Studies toward the First Stereoselective Total Synthesis of (±)-Quinolizidine 195C and Other Transformations

Abstract
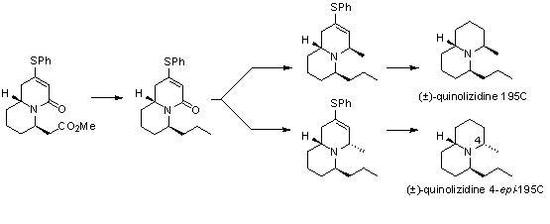
:
1. Introduction


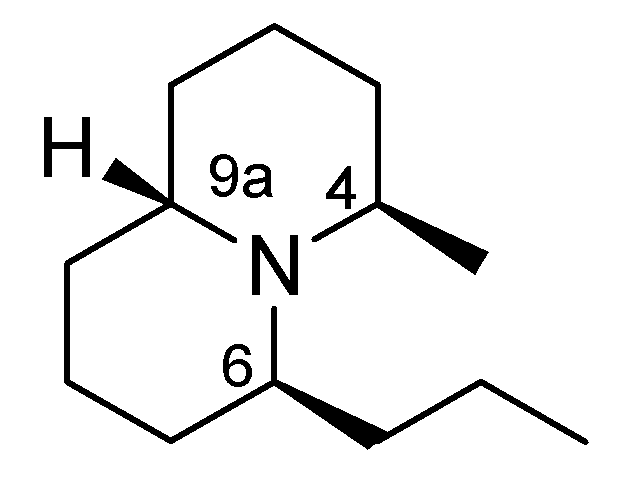
2. Results and Discussion



| Entry | Reaction Conditions | Results |
|---|---|---|
| 1 | MeMgBr (4 equiv), 65 °C, 3 h | NR a |
| 2 | MeLi (5 equiv), rt, 5 h | NR a |
| 3 | MeLi (5 equiv), 50 °C, 5 h | ND b |

{kind=link}
{kind=link}
{kind=link}
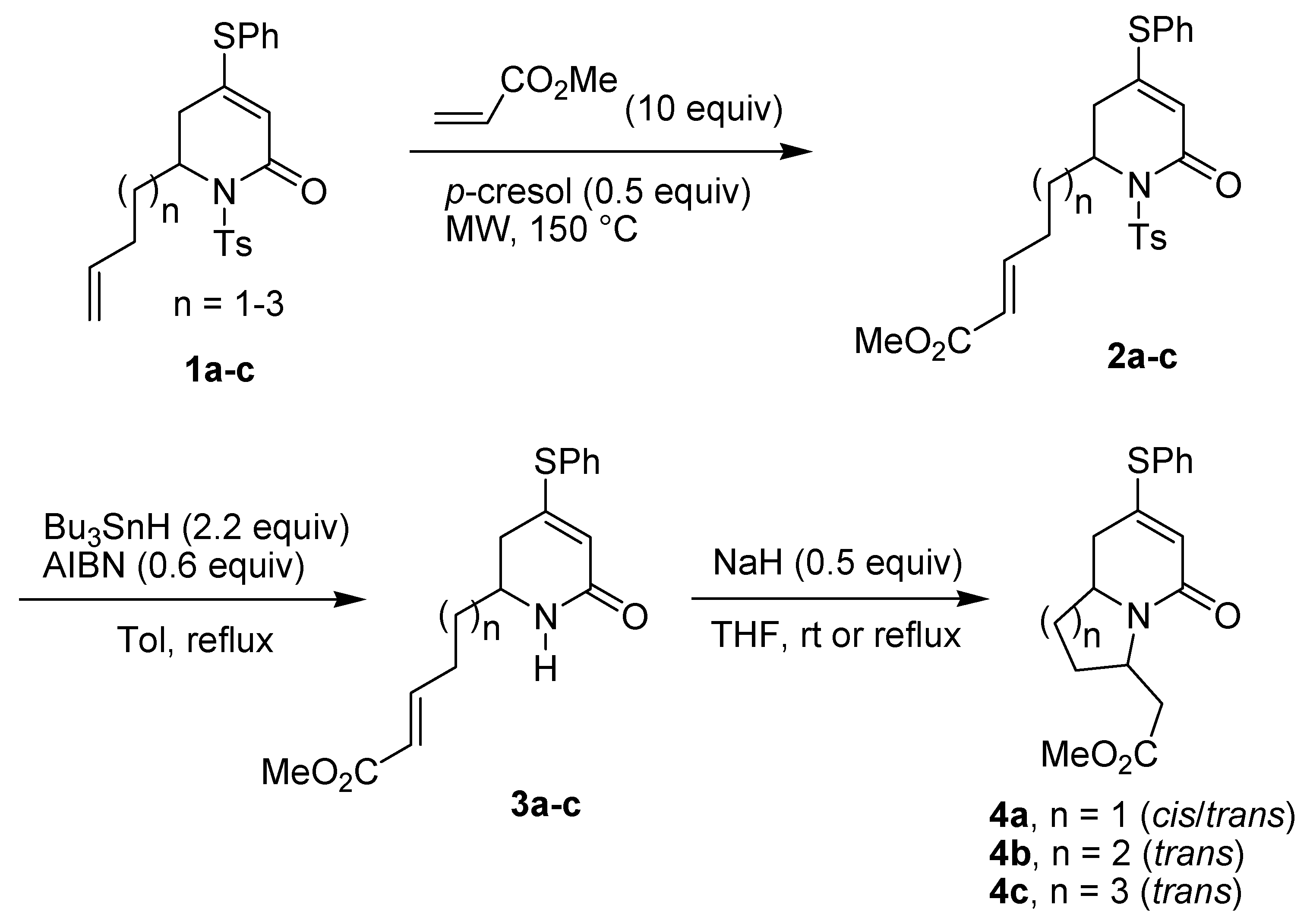{kind=link}
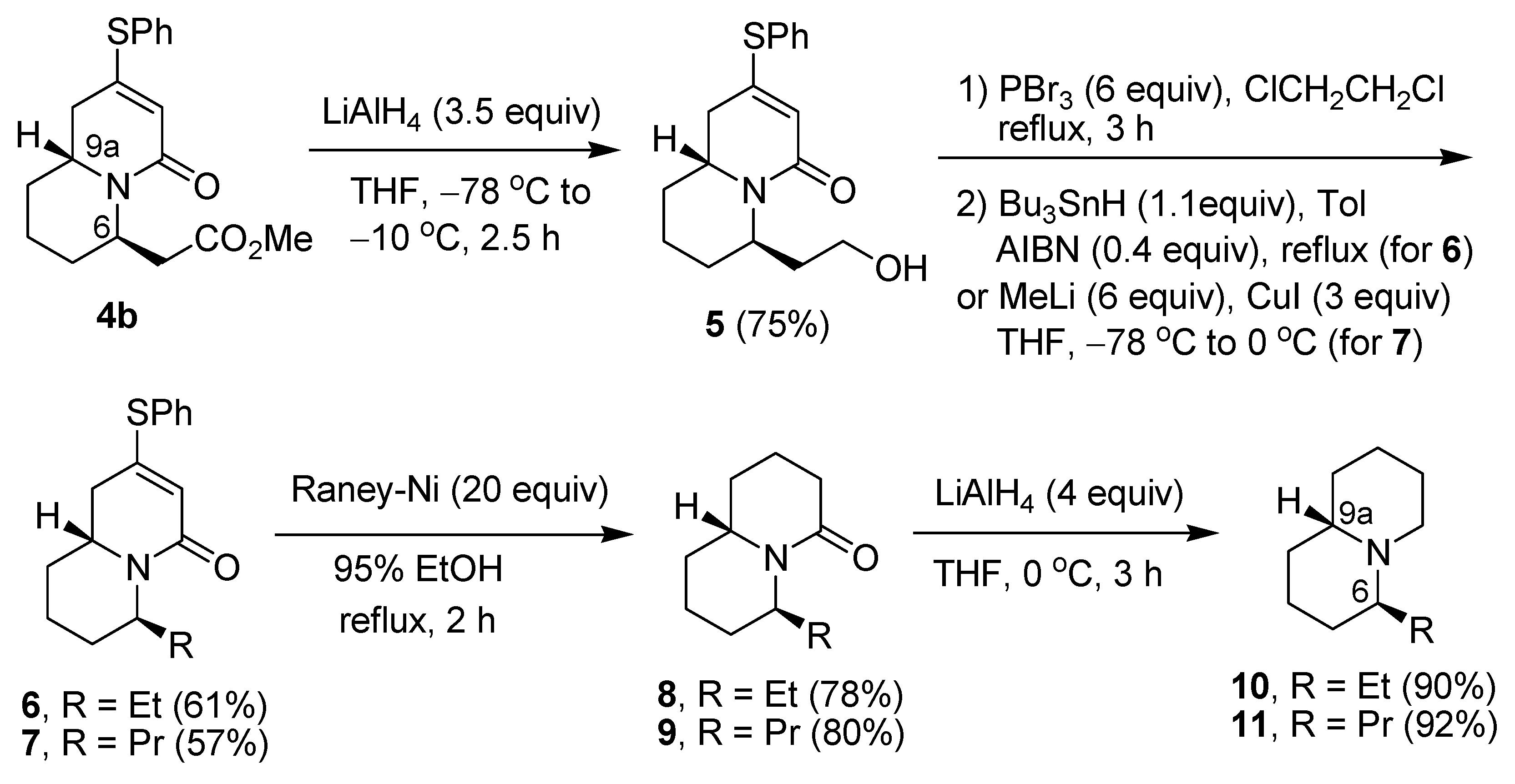{kind=link}
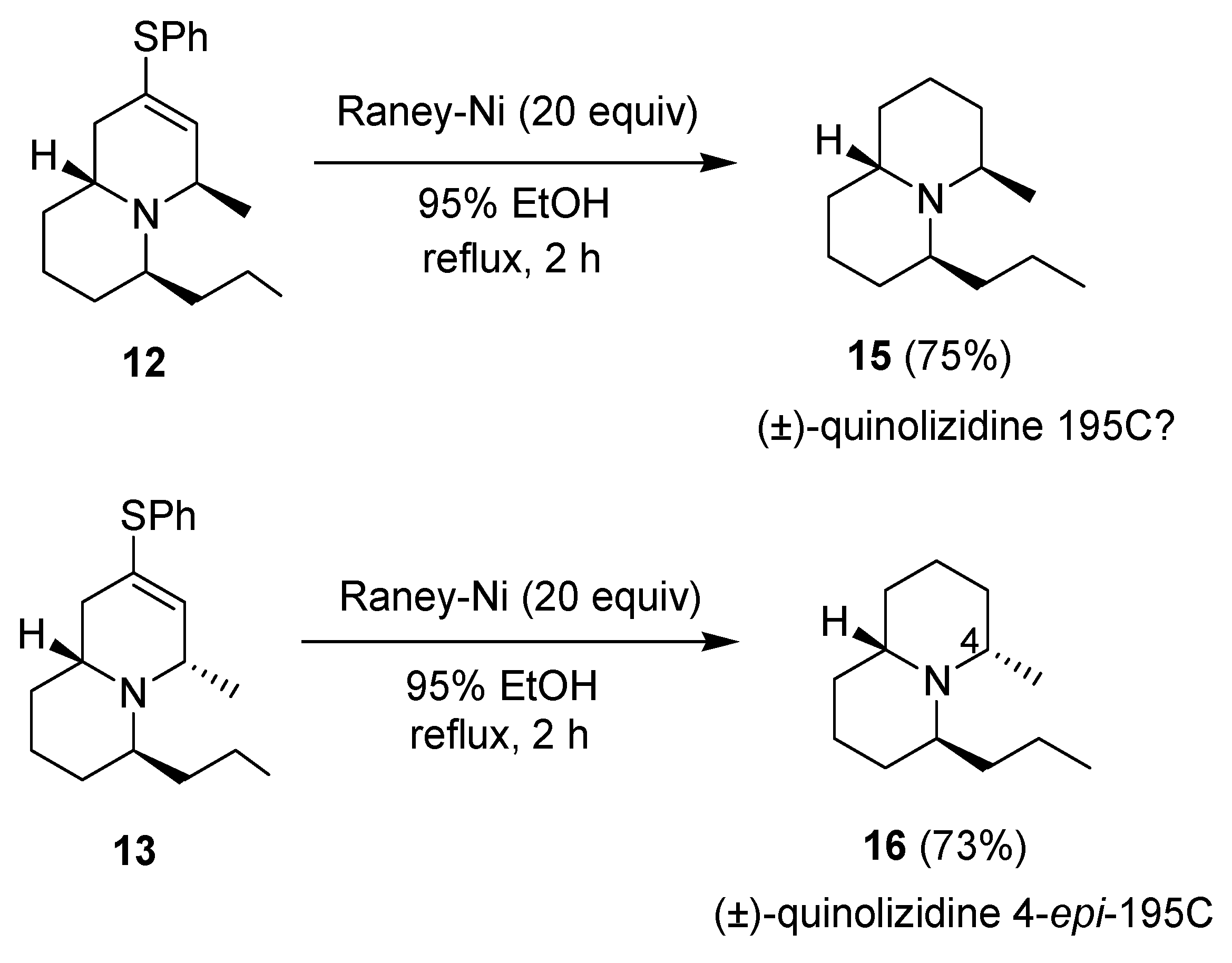{kind=link}
{kind=link}
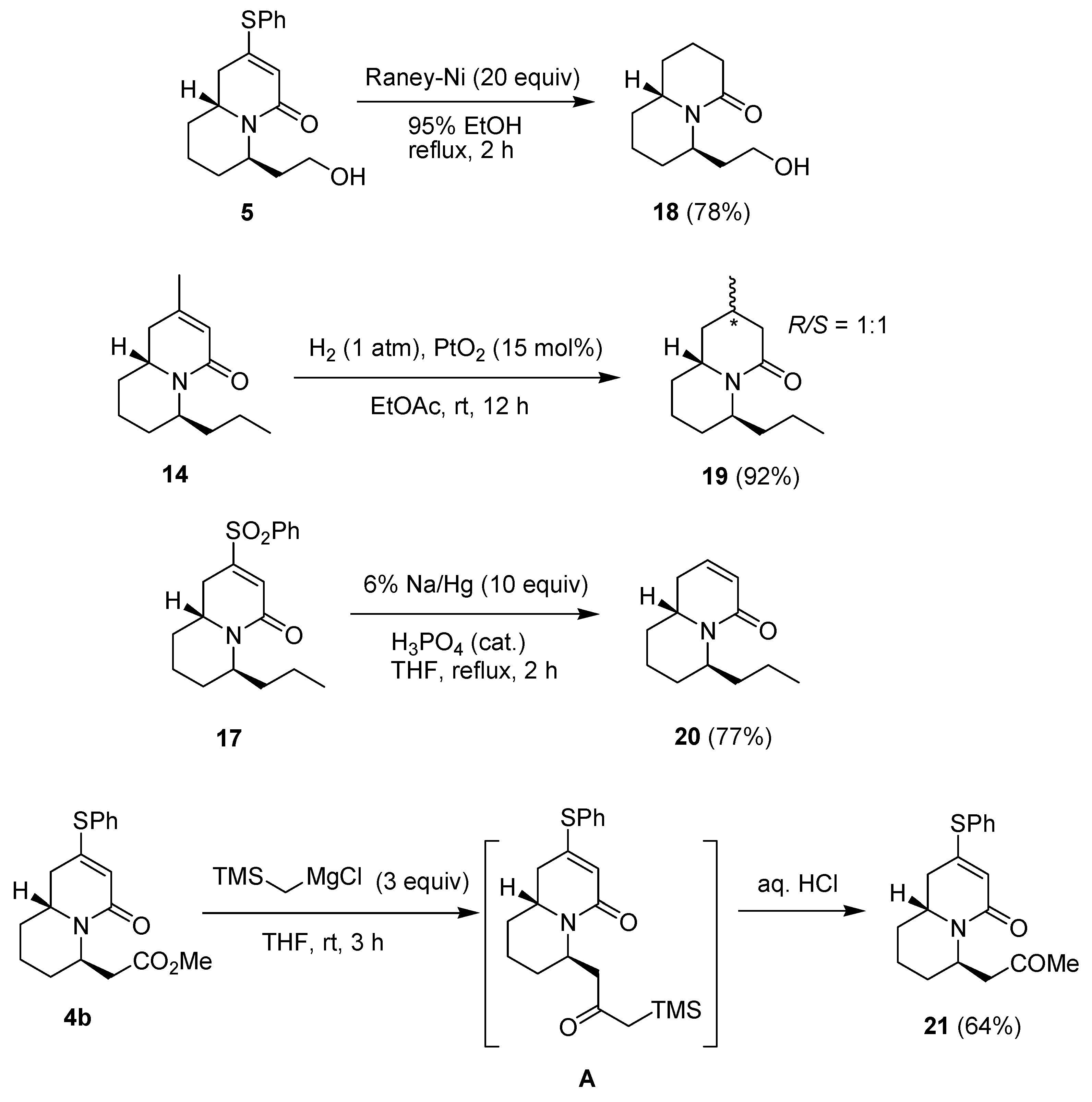{kind=link}
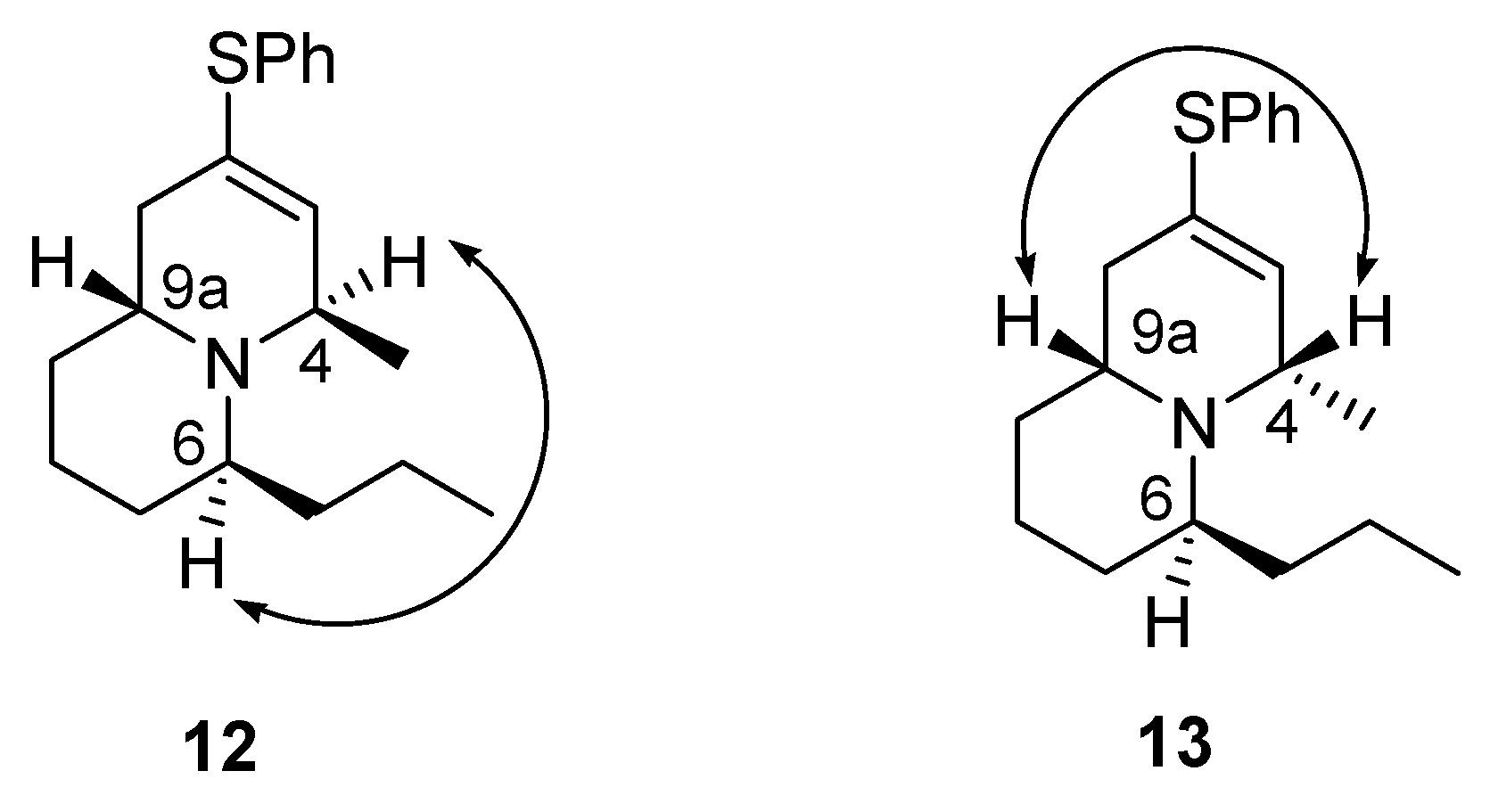
| Entry | Reaction Conditions | Products (%Yield) a | Ratio of12/13 b |
|---|---|---|---|
| 1 | NaBH4, 0 °C, 2.5 h | 12 (27), 13 (13), 14 (31) | 2:1 |
| 2 | NaB(OAc)3H, 0 °C, 2 h | 12 (29), 13 (14), 14 (27) | 2:1 |
| 3 | NaB(OAc)3H, rt, 2 h | 12/13 (33%), c 14 (30) | 1.5:1 |
| 4 | NaBH4, −50 °C, 2.5 h, then to 0 °C, 2 h | 12 (34), 13 (8), 14 (38) | 4:1 |
| 5 | NaB(OAc)3H, −50 °C to 0 °C, 2 h | 14 (34) | - |
