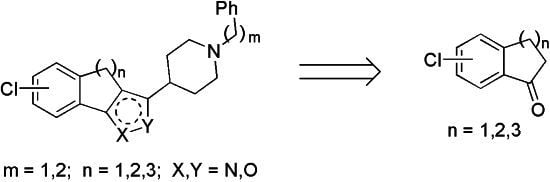
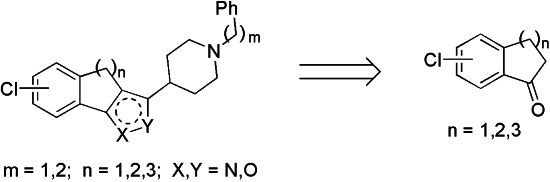
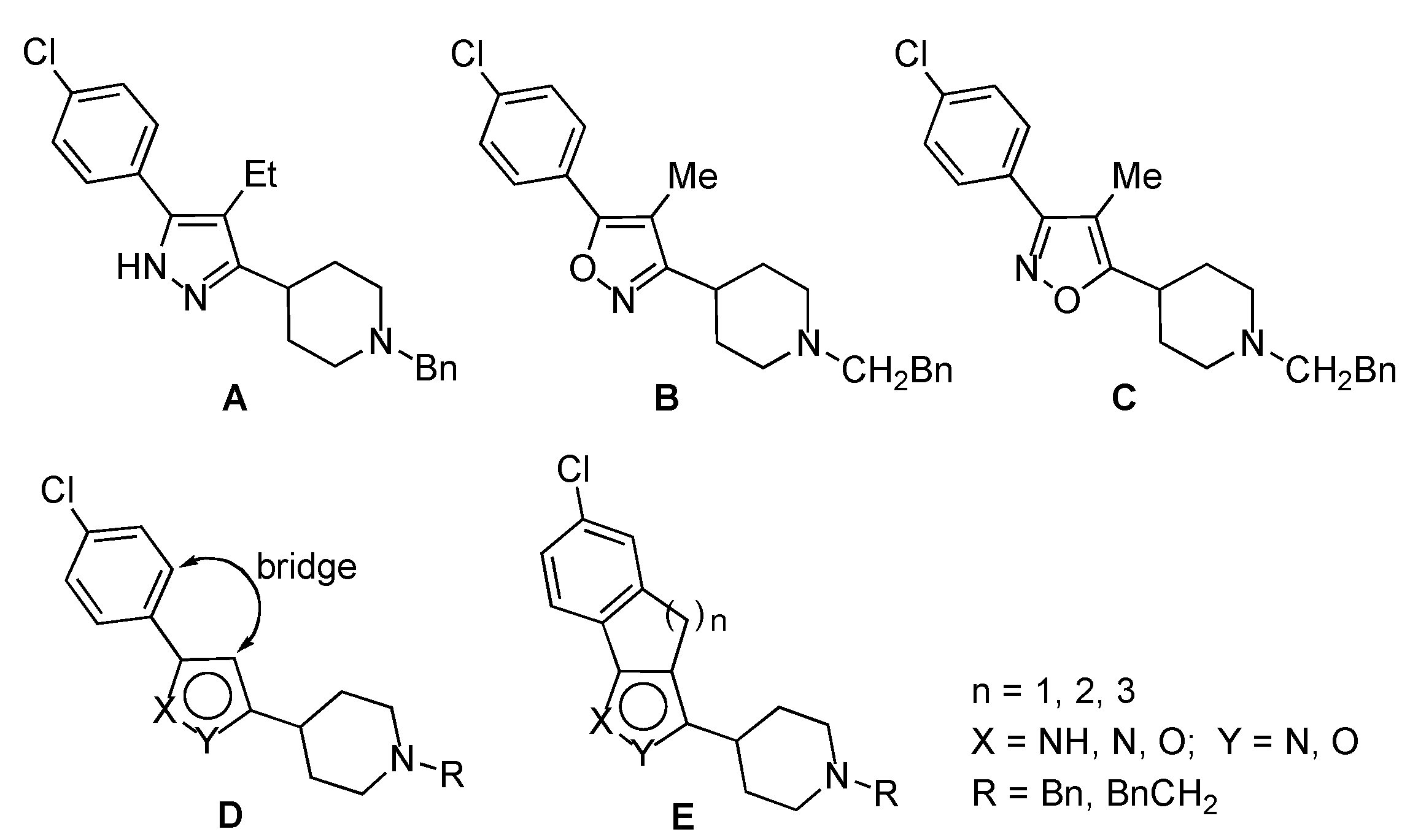
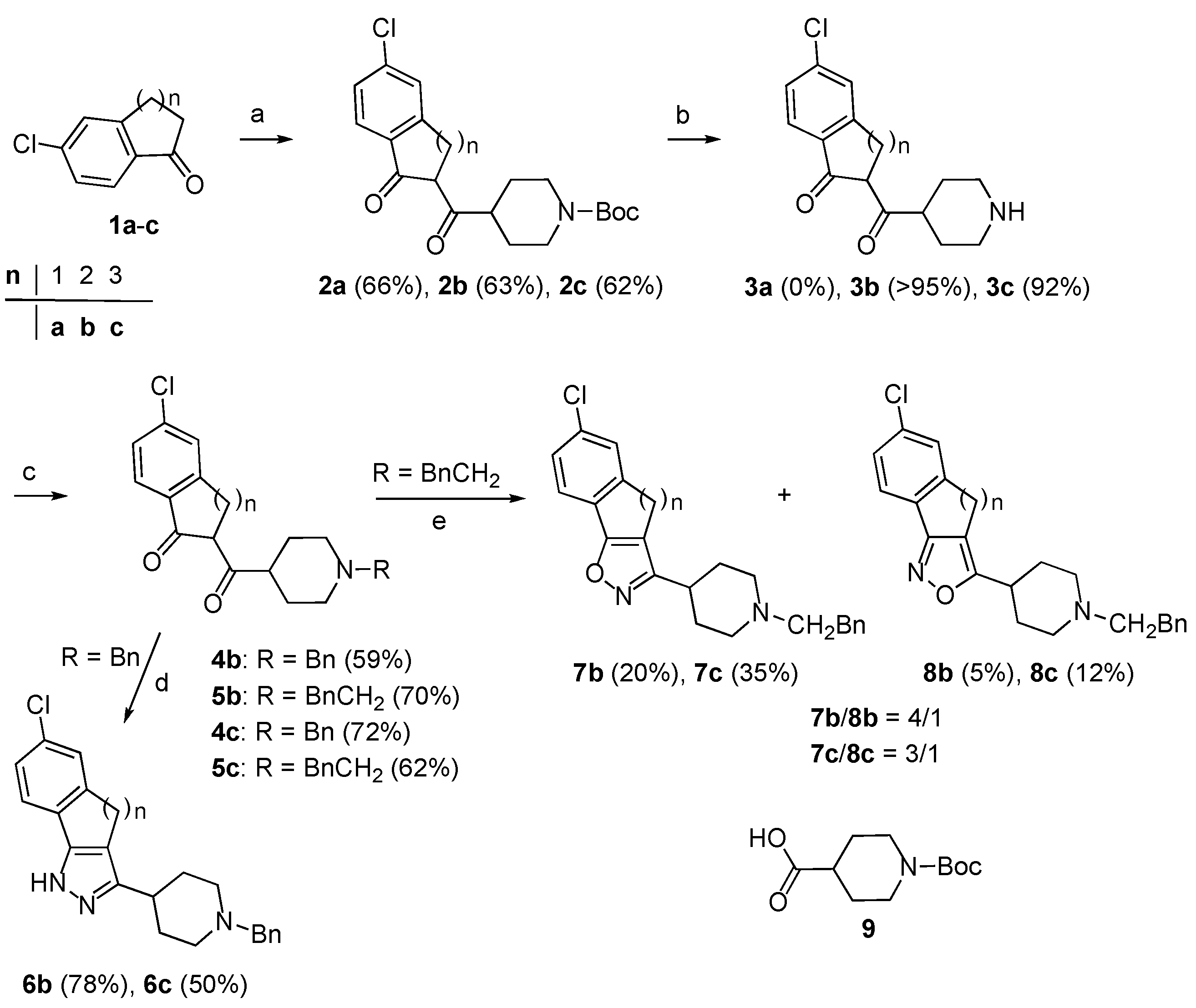
2. Results and Discussion
To begin the synthesis of the target derivatives
E (
Scheme 1), the known cyclic ketones
1a–
c [
6,
7] were acylated by reaction of the corresponding sodium enolate, obtained by reaction with sodium hydride, with the reagent formed by reaction of the
N-Boc protected isonipecotic acid
9 with 1,1′-carbonyldiimidazole [
8] (
Scheme 2). In this way, 1,3-dicarbonyl derivatives
2a–
c were obtained in 62–63% yields. Next, these compounds were submitted to
N-deprotection by treatment with trifluoroacetic acid in CH
2Cl
2. However, while
2b and
2c were easily deprotected giving compounds
3b and
3c in high yields (92–95%), the removal of the
N-Boc group from
2a failed. Further attempts to deprotect
2a with HCOOH, 3N HCl in AcOEt, CF
3COOH and Et
3SiH, and SnCl
4 in AcOEt all failed unexpectedly, therefore, alternative approaches to the target compounds
6a,
7a and
8a were investigated next (see below).
