 12.95. Found (%): C: 72.09; H: 4.58; N: 12.89.
(E)-(5-Methyl-2-oxo-1,3-dioxol)methyl-1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]-methyl]imidazole-4-acrylate (21b). General procedure 5 was employed for the preparation of 21b. Yield 84%; Rf = 0.52 (EtOAc); ESI-MS (m/z): 762.165 (M+H+), 484.44 (M+H+-ClTr), 277.76 (ClTr); 1Η-ΝΜR (DMSO-d6): δ 7.80 (s, 2H), 7.60–7.42 (m, 7H), 7.34–7.27 (m, 7H), 7.09 (s, 4H), 6.76 (s, 4H), 6.67 (s, 1H), 6.28 (d, 1H, J = 16.0 Hz), 5.12 (s, 2H), 5.02 (s, 2H), 2.16 (s, 3H). Anal. Calcd for C44H33N6O5Cl (%): C: 69.42; H: 4.37; N: 11.04. Found (%): C: 69.39; H: 4.32; N: 10.98.
(E)-1-[[2′-(1H-Tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-acrylic acid (22a). General procedure 6 was employed for the preparation of 22a. Yield 95%; Rf = 0.38 (4:1:1 n-butanol:acetic acid:H2O); ESI-MS (m/z): 373.44 (M+H+); 1Η-ΝΜR (DMSO-d6): δ 8.56 (s, 1H), 7.84 (s, 1H), 7.66 (s, 2H), 7.58 (s, 1H), 7.52 (s, 1H), 7.42 (d, 1H, J = 16.0 Hz), 7.26 (s, 2H), 7.11 (s, 2H), 6.40 (d, 1H, J = 16.0 Hz), 5.31 (s, 2H). Anal. Calcd for C20H16N6O2∙CF3COOH (%): C: 54.32; H: 3.52; N: 17.28. Found (%): C: 54.45; H: 3.41; N: 17.39.
(E)-(5-Methyl-2-oxo-1,3-dioxol)methyl-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-acrylate (22b). General procedure 6 was employed for the preparation of 22b. Yield 94%; Rf = 0.52 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 485.52 (M+H+); 1Η-ΝΜR (DMSO-d6): δ 8.10 (s, 1H), 7.76 (s, 1H), 7.65 (s, 2H), 7.57–7.51 (m, 3H), 7.24–7.20 (m, 2H), 7.11–7.07 (m, 2H), 6.38 (d, 1H, J = 16.0 Hz), 5.24 (s, 2H), 5.04 (s, 2H), 2.16 (s, 3H). Anal. Calcd for C25H20N6O5∙CF3COOH (%): C: 54.18; H: 3.54; N: 14.04. Found (%): C: 54.08; H: 3.41; N: 14.17.
1-[[2′-[[N-(2-Chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-propanoic acid (23a). General procedure 4 was employed for the preparation of 23a. Yield 93%; Rf = 0.33 (9:1 CHCl3:MeOH); ESI-MS (m/z): 652.19 (M+H+), 374.77 (M+H+-ClTr), 277.77 (ClTr); 1Η-ΝΜR (DMSO-d6): δ 7.82–7.69 (m, 1H), 7.64–7.44 (m, 6H), 7.40–7.27 (m, 7H), 7.10–7.02 (m, 4H), 7.09–6.66 (m, 6H), 5.00 (s, 2H), 2.60 (t, 2H, J = 7.2 Hz), 2.38 (t, 2H, J = 7.2 Hz). Anal. Calcd for C39H31N6O2Cl (%): C: 71.94; H: 4.80; N: 12.91. Found (%): C: 71.87; H: 4.86; N: 12.87.