| 6 | NaBH3CN, −50 °C to 0 °C, 2 h | 14 (36) | - |




3. Experimental
General
4. Conclusions
Supplementary Material
Acknowledgments
Conflicts of Interest
References
- Rubiralta, M.; Giralt, E.; Diez, E. Structure, Preparation, Reactivity and Synthetic Applications of Piperidine and its Derivatives; Elsevier: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Laschat, S.; Dickner, T. Stereoselective synthesis of piperidines. Synthesis 2000, 13, 1781–1813. [Google Scholar] [CrossRef]
- Buffat, M.G.P. Synthesis of piperidines. Tetrahedron 2004, 60, 1701–1729. [Google Scholar] [CrossRef]
- Merino, P.; Tejero, T.; Greco, G.; Marca, E.; Delso, I.; Gomez-SanJuan, A.; Matute, R. Recent advances on the synthesis of piperidines through ruthenium-catalyzed ring-closing metathesis (RCM) reactions. Heterocycles 2012, 84, 75–100. [Google Scholar] [CrossRef]
- Daly, J.W.; Spande, T.F. Alkaloids: Chemical and Biological Perspectives; Pelletier, S.W., Ed.; Wiley: New York, NY, USA, 1986; Volume 3, Chapter 1. [Google Scholar]
- Michael, J.P. Indolizidine and quinolizidine alkaloids. Nat. Prod. Rep. 2008, 25, 139–165. [Google Scholar] [CrossRef]