Compounds
3b,
c were converted in 59–72% yields into the related
N-benzyl and
N-phenylethyl derivatives
4b,
c and
5b,
c by reaction with benzyl chloride and 2-phenyl-1-iodoethane, respectively. With the key 1,3-dicarbonyl derivatives
4b,
c and
5b,
c in hand, their conversion into the desired derivatives
E was pursued according to the planned retrosynthetic scheme. Compounds
4b,
c and hydrazine in methanol were stirred at room temperature to afford the pyrazole derivatives
6b and
6c in good yields (78% and 50%, respectively). Treatment of
5b,
c with hydroxylamine hydrochloride in EtOH/AcOH at 80 °C gave isoxazoles
7b,
c and
8b,
c as mixtures of regioisomers in moderate to good yields. With
5b isoxazoles
7b and
8b were obtained in a 4/1 ratio, while
5c gave isoxazoles
7c and
8c in a 3.2/1 ratio [
9].
Scheme 2.
Synthesis of compounds 6b, 6c, 7b, 7c, 8b and 8c.
Scheme 2.
Synthesis of compounds 6b, 6c, 7b, 7c, 8b and 8c.
Reagents and conditions: (a) (i) NaH, DMF, rt; (ii) 9, 1,1'-carbonyldiimidazole, DMF, rt, 45 min; (iii) 120 °C, 6 h; (b) CF3COOH, CH2Cl2, rt, 2 h; (c) BnCl or BnCH2I. DMF, i-Pr2NEt, 25–60 °C, 12h; (d) H2N-NH2, MeOH, rt, 12 h; (e) NH2OH, EtOH, AcOH, 80 °C, 12 h.
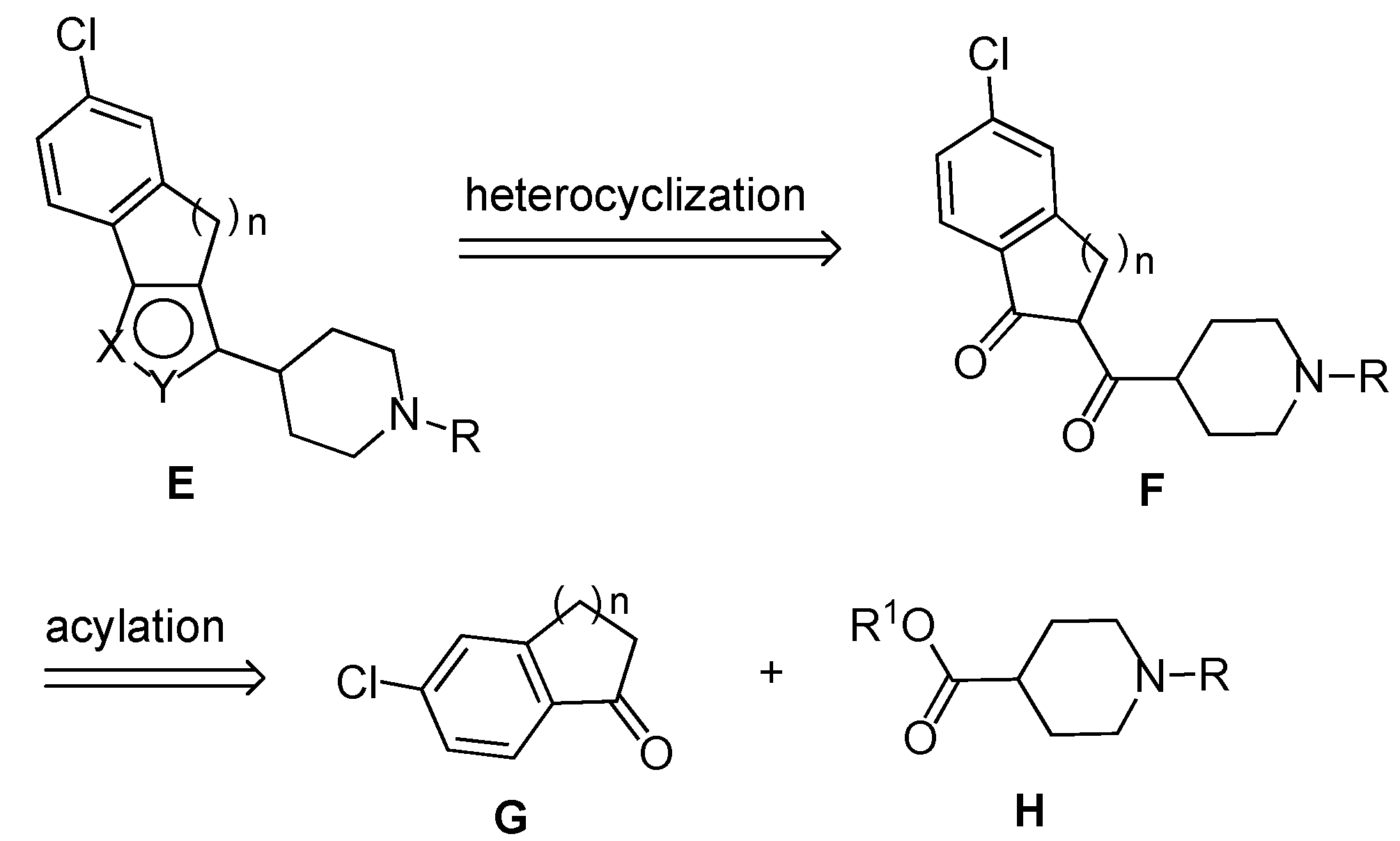
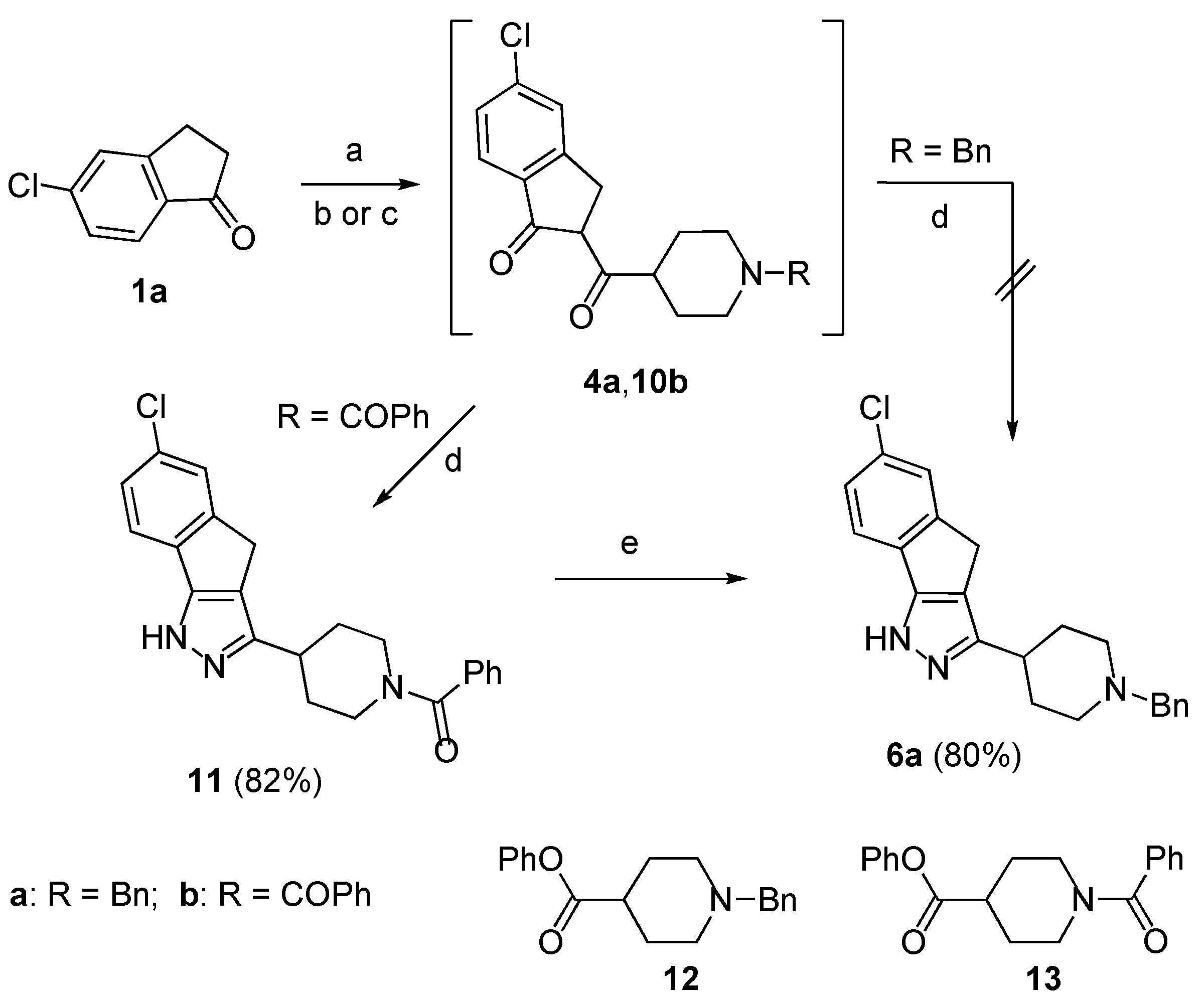
To obtain compound
6a the synthetic routes outlined in
Scheme 3 were followed. Firstly, the sodium enolate of the ketone
1a was reacted with phenyl 1-benzylpiperidine-4-carboxylate
12, but the 1,3-dicarbonyl intermediate
4a failed to give the expected pyrazole
6a by treatment with hydrazine in AcOH/MeOH at 80 °C. However, when the same enolate was treated with phenyl 1-(phenylcarbonyl)-piperidine-4-carboxylate
13 [
10], obtained by esterification with phenol of the parent acid (
Scheme 5), the formed 1,3-dicarbonyl
10b afforded by treatment with hydrazine in AcOH/MeOH at 80 °C the substituted pyrazole
11 in 82% yield. Finally, LiAlH
4 reduction of the carbonyl group to the methylene unit afforded the target pyrazole
6a in 80% yield (66% overall yield from
1a).
This satisfactory result appeared to open a way to isoxazoles
7a and
8a by simple replacing of the piperidine derivative
13 with the analogue
16 (
Scheme 4). However, the treatment of the 1,3-dicarbonyl intermediate
14, obtained in turn by reaction of
1a with
16, with hydroxylamine hydrochloride in EtOH/AcOH at 80 °C failed to afford the expected isoxazoles