(5-Μethyl-2-oxo-1,3-dioxol)methyl-1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]-methyl]imidazole-4-propanoate (23b). General procedure 5 was employed for the preparation of 23b. Yield 73%; Rf = 0.45 (9:1 CHCl3:MeOH); ESI-MS (m/z): 764.21 (M+H+), 486.56 (M+H+-ClTr), 277.76 (ClTr); 1Η-ΝΜR (CDCl3): δ 7.96 (d, 1H, J = 7.2 Hz), 7.49–7.31 (m, 9H), 7.22–7.18 (m, 5H), 7.13 (d, 2H, J = 8.0 Hz), 6.92-6.84 (m, 5H), 6.82 (s, 1H), 6.72 (d, 1H, J = 8.0 Hz), 4.88 (s, 2H), 4.81 (s, 2H), 2.83 (t, 2H, J = 7.2 Hz), 2.68 (t, 2H, J = 7.2 Hz), 2.16 (s, 3H). Anal. Calcd for C44H35N6O5Cl (%): C: 69.24; H: 4.62; N: 11.01. Found (%): C: 69.19; H: 4.67; N: 11.08.
1-[[2′-(1H-Tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoic acid (24a). General procedure 6 was employed for the preparation of 24a. Yield 92%; Rf = 0.33 (4:1:1 n-butanol:acetic acid:H2O); ESI-MS (m/z): 375.41 (M+H+); 1Η-ΝΜR (CD3OD): δ 8.89 (s, 1H), 7.71–7.67 (m, 2Η), 7.60–7.56 (m, 2H), 7.39 (s, 1H), 7.35 (d, 2H, J = 7.2 Hz), 7.20 (d, 2H, J = 7.2 Hz), 5.50 (s, 2H), 2.98 (t, 2H J = 7.2 Hz), 2.71 (t, 2H, J = 7.2 Hz). Anal. Calcd for C20H18N6O2∙CF3COOH (%): C: 54.10; H: 3.92; N: 17.21. Found (%): C: 54.17; H: 3.81; N: 17.32.
(5-Methyl-2-oxo-1,3-dioxol)methyl-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoate (24b). General procedure 6 was employed for the preparation of 24b. Yield 87%; Rf = 0.49 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 487.56 (M+H+); 1Η-ΝΜR (DMSO-d6): δ 9.06 (s, 1H), 7.67–7.12 (m, 9H), 5.35 (s, 2H), 4.94 (s, 2H), 2.87 (t, 2H, J = 7.2 Hz), 2.74 (t, 2H, J = 7.2 Hz), 2.12 (s, 3H). Anal. Calcd for C25H22N6O5∙CF3COOH (%): C: 54.00; H: 3.86; N: 13.99. Found (%): C: 53.91; H: 3.91; N: 14.11.
5-Chloro-1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-propanoic acid (25a). General procedure 4 was employed for the preparation of 25a. Yield 94%; Rf = 0.39 (9:1 CHCl3:MeOH); ESI-MS (m/z): 686.66 (M+H+), 408.92 (M+H+-ClTr), 277.81 (ClTr); 1Η-ΝΜR (CDCl3): δ 7.98–7.94 (m, 1H), 7.48-7.08 (m, 15H), 6.85 (d, 6H, J = 7.6 Hz), 6.71 (d, 1H, J = 8.0 Hz), 4.84 (s, 2H), 2.79 (bs, 2H), 2.58 (bs, 2H). Anal. Calcd for C39H30N6O2Cl2 (%): C: 68.31; H: 4.41; N: 12.26. Found (%): C: 68.08; H: 4.53; N: 12.57.
(5-Methyl-2-oxo-1,3-dioxol)methyl-5-chloro-1-[[2΄-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-propanoate (25b). General procedure 1 was employed for the preparation of 7. Yield 82%; Rf = 0.50 (EtOAc); ESI-MS (m/z): 798.72 (M+H+), 521.03 (M+H+-ClTr), 277.78 (ClTr); 1Η-ΝΜR (CDCl3): δ 7.95 (d, 1Η, J = 7.2 Hz), 7.48–7.34 (m, 6H), 7.34–7.32 (m, 5H), 7.23 (m, 5H), 7.11 (d, 2H, J = 8.0 Hz), 6.91–6.86 (m, 8H), 4.93 (s, 2H), 4.83 (s, 2H), 2.87 (t, 2H, J = 7.2 Hz), 2.74 (t, 2H, J = 7.2 Hz), 2.15 (s, 3H). Anal. Calcd for C44H34N6O5Cl2 (%): C: 66.25; H: 4.30; N: 10.54. Found (%): C: 66.24; H: 4.18; N: 10.33.