- Rueping, M.; Hubener, L. Enantioselective synthesis of quinolizidines and indolizidines via a catalytic asymmetric hydrogenation cascade. Synlett 2011, 1243–1246. [Google Scholar] [CrossRef]
- Belanger, G.; O’Brien, G.; Larouche-Gauthier, R. Rapid assembly of quinolizidines via consecutive nucleophilic cyclizations onto activated amides. Org. Lett. 2011, 13, 4268–4271. [Google Scholar] [CrossRef]
- Buchanan, G.S.; Dai, H.; Hsung, R.P.; Gerasyuto, A.I.; Scheinebeck, C.M. Asymmetric aza-[3+3] annulation in the synthesis of indolizidines: an unexpected reversal of regiochemistry. Org. Lett. 2011, 13, 4402–4405. [Google Scholar] [CrossRef]
- Lazzaroni, R.; Settambolo, R. Synthesis of indolizidines from optically pure α-amino acids via stereocontrolled rhodium-catalyzed hydroformylation of N-allylpyrroles. Chirality 2011, 23, 730–735. [Google Scholar] [CrossRef]
- Yang, D.; Micalizio, G.C. A convergent stereoselective synthesis of quinolizidines and indolizidines: Chemoselective coupling of 2-hydroxymethyl-substituted allylic silanes with imines. J. Am. Chem. Soc. 2009, 131, 17548–17549. [Google Scholar] [CrossRef]