15. This unexpected result prompted us to verify another route based on the on the use of the
N-benzylpyperidine
17 that was obtained by esterification with phenol of the parent acid (
Scheme 5). We were pleased to find that the1,3-dicarbonyl intermediate
5a, formed by reaction of the enolate of the ketone
1a with
17, could be directly converted in the usual way into a mixture of isoxazoles
7a and
8a in 41% and 12% yield, respectively (
Scheme 4).
Scheme 3.
Synthesis of compound 6a.
Scheme 3.
Synthesis of compound 6a.
Reagents and conditions: (a) (i) NaH, benzene, rt, (b) 12, reflux, 3.5 h; (c) 13, reflux, 3.5 h; (d) H2N-NH2, AcOH, EtOH, 80 °C, 4 h; (e) LiAlH4, THF, rt, 12 h.
Scheme 4.
Synthesis of compounds 7a and 8a.
Scheme 4.
Synthesis of compounds 7a and 8a.
Reagents and conditions: (a) (i) NaH, benzene, rt, (ii) 16, reflux; (b) NH2OH, AcOH, EtOH, 80 °C; (c) (i) NaH, benzene, rt, (ii) 17, reflux, 4 h; (b) NH2OH, AcOH, EtOH, 80 °C, 7 h.
Scheme 5.
Synthesis of compounds 13 and 17.
Scheme 5.
Synthesis of compounds 13 and 17.
Reagents and conditions: (a) PhOH, EDC, DMAP, CH2Cl2, 40 °C, 14 h.
3. Experimental
3.1. General
All reagents and solvents were purchased from commercial suppliers and used as received. Low boiling petroleum ether corresponds to the fraction collected between 40 and 60 °C. THF was distilled from sodium-benzophenone ketyl and degassed thoroughly with dry nitrogen directly before use. Melting points were determined on a Büchi 510 capillary apparatus and are uncorrected. IR spectra were recorded on a J ASCO FT/IR-460 plus equipment.The NMR spectra were obtained with a Varian VXR-300 spectrometer at 200 MHz for
1H and 50 MHz for
13C. Chemical shifts are reported in ppm downfield from internal Me
4Si in CDCl
3. The following abbreviations were used to describe peak patterns where appropriate: singlet (s), doublet (d), triplet (t), multiplet (m) and broad resonances (br). Elemental analyses were performed on a Perkin-Elmer 240 B analyser. TLC was performed on Merck silica gel 60 TLC plates F254 and visualized using UV or phosphomolibdic acid. Flash chromatography was carried out on silica gel (40–60 mesh). The chloroketone
1a was a commercial compound. 6-Chloro-3,4-dihydronaphthalen-1-one (
1b) [
6], 7-chloro-2,3,4,5-tetrahydrobenzocyclo-heptan-1-one (
1c) [
7],
N-Boc-nipecotic acid [
8] and the piperidines
18 [
10] and
19 [
11] were obtained following the corresponding literature procedures.