5-Bromo-1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-propanoic acid (25c). General procedure 4 was employed for the preparation of 25c. Yield 91%; Rf = 0.37 (9:1, CHCl3:MeOH); ESI-MS (m/z): 731.11 (M+H+), 453.36 (M+H+-ClTr), 277.76 (ClTr); 1Η-ΝΜR (CDCl3): δ 8.04 (d, 1H, J = 7.6 Hz), 7.67–7.28 (m, 15H), 7.09 (d, 2H, J = 8.0 Hz), 6.97 (d, 4H, J = 7.6 Hz), 6.87 (d, 1H, J = 8.0 Hz), 5.12 (s, 1H), 2.96–2.92 (m, 2H), 2.81–2.78 (m, 2H). Anal. Calcd for C39H30N6O2ClBr (%): C: 64.16; H: 4.14; N: 11.51. Found (%): C: 64.11; H: 4.21; N: 11.44.
(5-Methyl-2-oxo-1,3-dioxol)methyl-5-bromo-1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-propanoate (25d). General procedure 5 was employed for the preparation of 25d. Yield 72%; Rf = 0.48 (EtOAc); ESI-MS (m/z): 843.18 (M+H+), 565.43 (M+H+-ClTr), 277.78 (ClTr); 1Η-ΝΜR (CDCl3): δ 8.03 (d, 1H, J = 7.2 Hz), 7.65–7.33 (m, 13H), 7.24 (d, 2H, J = 7.6 Hz), 6.99 (m, 4H), 6.87 (d, 1H, J = 7.6 Hz), 5.07 (s, 2H), 4.93 (s, 2H), 2.94 (t, 2H, J = 6.4 Hz), 2.82 (t, 2H, J = 6.4 Hz), 2.24 (s, 3H). Anal. Calcd for C44H34N6O5ClBr (%): C: 62.75; H: 4.07; N: 9.98. Found (%): C: 62.68; H: 4.15; N: 9.91.
5-Iodo-1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-propanoic acid (25e). General procedure 4 was employed for the preparation of 25e. Yield 95%; Rf = 0.37 (9:1 CHCl3:MeOH); ESI-MS (m/z): 778.13 (M+H+), 500.35 (M+H+-ClTr), 277.76 (ClTr); 1Η-ΝΜR (CDCl3): δ 7.97 (d, 1H, J = 7.6 Hz), 7.72 (s, 1H), 7.49–7.17 (m, 15H), 6.93 (d, 2H, J = 7.6 Hz ), 6.88 (d, 4H, J = 7.6 Hz), 6.73 (d, 1H, J = 7.2 Hz), 4.98 (s, 2H), 2.88 (t, 2H, J = 7.2 Hz), 2.73 (t, 2H, J = 7.2 Hz). Anal. Calcd for C39H30N6O2ClI (%): C: 60.28; H: 3.89; N: 10.82. Found (%): C: 60.21; H: 3.96; N: 10.73.
(5-Methyl-2-oxo-1,3-dioxol)methyl-5-iodo-1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]imidazole-4-propanoate (25f). General procedure 5 was employed for the preparation of 25f. Yield 74%; Rf = 0.51 (EtOAc); ESI-MS (m/z): 890.21 (M+H+), 612.45 (M+H+-ClTr), 276.88 (ClTr); 1Η-ΝΜR (CDCl3): δ 7.77 (d, 1H, J = 7.6 Hz), 7.45–7.22 (m, 8Η), 7.15–6.80 (m, 14H), 4.86 (s, 2H), 4.71 (s, 2H), 2.71 (t, 2H, J = 7.2 Hz), 2.59 (t, 2H, J = 7.2 Hz), 2.02 (s, 3H); Anal. Calcd for C44H34N6O5ClI (%): C: 59.44; H: 3.85; N: 9.45. Found (%): C: 59.36; H: 3.94; N: 4.37.