- Amorde, S.M.; Jewett, I.T.; Martin, S.F. Iminium ion cascade reactions: stereoselective synthesis of quinolizidines and indolizidines. Tetrahedron 2009, 65, 3222–3231. [Google Scholar] [CrossRef]
- Furman, B.; Lipner, G. Rhodium-catalyzed intramolecular conjugate addition of vinylstannanes to dihydro-4-pyridones: A simple method for stereoselective construction of 1-azabicyclic alkaloids. Tetrahedron 2008, 64, 3464–3470. [Google Scholar] [CrossRef]
- Coldham, I.; Jana, S.; Watson, L.; Pilgram, C.D. Cascade cyclization intermolecular dipolar cycloaddition by multi-component couplings–synthesis of indolizidines and pyrrolizidines. Tetrahedron Lett. 2008, 49, 5408–5410. [Google Scholar] [CrossRef]
- Pattenden, L.C.; Adams, H.; Smith, S.A.; Harrity, J.P.A. Development of a [3+3] approach to tetrahydropyridines and its application in indolizidine alkaloid synthesis. Tetrahedron 2008, 64, 2951–2961. [Google Scholar] [CrossRef]
- Lee, E.E.; Rovis, T. Enantioselective synthesis of indolizidines bearing quaternary substituted stereocenters via rhodium-catalyzed [2+2+2] cycloaddition of alkenyl isocyanates and terminal alkynes. Org. Lett. 2008, 10, 1231–1234. [Google Scholar] [CrossRef]