3.2. General Procedure for the Synthesis of the Compounds 2a–2c
A solution of 1-(tert-butoxycarbonyl)piperidine-4-carboxylic acid (9, 2.85 g, 12.45 mmol) and 1,1′-carbonyldiimidazole (2.29 g, 14.11 mmol) in DMF (3 mL) was stirred at room temperature for 45 min. This solution was added dropwise to a solution prepared by stirring for 20 min the suitable ketone 1a, 1b or 1c (7.64 mmol) with NaH (60% in oil, 0.93 g, 23.20 mmol) in DMF (20 mL). The resulting mixture was heated for the appropriate time. After cooling, H2O was added and the mixture was extracted with Et2O (3 × 30 mL). The organic phase was dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography.
tert-Butyl 4-(5-chloro-1-oxo-2,3-dihydro-1H-indene-2-carbonyl)piperidine-1-carboxylate (2a). According to the general procedure, the reaction between 1a and 9 was carried out at 30 °C for 7 h. The residue was purified by flash chromatography (petroleum ether/EtOAc = 9:1) affording 2a: yield 66%; red solid; Mp 102–103 °C. Rf = 0.10 (petroleum ether/AcOEt = 9:1). 1H-NMR: δ 1.48 (s, 9H), 1.60–1.95 (m, 4H), 2.40–2.64 (m, 1H), 2.64–2.98 (m, 2H), 3.62 (s, 2H), 4.13–4.33 (m, 2H), 7.40 (d, 1H, J = 8.0 Hz), 7.48 (s, 1H), 7.74 (d, 1H, J = 8.0 Hz), 13.80 (brs, 1H). 13C-NMR: δ 27.9 (CH2), 28.4 (3 × CH3), 29.8 (2 × CH2), 41.8 (CH), 43.4 (2 × CH2), 79.7 (C), 108.8 (C), 124.2 (CH), 126.0 (CH), 128.1 (CH), 136.7 (C), 139.1 (C), 148.6 (C), 154.7(CO), 182.7 (CO), 191.3 (COH). IR: (nujol) ν 1703 (CO), 1655 (CO), 1605 (CO) cm−1. Anal. Calcd for C20H24ClNO4: C, 63.57; H, 6.40; N, 3.71. Found: C, 63.65; H, 6.49; N, 3.81.
tert-Butyl 4-(6-chloro-1-oxo-1,2,3,4-tetrahydronaphthalene-2-carbonyl)piperidine-1-carboxylate (2b). According to the general procedure, the reaction between 1b and 9 was carried out at 110 °C for 7 h. The residue was purified by flash chromatography (petroleum ether/EtOAc = 8:2) affording 2b: yield 63%; red solid; Mp 120–121 °C. Rf = 0.43 (petroleum ether/AcOEt = 8:2). 1H-NMR: δ 1.47 (s, 9H), 1.59–1.87 (m, 5H), 2,67 (d, 2H, J = 7.4 Hz), 2.80–2.88 (m, 4H), 4.18 (d, 2H, J = 7.4 Hz), 7.24 (d, 1H, J = 10.2 Hz), 7.31 (s, 1H), 7.87 (d, 1H, J = 8.2 Hz), 16.65 (s, 1H). 13C-NMR: δ 21.9 (CH2), 27.8 (CH2), 28.4 (3 × CH3), 29.3 (2 × CH2), 41.4 (CH), 43.3 (2 × CH2), 79.6 (C), 104.6 (C), 126.2 (CH), 127.2 (CH), 127.3 (CH), 127.5 (C), 136.7 (C), 142.2 (C), 157.3 (CO), 179.0 (CO), 197.8 (COH). IR: (nujol) ν 1707 (CO), 1650 (CO), 1611 (CO) cm−1. Anal. Calcd for C21H26ClNO4: C, 64.36; H, 6.69; N, 3.57. Found: C, 64.88; H, 6.65; N, 3.59.
tert-Butyl 4-(2-chloro-5-oxo-6,7,8,9-tetrahydro-5H-benzo[7]annulene-6-carbonyl)piperidine-1-carboxylate (2c). According to the general procedure the reaction between 1c and 9 was carried out at 70 °C for 7 h. The residue was purified by flash chromatography (petroleum ether/EtOAc = 9:1) affording 2c: yield 63%; yellow solid; Mp 134–136 °C. Rf = 0.31 (petroleum ether/AcOEt = 9:1). 1H-NMR: δ 1.48 (s, 9H), 1.53–1.92 (m, 7H), 1.92–2.13 (m, 1H), 2.18 (t, 2H, J = 6.8 Hz), 2.55–2.90 (m, 3H), 4.10–4.31 (m, 2H), 7.21 (s, 1H), 7.27–7.42 (m, 1H), 7.57 (d, 1H, J = 8.2 Hz), 16.78 (s, 1H). 13C-NMR: δ 22.7 (CH2), 28.3 (3 × CH2), 28.5 (CH3), 31.1 (CH2), 31.3 (2 × CH2), 40.9 (CH), 43.2 (2 × CH2), 79.5 (C), 108.2 (C), 126.8 (CH), 128.7 (CH), 129.0 (CH), 131.0 (C), 136.4 (C), 141.4 (C), 154.5 (CO), 186.2 (CO), 194.4 (COH). IR: (nujol) ν 1706 (CO), 1652 (CO), 1613 (CO) cm−1. Anal. Calcd for C22H28ClNO4: C, 65.10; H, 6.95; N, 3.45. Found: C, 66.08; H, 6.98; N, 3.42.
3.3. General Procedure for the Synthesis of Compounds 3b, 3c
A solution of CF3COOH (1.46 g, 12.8 mmol) in CH2Cl2 (4.6 mL) was added dropwise to a solution of the 1,3-dicarbonyl compound 2b or 2c (1.28 mmol) in CH2Cl2 (9.2 mL). After stirring 2 h at room temperature, CH2Cl2 was added. The resulting mixture was washed two times with a 10% solution of K2CO3 and then with H2O. The organic phase was dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography.
6-Chloro-2-(piperidine-4-carbonyl)-3,4-dihydronaphthalen-1(2H)-one (3b). Compound 2b was converted into the title product 3b according to the general procedure. The residue was purified by flash chromatography (CHCl3/MeOH = 8:2) affording 3b: yield 63%; yellow solid; Mp 150–154 °C. Rf = 0,10 (CHCl3/MeOH 8:2); 1H-NMR: δ 2.62–2.85 (m, 4H), 2.58–2.77 (m, 4H), 2.80–2.95 (m, 4H), 3.10-3.34 (m, 2H), 7.21 (s, 1H), 7.32 (d, 1H, J = 8.2 Hz), 7.86 (d, 1H, J = 8.2 Hz), 8.52–9.20 (brs, 1H). 13C-NMR: δ 21.9 (CH2), 27.5 (CH2), 31.3 (2 × CH2), 42.0 (CH), 45.1 (2 × CH2), 109.2 (C), 126.3 (CH), 127.4 (CH), 129.1 (CH), 137.0 (C), 137.2 (C), 144.5(C), 189.0 (CO), 192.4 (COH) IR: (nujol) ν 3453 (NH), 1701 (CO), 1680 (CO) cm−1. Anal. Calcd for C16H18ClNO2: C, 65.86; H, 6.22; N, 4.80. Found: C, 65.56; H, 6.26; N, 4.83.