5-Chloro-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoic acid (26a). General procedure 6 was employed for the preparation of 26a. Yield 90%; Rf = 0.32 (4:1:1 n-butanol:acetic acid:H2O); ESI-MS (m/z): 409.31 (M+H+); 1Η-ΝΜR (CD3OD): δ 8.62 (s, 1H), 7.70 (d, 2H, J = 7.2 Hz), 7.62–7.56 (m, 2H), 7.27 (d, 2H, J = 7.6 Hz), 7.19 (d, 2H, J = 7.6 Hz), 5.38 (s, 2H), 2.94 (t, 2H, J = 7.2 Hz), 2.69 (t, 2H, J = 7.2 Hz). Anal. Calcd for C20H17N6O2Cl∙CF3COOH (%): C: 50.54; H: 3.47; N: 16.07. Found (%): C: 50.43; H: 3.58; N: 16.17.
(5-Methyl-2-oxo-1,3-dioxol)methyl-5-chloro-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoate (26b). General procedure 6 was employed for the preparation of 26b. Yield 89%; Rf = 0.56 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 522.00 (M+H+); 1Η-ΝΜR (DMSO-d6): δ 8.24 (s, 1H), 7.71–7.54 (m, 4H), 7.17-7.08 (m, 4H), 5.24 (s, 2H), 4.92 (s, 2H), 2.76 (t, 2H, J = 7.6 Hz), 2.69 (t, 2H, J = 7.6 Hz), 2.13 (s, 3H). Anal. Calcd for C25H21N6O5Cl∙CF3COOH (%): C: 51.07; H: 3.49; N: 13.24. Found (%): C: 51.13; H: 3.41; N: 13.35.
5-Bromo-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoic acid (26c). General procedure 6 was employed for the preparation of 26c. Yield 93%; Rf = 0.31 (4:1:1 n-butanol:acetic acid:H2O); ESI-MS (m/z): 454.37 (M+H+); 1Η-ΝΜR (CD3OD): δ 7.67–7.55 (m, 5H), 7.12 (s, 4H), 5.21 (s, 2H), 2.83 (t, 2H, J = 7.2 Hz), 2.60 (t, 2H, J = 7.2 Hz). Anal. Calcd for C20H17N6O2Br∙CF3COOH (%): C: 46.58; H: 3.20; N: 14.81. Found (%): C: 46.63; H: 3.32; N: 14.69.
(5-Methyl-2-oxo-1,3-dioxol)methyl-5-bromo-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoate (26d). General procedure 6 was employed for the preparation of 26d. Yield 91%; Rf = 0.55 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 566.45 (M+H+); 1Η-ΝΜR (DMSO-d6): δ 7.91 (s, 1H), 7.67–7.64 (m, 2H), 7.58–7.53 (m, 2H), 7.09 (s, 4H), 5.17 (s, 2H), 4.92 (s, 2H), 2.72–2.63 (m, 4H), 2.13 (s, 3H). Anal. Calcd for C25H21N6O5Br∙CF3COOH (%): C: 47.73; H: 3.26; N: 12.37. Found (%): C: 47.83; H: 3.34; N: 13.27.
5-Iodo-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoic acid (26e). General procedure 6 was employed for the preparation of 26e. Yield 91%, Rf = 0.29 (4:1:1 n-butanol:acetic acid:H2O); ESI-MS (m/z): 501.38 (M+H+); 1Η-ΝΜR (CD3OD): δ 8.79 (s, 1H), 7.68–7.64 (m, 2H), 7.56–7.31 (m, 4H), 7.19 (d, 2H, J = 8.0 Hz), 5.35 (s, 2H), 2.95 (t, 2H, J = 7.2 Hz), 2.68 (t, 2H, J = 7.2 Hz). Anal. Calcd for C20H17N6O2I∙CF3COOH (%): C: 43.01; H: 2.95; N: 13.68. Found (%): C: 43.13; H: 2.87; N: 13.79.
(5-Methyl-2-oxo-1,3-dioxol)methyl-5-iodo-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]imidazole-4-propanoate (26f). General procedure 6 was employed for the preparation of 26f. Yield 90%; Rf = 0.56 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 613.44 (M+H+); 1Η-ΝΜR (CD3OD): δ 8.00 (s, 1H), 7.66–7.56 (m, 4H), 7.15 (bs, 4Η), 5.22 (s, 2H), 4.88 (s, 2H), 2.86 (t, 2H, J = 7.2 Hz), 2.70 (t, 2H, J = 7.2 Hz), 2.16 (s, 3H). Anal. Calcd for C25H21N6O5I∙CF3COOH (%): C: 44.64; H: 3.05; N: 11.57. Found (%): C: 44.72; H: 3.14; N: 11.65.