- Buonora, P.; Olsen, J.-C.; Oh, T. Recent developments in imino Diels–Alder reactions. Tetrahedron 2001, 57, 6099–6138. [Google Scholar] [CrossRef]
- Heintzelman, G.R.; Meigh, I.R.; Mahajan, Y.R.; Weinreb, S.M. Diels-Alder reactions of imino dienophiles. Org. React. 2005, 65, 141. [Google Scholar]
- Maison, W. Science of Synthesis; Enders, D., Schaumann, E., Eds.; Georg Thieme Verlag KG: Stuttgart, Germany, 2009; Volume 40a, p. 343. [Google Scholar]
- Chou, S.S.P.; Hung, C.C. Aza-Diels–Alder reactions and synthetic applications of thio-substituted 1,3-dienes with arylsulfonyl isocyanates. Tetrahedron Lett. 2000, 41, 8323–8326. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Hung, C.C. Synthesis and applications of tetrahydro-2-pyridinones via aza-Diels-Alder reactions of thio-substituted 1,3-dienes with arylsulfonyl isocyanates. Synthesis 2001, 2450–2462. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Chiu, H.C.; Hung, C.C. Synthesis of 6-substituted tetrahydropyridinones and cyclization to indolizidine and quinolizidine structures. Tetrahedron Lett. 2003, 44, 4653–4655. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Ho, C.W. Synthesis and transformations of sulfur-substituted indolizidines and quinolizidines. Tetrahedron Lett. 2005, 46, 8551–8554. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Liang, C.F.; Lee, T.M.; Liu, C.F. Synthesis of sulfur-substituted quinolizidines and pyrido[1,2-a]azepines by ring-closing metathesis. Tetrahedron 2007, 63, 8267–8273. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Liu, C.F. A rapid, solvent-free phase-transfer catalysis procedure for N-alkylation of dihydropyridones and construction of the indolizidine, quinolizidine and pyridoazepine structures. J. Chin. Chem. Soc. 2010, 57, 811–819. [Google Scholar]
- Chou, S.S.P.; Cai, Y.L. Synthesis and applications of sulfur-substituted cis-hexahydro-2-quinolinones. Tetrahedron 2011, 67, 1183–1186. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Chung, Y.C.; Chen, P.A.; Chiang, S.L.; Wu, C.J. Synthetic applications of sulfur-substituted indolizidines and quinolizidines. J. Org. Chem. 2011, 76, 692–695. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Yang, T.H.; Wu, W.S.; Chiu, T.H. Stereospecific synthesis of trans-5,6-dihydropyridinones. Synthesis 2011, 759–763. [Google Scholar]
- Chou, S.S.P.; Wu, C.J.J. Ring opening of dihydro-2-pyridones and intramolecular Diels-Alder reactions. Tetrahedron 2012, 68, 1185–1191. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Lu, C.L.; Hsu, Y.H. Synthesis of triazolyl-substituted quinolizidine imides. J. Chin. Chem. Soc. 2012, 59, 365–372. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Wu, C.J.J. Chiral synthesis of indolizidines 209D and 167B via asymmetric oxidation of sulfides and sulfoxides. Tetrahedron 2012, 68, 5025–5030. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Chiang, S.L.; Huang, G.L.; Chiang, B.S.; Yu, Y.C. New synthesis and reactions of indolizidine 167E and indolizidine derivatives. Tetrahedron 2013, 69, 274–283. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Huang, J.L. Tandem cross metathesis and intramolecular aza-Michael reaction to synthesize bicyclic piperidines and indolizidine 167E. Tetrahedron Lett. 2012, 53, 5552–5554. [Google Scholar] [CrossRef]
- Jones, T.H.; Gorman, J.S.T.; Snelling, R.R.; Delabie, J.H.C.; Blum, M.S.; Garraffo, H.M.; Jain, P.; Daly, J.W.; Spande, T.F. Further alkaloids common to ants and frogs: decahydroquinolines and a quinolizidine. J. Chem. Ecol. 1999, 25, 1179–1193. [Google