2-Chloro-6-(piperidine-4-carbonyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (3c). Compound 2c was converted into the title product 3c according to the general procedure. The residue was purified by flash chromatography (CHCl3/MeOH = 8:2) affording 3c: yield 63%; white solid; Mp 138–142 °C. Rf = 0,11 (CHCl3/MeOH = 8:2); 1H-NMR: δ 1.60–1.92 (m, 4H), 1.92–2.10 (m, 2H), 2.18 (t, 2H, J = 6.2 Hz), 2.58–2.85 (m, 5H), 2.90–2.98 (m, 1H), 3.12–3.28 (m, 2H), 7.21 (s, 1H), 7.33 (d, 1H, J = 8.0 Hz), 7.56 (d, 1H, J = 8.0 Hz), 8.00–9.00 (brs, 1H). 13C-NMR: δ 22.8 (CH2), 28.7 (CH2), 31.1 (2 × CH2), 31.5 (CH), 40.6 (CH2), 45.1 (2 × CH2), 108.2 (C), 126.8 (CH), 128.7 (CH), 129.0 (CH), 136.0 (C), 136.8 (C), 141.5 (C), 188.0 (CO), 194.4 (COH). IR: (nujol) ν 3,430 (NH), 1,703 (CO), 1,680 (CO) cm−1. Anal. Calcd for C17H20ClNO2: C, 66.77; H, 6.59; N, 4.58. Found: C, 67.37; H, 6.64; N, 4.53.
3.4. General Procedure for the Synthesis of the Compounds 4b, 4c and 5b 5c
To a solution of the 1,3-dicarbonyl compound 3b or 3c (3.27 mmol) in DMF (18.25 mL) was added i-Pr2NEt (0.59 g, 4.58 mmol) and then the appropriate halide (1.1 eq). The mixture was then stirred at room temperature or heated under reflux for the necessary time. Water was added and the mixture was extracted with AcOEt. The organic phase was washed with brine, dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography.
2-(1-Benzylpiperidine-4-carbonyl)-6-chloro-3,4-dihydronaphthalen-1(2H)-one (4b). A solution of the ketone 3b and benzyl chloride in DMF was stirred at room temperature for 12 h. After workup the residue was purified by flash chromatography (petroleum ether/EtOAc = 1:1) affording 4b: yield 59%; brown oil; Rf = 0.46 (petroleum ether/EtOAc = 1:1); 1H-NMR: δ 1.51–1.80 (m, 4H), 1.90 (d, 2H, J = 11 Hz), 2.03 (d, 2H, J = 13.2 Hz), 2.58–2.76 (m, 1H), 2.84 (t, 2H, J = 7.4 Hz), 3.00 (d, 2H, J = 9.6 Hz), 3.55 (s, 2H), 7.20 (s, 1H), 7.29–7.40 (m, 6H), 7.86 (d, 1H, J = 8.6 Hz), 16.68 (s, 1H). 13C-NMR: δ 22.8 (CH2) , 28.6 (CH2), 32.1 (2 × CH2), 33.5 (CH), 45.1 (2 × CH2), 64.5 (CH2), 118.4 (C), 126.7 (CH), 126.9 (CH), 127.5 (CH), 128.3 (2 × CH), 128.6 (CH), 128.9 (CH), 129.1 (C), 129.3 (CH), 131.2 (C), 139.6 (C), 142.3 (C), 184.9 (CO), 195.3 (COH) IR: (nujol) ν 1,710 (CO), 1,682 (CO) cm−1. Anal. Calcd for C23H24ClNO2: C, 72.34; H, 6.33; N, 3.67. Found: C, 72.41; H, 6.38; N, 3.75.
6-(1-Benzylpiperidine-4-carbonyl)-2-chloro-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (4c). A solution of the ketone 3c and benzyl chloride in DMF was stirred at room temperature for 12 h. After workup, the residue was purified by flash chromatography (petroleum ether/EtOAc = 1:1) affording 4c: yield 72%; brown oil; Rf = 0.37 (petroleum ether/EtOAc = 1:1); 1H-NMR: δ 1.61–2.24 (m, 8H), 2.53–2.76 (m, 2H), 2.76–3.11 (m, 5H), 3.54 (s, 2H), 7.00–7.48 (m, 6H), 7.55 (d, 1H, J = 8.2 Hz), 8.00 (s, 1H), 16.8 (s, 1H). 13C-NMR: δ 22.9 (CH2), 28.5 (CH2), 31.0 (2 × CH2), 31.4 (CH2), 31,6 (CH), 52.8 (2 × CH2), 62.9 (CH2), 108.4 (C), 126.6 (CH), 126.8 (CH), 127.2 (CH), 127.5 (CH), 128.0 (CH), 128.3 (2 × CH), 128.7 (CH), 129.3 (C), 131.0 (C), 139.6 (C), 145.1 (C), 194.9 (CO), 195.3 (COH). Anal. IR: (nujol) ν 1,705 (CO), 1,682 (CO) cm−1. Calcd for C24H26ClNO2: C, 72.81; H, 6.62; N, 3.54. Found: C, 72.21; H, 6.65; N, 3.57.
6-Chloro-2-(1-phenethylpiperidine-4-carbonyl)-3,4-dihydronaphthalen-1(2H)-one (5b). A solution of the ketone 3b and phenylethyl iodide in DMF was heated at 60 °C for 12 h. After workup, the residue was purified by flash chromatography (petroleum ether/EtOAc = 2:8) affording 5b: yield 70%; brown oil;Rf = 0.42 (petroleum ether/EtOAc = 1:1); 1H-NMR: δ 1.26–2.53 (m, 11H), 2.54–2.75 (m, 2H), 2.75–2.99 (m, 2H), 3.04–3.23 (m, 2H), 7.08–7.45 (m, 6H), 7.49 (m, 1H), 7.71 (d, 1H, J = 9.0 Hz), 14,27 (s, 1H). 13C-NMR: δ 22.8 (CH2), 28.7 (CH2), 32.1 (CH2), 32.6 (2 × CH2), 33.5 (CH), 45.3 (2 × CH2), 64.5 (CH2) , 117.9 (C), 126.6 (CH), 128.3 (CH), 128.8 (CH), 128.9 (2 × CH), 129.1 (2 × CH), 129.2 (CH), 131.2 (C), 139.6 (C), 141.5 (C), 142.3 (C), 194.9 (CO), 195.3 (COH) Anal. IR: (nujol) ν 1,700 (CO), 1,681 (CO) cm−1. Calcd for C24H26ClNO2: C, 72.81; H, 6.62; N, 3.54. Found: C, 72.11; H, 6.66; N, 3.58.
2-Chloro-6-(1-phenethylpiperidine-4-carbonyl)-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one (5c). A solution of the ketone 3c and phenylethyl iodide in DMF was heated at 60 °C for 12 h. After workup, the residue was purified by flash chromatography (CHCl3/acetone = 9:1) affording 5c: yield 62%; brown oil; Rf = 0.33 (CHCl3/acetone = 9:1); 1H-NMR: δ 1.72–1.89 (m, 3H), 1.95–2.28 (m, 7H), 2.53–2.92 (m, 7H), 3.12 (d, 2H, J = 9.6), 7.20 (s, 1H), 7.23–7.25 (m, 6H), 7.56 (d, 1H, J = 8.4 Hz), 16.7 (s, 1H). 13C-NMR: δ 22.8 (CH2), 28.8 (CH2), 31.2 (CH2), 31.7 (2 × CH2), 33.5 (CH2), 41.0 (CH), 53.2 (2 × CH2), 60.7 (CH2), 108.3 (CH), 126.0 (CH), 126.6 (CH), 126.8 (CH), 127.5 (2 × CH), 128.3 (CH), 128.6 (2 × CH), 128.7 (C), 129.1 (C), 131.0 (C), 141.5 (C), 187.9 (CO), 195.3 (COH). IR: (nujol) ν 1,699 (CO), 1,676 (CO) cm−1. Anal. Calcd for C25H28ClNO2: C, 73.25; H, 6.88; N, 3.42. Found: C, 73.76; H, 6.84; N, 3.46.