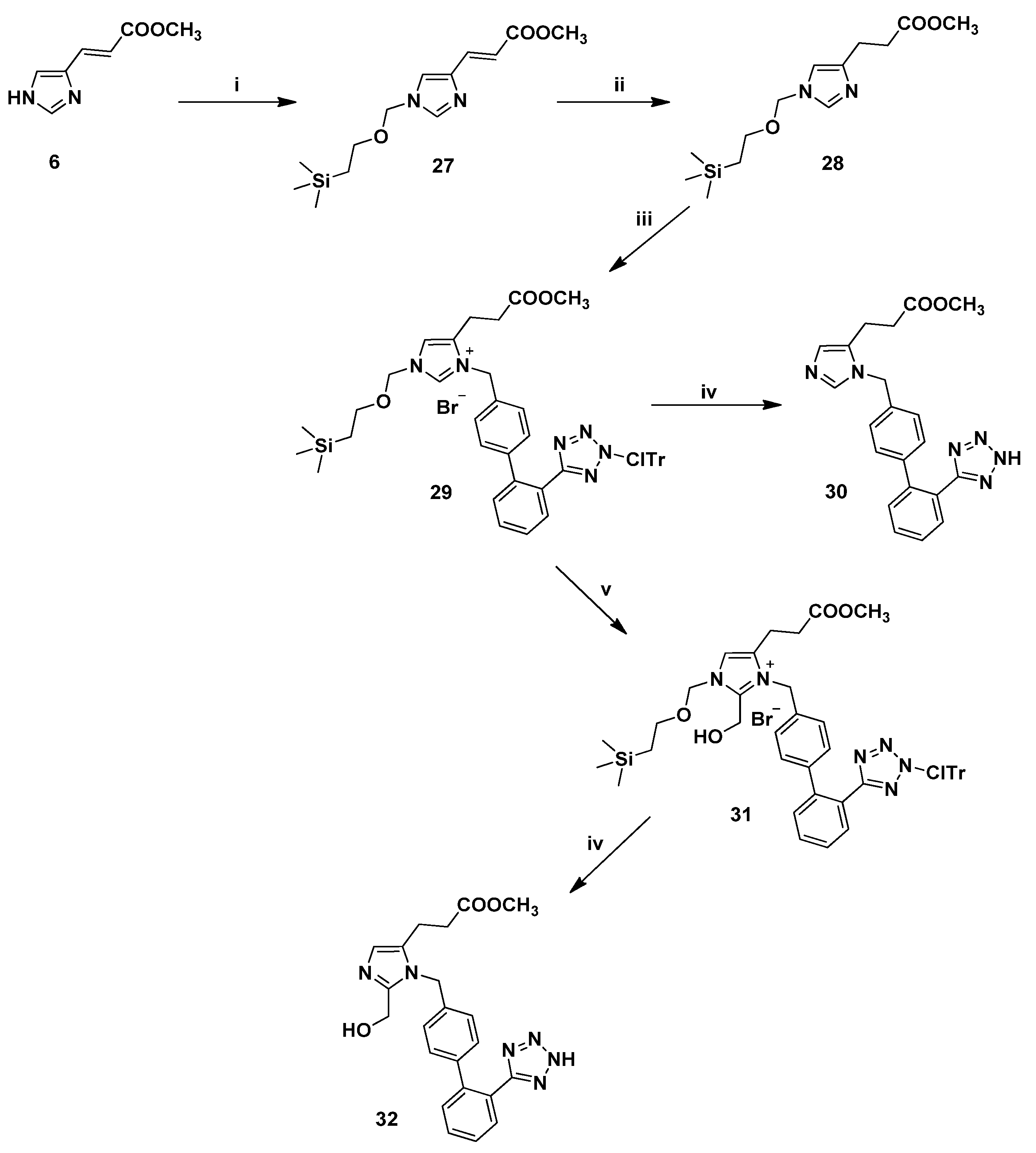
3.2.9. Synthesis of 1,5-Disubstituted Benzyl Analogues 30, 32
(E)-Methyl 3-[1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-imidazole-4-yl]acrylate (27). To a solution of (E)-urocanic methyl ester (6) (0.61 g, 4.03 mmol) in dry DMF (15.0 mL) under argon atmosphere at 0 °C was added dry NaH (powdered 95%, 0.12 g, 4.84 mmol) and the suspension was left at the same temperature for 15 min. Then, SEM-Cl (0.68 mL, 4.84 mmol) was added in three portions and the reaction mixture was allowed to warm to rt for 4 h. The reaction was quenched with 0.5 N NaOH (15 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were washed with brine (× 2), dried (Na2SO4) and concentrated in vacuo. Purification by flash column chromatography (7:3 EtOAc:hexanes) afforded 27. Yield 78%; Rf = 0.35 (8:2 EtOAc:hexanes); ESI-MS (m/z): 283.29 (M+H+); 1Η-ΝΜR (CDCl3): δ 8.58 (s, 1H), 7.52 (d, 1H, J = 16.0 Hz), 7.35 (s, 1H), 6.68 (d, 1H, J = 16.0 Hz), 5.42 (s, 2H), 3.82 (s, 3H), 3.58 (t, 2H, J = 8.0 Hz), 0.96 (t, 2H, J = 8.0 Hz), 0.01 (s, 9H); 13C-NMR (CDCl3): δ 166.64, 137.98, 134.48, 130.47, 121.21, 120.59, 50.85, 76.79, 68.14, 51.95, 18.22, 1.44. Anal. Calcd for C13H22N2O3Si (%): C: 55.29; H: 7.85; N:9.92. Found (%): C:55.36; H:7.72; N:9.87.