3.5. General Procedure for the Synthesis of Compounds 6b, 6c
A solution of the 1,3-dicarbonyl compound 4b or 4c (0,68 mmol) and hydrazine hydrate (0.32 g, 6,39 mmol) in MeOH (9 mL) was stirred overnight at room temperature. Water was added and the mixture was extracted with ethyl acetate. The organic phase was dried over anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography.
3-(1-Benzylpiperidin-4-yl)-7-chloro-4,5-dihydro-1H-benzo[g]indazole (6b). Compound 4b was converted into the title product 6b according to the general procedure. After workup, the residue was purified by flash chromatography (CHCl3/acetone = 9:1) affording 6b: yield 78%; yellow solid; Mp 173–174 °C; Rf = 0.51 (CH2Cl2/MeOH = 95:5); 1H-NMR: δ 1.80–2.27 (m, 6H), 2.71–2.82 (m, 3H), 2.91 (t, 2H, J = 7.2 Hz), 3.01 (t, 2H, J = 7.2 Hz), 3.56 (s, 2H), 7.20–7.40 (m, 7H), 7.64–7.71 (m, 1H). 13C-NMR: δ 18.9 (CH2), 29.6 (CH2), 31.3 (2 × CH2), 33.7 (CH), 53.6 (2 × CH2), 63.3 (CH2), 111.2 (C) 123.2 (CH), 126.9 (CH), 127.1 (CH), 127.4 (CH), 127.5 (C), 128.2 (2 × CH), 129.2 (2 × CH), 132.7 (C), 137.8 (C), 138.3 (C). 142.3 (CN), 142.8 (CN). Anal. Calcd for C23H24ClN3: C, 73.10; H, 6.40; N, 11.12. Found: C, 73.91; H, 6.43; N, 11.07.
3-(1-Benzylpiperidin-4-yl)-8-chloro-1,4,5,6-tetrahydrobenzo[3,4]cycloepta[2,1-c]pyrazole (6c). Compound 4c was converted into the title product 6c according to the general procedure. After elaboration, the residue was purified by flash chromatography (CHCl3/acetone = 9:1) affording 6c: yield 50%; yellow solid; Mp 165–166 °C; Rf = 0.51 (CH2Cl2/MeOH = 95:5); 1H-NMR: δ 1.68–2.32 (m, 8H), 2.51–2.90 (m, 5H), 3.04 (d, 2H, J = 9.8 Hz), 3.59 (s, 2H), 7.10–7.42 (m, 7H), 7.60–7.72 (m, 1H), 9.10–10,01 (brs, 1H). 13C-NMR: δ 24.1 (CH2), 26.9 (CH2), 29.7 (CH2), 31.0 (2 × CH2), 34.8 (CH), 53.8 (2 × CH2), 63.2 (CH2), 112.5 (C), 125.7 (CH), 126.4 (CH), 127.1 (CH), 127.4 (CH), 127.5 (2 × CH), 128.2 (2 × CH), 129.3 (2 × C), 129.6 (C), 134.5 (C) 141.3 (CN, 142.8 (CN). Anal. Calcd for C24H26ClN3: C, 73.55; H, 6.69; N, 10.72 Found: C, 73.25; H, 6.88; N, 10.42.
3.6. 1H-1-Oxa-2-aza-7-chloro-3-(1-phenethylpiperidin-4-yl)-4,5-dihydronaphto[2,1-d]isoxazole (7b) and 1H-1-oxa-2-aza-8-chloro-3-(1-phenethylpiperidin-4-yl)-5,6-dihydro-4H-benzo[3,4]cycloepta[1,2-d]isoxazole (7c)
A solution of the 1,3-dicarbonyl compound 5b or 5c (2.44 mmol) and hydroxylamine hydrochloride (1.02 g, 14.64 mmol) in EtOH (12.2 mL) containing 4 drops of AcOH was heated under reflux for 24 h. Water was added and the mixture was extracted with CHCl3. The organic phase was dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (CHCl3/MeOH = 97:3) to give 7b or 7c.
Compound 7b. Yield 20%; brown solid; Mp 170–172 °C; Rf = 0.11 (CHCl3/MeOH = 97:3); 1H-NMR (DMSO-d6): δ 1.50–1.80 (m, 9H), 1.42–2.10 (m, 4H), 2.48 (t, 2H, J = 7.0 Hz), 2.69 (t, 2H, J = 7.0 Hz), 7.12–7.42 (m, 8H). 13C-NMR: δ 28.4 (CH2), 29.5 (CH2), 32.7 (CH2), 38.2 (2 × CH2), 40.7 (CH), 52.7 (2 × CH2), 59.7 (CH2), 117.8 (C), 125.0 (CH), 125.4 (CH), 125.8 (CH), 126.5 (CH), 127.9 (2 × CH), 128.2 (2 × CH), 128.6 (C), 130.9 (C), 136.4 (C), 144.1 (C), 157.3 (CN), 166.1 (CO). Anal. Calcd for C24H25ClN2O: C, 73.36; H, 6.41; N, 7.13. Found: C, 73.96; H, 6.44; N, 7.10.
Compound 7c. yield 35%; brown solid; Mp 114–117 °C; Rf = 0.24 (CHCl3/MeOH = 8:2); 1H-NMR (DMSO-d6): δ 1.82–2.10 (m, 4H), 2.11–2.33 (m, 4H), 2.60–2.76 (m, 5H), 2.70–2.98 (m, 4H), 3.10–3.21 (m, 2H), 7.12–7.20 (m, 7H), 7.88 (d, 1H, J = 8.2 Hz). 13C–NMR: δ 23.9 (CH2), 24.5 (CH2), 29.8 (2 x CH2), 33.6 (CH2), 35.2 (CH), 53.5 (2 x CH2), 60.7 (CH2), 113.8 (C), 126.1 (CH), 126.7 (CH), 128.1 (CH), 128.2 (CH), 128.3 (2 x CH), 128.6 (2 x CH), 129.6 (C), 134.7 (C), 141.5 (C), 142.1 (C), 161.2 (CN), 166.6 (CO) Anal. Calcd for C25H27ClN2O: C, 73.79; H, 6.69; N, 6.88. Found: C, 73.25; H, 6.71; N, 6.91.
3.7. 2H-1-Aza-2-oxa-7-chloro-3-(1-phenethylpiperidin-4-yl)-4,5-dihydronaphto[1,2-c]isoxazole (8b) and 2H-1-aza-2-oxa-8-chloro-3-(1-phenethylpiperidin-4-yl)-5,6-dihydro-4H-benzo[3,4]cycloepta[2,1-c]isoxazole (8c)
A solution of the 1,3-dicarbonyl compound 5b or 5c (2.44 mmol) and hydroxylamine hydrochloride (1.02 g, 14.64 mmol) in EtOH (12.2 mL) containing 15 drops of AcOH was heated under reflux for 24 h. After cooling, H2O was added and the mixture was extracted with CHCl3. The organic phase was dried over Na2SO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (CHCl3/acetone = 8:2) to give 8b or 8c.