Methyl 3-[1-[(2-(trimethylsilyl)ethoxy)methyl]-1H-imidazole-4-yl]propanoate (28). General procedure 2 was employed for the preparation of 28. Yield 91%; Rf = 0.44 (9:1 CHCl3:MeOH); ESI-MS (m/z): 284.99 (M+H+); 1Η-ΝΜR (CDCl3): δ 7.91 (s, 1H), 6.88 (s, 1H), 5.25 (s, 2H), 3.67 (s, 3H), 3.50 (t, 2H, J = 8.0 Hz), 3.00 (t, 2H, J = 7.2 Hz), 2.72 (t, 2H, J = 7.2 Hz), 0.91 (t, 2H, J = 8.0 Hz), 0.01 (s, 9H); 13C-NMR (CDCl3): δ 173.13, 136.14, 116.19, 76.88, 67.08, 120.59, 51.65, 33.27, 22.27, 17.89, 1.44. Anal. Calcd for C13H24N2O3Si (%): C:54.90; H:8.51; N:9.85. Found (%): C:54.81; H:8.61; N:9.74.
Methyl 1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]-3-[(2-(trimethylsilyl)-ethoxy)methyl]imidazolium-5-propanoate bromide (29). To a stirred solution of 28 (2.0 g, 7.03 mmol) in dry CH2Cl2 (20.0 mL) under argon was added the alkylating agent 4 (4.31 g, 7.73 mmol) in one portion and the resulting mixture was heated under reflux for 3 h. Upon completion (disappearance of starting material confirmed by RP-HPLC), the solvent was concentrated, followed by chromatographic purification (97:3 CHCl3:MeOH) to afford 29 as a white powder. Yield 81%; Rf = 0.40 (9:1 CHCl3:MeOH); ESI-MS (m/z): 796.19 (M+-Br); 1Η-ΝΜR (CDCl3): δ 7.93 (dd, 1H, J = 1.6, 7.6 Hz), 7.52–7.46 (m, 3H), 7.40–7.13 (m, 13H), 7.11 (s, 1H), 6.94 (d, 6H, J = 7.6 Hz), 5.67 (s, 2H), 5.46 (s, 2H), 3.75–3.64 (m, 5H), 2.75 (t, 2H, J = 7.2 Hz), 2.47 (t, 2H, J = 7.2 Hz), 0.94 (t, 2H, J = 8.0 Ηz), 0.002 (s, 9H). Anal. Calcd for C46H48N6O3SiBr (%): C:63.04; H:5.52; N:9.59. Found (%): C:62.93; H:5.57; N:9.51.