Compound 8b. Yield 5%; brown solid; Mp 165–166 °C; Rf = 0.24 (CHCl3/acetone = 97:3); 1H-NMR (CDCl3/DMSO-d6): δ 1.43–1.70 (m, 4H), 1.71–1.93 (m, 4H), 1.95–2.10 (m, 1H), 2.42–2.65 (m, 4H), 2.52 (t, 2H, J = 7.2 Hz), 2.65 (t, 2H, J = 7.2 Hz), 7.05–7.38 (m, 5H), 7.42–7.84 (m, 2H), 8.32 (d, 1H, J = 9.2 Hz). 13C–NMR (CDCl3/DMSO-d6): δ 28.3 (CH2), 29.5 (CH2), 32.6 (CH2), 38.2 (2 × CH2), 40.7 (CH), 53.1 (2 × CH2), 59.8 (CH2), 118.0 (C), 120.2 (CH), 124.1 (CH), 125.4 (CH), 125.0 (CH), 125.8 (2 × CH), 127.9 (2 × CH), 128.6 (C), 131.2 (C), 134.8 (C), 140.3 (C), 161.2 (CN), 167.9 (CO). Anal. Calcd for C24H25ClN2O: C, 73.36; H, 6.41; N. 7.13. Found: C, 72.86; H, 6.44; N. 7.01.
Compound 8c. Yield 12%; light brown solid; Mp 125–126 °C; Rf = 0.47 (CHCl3/acetone = 8:2); 1H-NMR: δ 1.95–2.38 (m, 8H), 2.52–2.78 (m, 5H) 2.78–3.01 (m, 4H), 3.15 (d, 2H, J = 9.6 Hz), 7.08–7.40 (m, 7H), 7.89 (d, 1H, J = 8.4 Hz). 13C-NMR: δ 20.7 (CH2), 27.0 (CH2), 29.2 (CH2), 33.0 (2 × CH2), 33.2, (CH2), 39.3 (CH), 53.1 (2 × CH2), 60.4 (CH2), 111.2 (C), 126.3 (CH), 126.7 (CH), 126.8 (CH), 127.8 (CH), 128.5 (2 × CH), 128.7 (2 × CH2), 129.2 (C), 129.7 (C), 135.2 (C), 142.7 (C), 161.8 (CN), 170.4 (CO). Anal. Calcd for C25H27ClN2O: C, 73.79; H, 6.69; N, 6.88. Found: C, 73.19; H, 6.62; N, 6.83.
(4-(6-Chloro-1,4-dihydroindeno[1,2-c]pyrazol-3-yl)piperidin-1-yl)(phenyl)metanone (11). 5-Chloro-2,3-dihydro-1H-inden-1-one 1a (0.2 g, 1.23 mmol) and NaH (60% in oil, 0.12 g, 3.08 mmol) were added in sequence to a solution of phenyl 1-(phenylcarbonyl)piperidine-4-carboxylate 13 (0.33 g, 1.23 mmol) and the resulting mixture was heated under reflux for 3.5 h. After cooling a 50% aqueous solution of acetic acid was added and the resulting mixture was concentrated under reduced pressure. The residue was taken up in EtOH (4 mL) and AcOH (0.21 mL, 3.69 mmol). Hydrazine hydrate (0.09 mL, 1.85 mmol) was added and the resulting mixture was heated under reflux for 4 h. After cooling, the solvent was evaporated under reduced pressure and the residue was taken up in CH2Cl2. The organic phase was dried over Na2SO4, filtered and the solvent removed under reduced pressure. The residue was purified by flash chromatography (CHCl3/acetone = 95:5) affording 11: yield 82%; yellow solid; Mp 167–170 °C; Rf = 0,24 (CHCl3/MeOH = 95:5); 1H-NMR: δ 1.58–2.21 (m, 6H), 2.90–3.21 (m, 3H), 3.63 (s, 2H), 7.33 (d, 1H, J = 8.0 Hz), 7.37–7.50 (m, 6H), 7.6 (d, 1H, J = 8.0 Hz). 13C-NMR: δ 27.3 (CH2), 29.1 (2 × CH2), 29.4 (CH), 34.2 (2 × CH2), 120.7 (C), 126.2 (CH), 126.9 (CH), 127.3 (CH), 128.5 (2 × CH), 129.8 (2 × CH2), 129.9 (CH), 132.1(C), 132.7 (C), 133.8 (C), 141.6 (C), 150.1 (CN) 157.3 (CN), 170.5 (CO). Anal. Calcd for C22H20ClN3O: C, 69.93; H, 5.33; N, 11.12. Found: C, 70.34; H, 5.31; N, 11.17.
3-(1-Benzylpiperidin-4-yl)-6-chloro-1,4-dihydroindeno[1,2-c]pyrazole (6a). A solution of the amide 11 (0.14 g, 0.37 mmol) in THF (2 mL) was added dropwise to a suspension of LiAlH4 (56.0 mg, 1.48 mmol) in THF (2 mL) at 0 °C. After stirring at room temperature for 12 h the mixture was diluted with Et2O (2,5 mL) and then NaOH (1 M, 0.1 mL) e H2O (0.3 mL) were added. The formed solid was filtered and diluted with CH2Cl2. The organic phase was dried over anhydrous Na2SO4, filtered and the solvent was removed under reduced pressure. The residue was purified by flash chromatography (CHCl3/acetone = 95:5) affording 6a: yield 80%; yellow solid; Mp 146–148 °C; Rf = 0.25 (CHCl3/MeOH 95:5); 1H-NMR: 1.75–2.16 (m, 6H), 2.60–2.80 (m, 1H), 2.95 (m, 2H), 3.50 (m, 1H), 3.53 (s, 2H), 3.59 (s, 2H), 7.17–7.37 (m, 6H), 7.42 (s, 1H), 7.61 (d, 1H, J = 8.0 Hz), 9.25–10.35 (brs, 1H). 13C-NMR: δ 28.7 (CH2), 30.3 (2 × CH2), 31.0 (CH), 52.5 (2 × CH2), 62.3 (CH2), 119.7(C), 125.3 (CH), 126.2 (CH), 126.6 (CH), 127.6 (2 × CH), 128.8 (2 × CH), 130.8 (CH), 133.0 (C), 136.5 (C), 141.1 (C), 141.4 (C) 143.6 (CN) 149.7 (CN). Anal. Calcd for C22H22ClN3: C, 72.62; H, 6.09; N, 11.55. Found: C, 72.70; H, 6.15; N, 11.59.
3.8. 1H-1-Oxa-2-aza-6-chloro-3-(1-phenethylpiperidin-4-yl)-1,4dihydroindeno[2,1-d]isoxazole (7a) and 2H-1-aza-2-oxa-6-chloro-3-(1-phenethylpiperidin-4-yl)-1,4dihydroindeno[1,2-c]isoxazole (8a)
To a solution of phenyl 1-(phenylethyl)piperidine-4-carboxylate 17 (0.50 g, 1.23 mmol) were added in sequence 5-chloro-2,3-dihydro-1H-inden-1-one 1a (0.2 g, 1.23 mmol) and NaH (60% in oil, 0.12 g, 3.08 mmol). The resulting mixture was heated under reflux for 4 h. After cooling a 50% aqueous solution of acetic acid was added and the resulting mixture was concentrated under reduced pressure. To the residue was taken up in EtOH (5 mL), AcOH (0.21 mL, 3.69 mmol) and hydroxylamine hydrochloride (0.128 mg, 1.85 mmol) was added. The resulting mixture was heated under reflux for 8 h. The solvent was evaporated under reduced pressure and the residue was taken up in CH2Cl2. The organic phase was dried over Na2SO4,, filtered and the solvent removed under reduced pressure. The residue was purified by flash chromatography (CHCl3/acetone = 8:2) to give 7a and 8a.
Compound 7a. Yield 12%; brown solid; Mp 149–151 °C; Rf = 0,20 (CHCl3/acetone = 8:2); 1H-NMR: δ 1.60–2.04 (m, 9H), 2.49 (t, 2H, J = 7.2 Hz), 2.62 (t, 2H, J = 7.2 Hz), 3.55 (s, 2H) 7.23–7.42 (m, 8H). 13C-NMR: δ 28.8 (CH2), 30.6 (CH2), 31.1 (2 × CH2), 31.7 (CH), 52.5 (2 × CH2), 62.4 (CH2), 119.7 (C), 125.4 (CH), 126.2 (CH), 126.7 (CH), 127.8 (2 × CH), 128.8 (2 × CH), 130.9 (CH), 134.0 (C), 136.0 (C), 142.5 (C), 153.7 (C), 161.0 (CN), 166.4 (CO). Anal. Calcd for C23H23ClN2O: C, 72.91; H, 6.12; N, 7.39. Found: C, 73.12; H, 6.10; N, 7.43.
Compound 8a. Yield 41%; brown solid; Mp 157–160 °C; Rf = 0.41 (CHCl3/MeOH 8:2); 1H-NMR (DMSO-d6): δ 1.82–2.10 (m, 5H), 2.11–2.33 (m, 4H), 2.60–2.70 (m, 2H), 2.73–2.88 (m, 2H), 3.57 (s, 2H), 7.12–7.28 (m, 8H). 13C-NMR: δ 28.8 (CH2), 30.5 (CH2), 31.2 (2 x CH2), 31.4 (CH), 53.2 (2 × CH2), 62.3 (CH2), 119.6 (C), 125.3 (CH), 126.5 (CH), 126.6 (CH), 127.8 (2 × CH2), 128.8 (2 × CH2), 130.9 (CH) 134.1 (C), 136.5 (C), 142.4 (C), 151.9 (C), 162.3 (CN) 169.2 (CO). Anal. Calcd for C23H23ClN2O: C, 72.91; H, 6.12; N, 7.39. Found: C, 73.10; H, 6.14; N, 7.46.
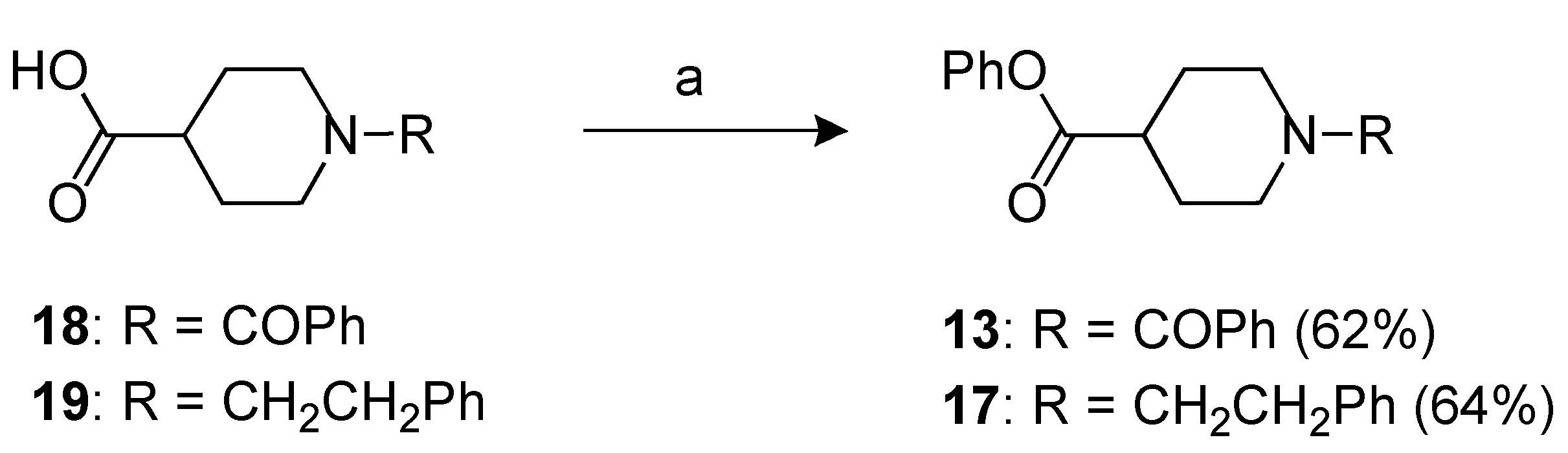
3.9. Phenyl 1-benzoylpiperidine-4-carboxylate (13) and phenyl 1-phenethylpiperidine-4-carboxylate (17)
A mixture of the acid 18 or 19 (2.19 mmol) in CH2Cl2 (40 ml), 1-3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (0.84 g, 4.38 mmol), dimethylaminopiridine (0.54 g, 4.38 mmol) and phenol (0.62 g, 6.57 mmol) was heated under reflux for 14 h. After cooling, the reaction mixture was diluted with CH2Cl2 and washed with a saturated NH4Cl solution (3 × 20 mL). The separated organic phase was dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the residue was purified by flash chromatography affording the product 13 or 17.
Compound 13. purified by flash chromatography by using as the eluent petroleum ether/EtOAc = 1:1); yield 62%; yellow oil; Rf = 0.46 (petroleum ether/EtOAc = 1:1); 1H-NMR: δ 1.65–2.10 (m, 6H), 2.70–2.90 (m, 1H), 3.10–3.25 (m, 2H), 7.00–7.45 (m, 10H). 13C-NMR: δ 28.2 (2 × CH2), 41.1 (CH), 46.8 (2 × CH2), 121.3 (2 × CH), 125.9 (CH), 126.8 (2 × CH), 128.4 (2 × CH), 129.4 (2 × CH), 129.6 (CH), 135.8 (C), 150.4 (C), 170.4 (CO), 172.6 (CO). IR: (nujol) ν 1,752 (CO), 1,628 (CO) cm−1. Anal. Calcd for C19H19NO3: C, 73.77; H, 6.19; N, 4.53. Found: C, 73.85; H, 6.25; N, 4.46.
Compound 17. purified by flash chromatography by using as the eluent CHCl3/MeOH = 9:1; yield 64%; yellow oil; Rf = 0,27 (CHCl3/MeOH = 9:1); 1H-NMR: δ 1.80–2.20 (m, 4H), 2.56–2.65 (m, 2H), 2.80–2.90 (m, 3H), 3.00–3.15 (m, 2H), 3.43 (t, 2H, J = 5.4 Hz), 7.06 (d, 2H, J = 8.4 Hz), 7.20-7.42 (m, 8H). 13C-NMR: δ 28.2, (2 x CH2) 33.8 (CH2), 41.1 (CH), 46.8 (2 x CH2), 57.4 (CH2), 122.5 (2 × CH), 126.9 (2 × CH), 127.3 (CH), 128.4 (2 × CH), 129.0 (2 × CH), 129.5 (CH), 135.9 (C), 151.4 (C), 170.4 (CO). IR: (nujol) ν 1,750 (CO) cm−1. Anal. Calcd for C20H23NO2: C, 77.64; H, 7.49; N, 4.53. Found: C, 77.75; H, 7.42; N, 4.58.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