Methyl 1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]-3-[(2-(trimethylsilyl)ethoxy)methyl]imidazole-5-propanoate (30). General procedure 6 was employed for the preparation of 30. Yield 90%; Rf = 0.53 (8.5:1.5 CHCl3:MeOH); ESI-MS (m/z): 389.51 (M+H+); 1Η-ΝΜR (DMSO-d6): δ 9.13 (s, 1H), 7.68 (d, 1H, J = 8.0 Hz), 7.59 (d, 1H, J = 7.6 Hz), 7.54 (d, 1H, J = 7.6 Hz), 7.50 (s, 1H), 7.21 (d, 2H, J = 8.0 Hz), 7.14 (d, 1H, J = 8.0 Hz), 5.48 (s, 2H), 3.60 (s, 3H), 2.77 (t, 2H, J = 7.2 Hz), 2.64 (t, 2H, J = 7.2 Hz). Anal. Calcd for C21H20N6O2∙CF3COOH (%): C:54.98; H:4.21; N:16.73. Found (%): C:54.91; H:4.13; N:16.61.
Methyl 2-hydroxymethyl-1-[[2′-[[N-(2-chlorotrityl)]-1H-tetrazol-5-yl]biphenyl-4-yl]methyl]-3-[(2-(trimethylsilyl)ethoxy)methyl]imidazolium-5-propanoate bromide (31). In a sealed tube were sequentially added 29 (2.0 g, 2.38 mmol), DMF (1.0 mL), 37% formalin (2.65 mL, 35.63 mmol) and diisopropylethylamine (2.02 mL, 11.90 mmol). The resulting mixure was stirred at 85 °C until HPLC showed no starting material left (ca. 1 h). Then, the mixture was quenched with 5% aqueous citric acid (10 mL), extracted with CH2Cl2 and the combined organic phases were washed with brine, dried (Na2SO4), filtered and concentrated in vacuo. Purification by flash column chromatography (96:4 CHCl3:MeOH) afforded 31. Yield 91%; Rf = 0.28 (9:1 CHCl3:MeOH); ESI-MS (m/z): 826.32 (M+-Br); 1Η-ΝΜR (CDCl3): δ 7.95 (d, 1H, J = 6.8 Hz), 7.52–7.42 (m, 3H), 7.40–7.26 (m, 9H, ), 7.19 (s, 1H), 7.15 (d, 2H, J = 8.0 Hz), 6.94–6.89 (m, 8H), 5.76 (s, 2H), 5.52 (s, 2H), 4.72 (s, 2H), 3.70–3.62 (m, 5H), 2.76 (t, 2H, J = 6.8 Hz), 2.54 (t, 2H, J = 6.8 Hz), 0.95 (t, 2H, J = 8.0 Ηz), 0.001 (s, 9H). Anal. Calcd for C47H50N6O4SiBr (%): C:62.28; H:5.56; N:9.27. Found (%): C:62.37; H:5.43; N:9.19.
Methyl 2-hydroxymethyl-1-[[2′-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]-3-[(2-(trimethylsilyl)ethoxy)-methyl]imidazole-5-propanoate (32). General procedure 6 was employed for the preparation of 32. Yield 88%; Rf = 0.32 (8:2 CHCl3:MeOH); ESI-MS (m/z): 419.56 (M+H+); 1Η-ΝΜR (DMSO-d6): δ 7.67 (dd, 2H, J = 2.0, 7.6 Hz), 7.59 (d, 1H, J = 7.6 Hz), 7.52 (d, 1H, J = 7.6 Hz), 7.44 (s, 1H), 7.12 (s, 4H), 6.26 (bs, 1H), 5.45 (s, 2H), 4.74 (s, 2H), 3.58 (s, 3H), 2.71 (t, 2H, J = 6.8 Hz), 2.61 (t, 2H, J = 6.8 Hz). Anal. Calcd for C22H22N6O3∙CF3COOH (%): C:54.14; H:4.35; N:15.78. Found (%): C:54.23; H:4.25; N:15.86.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


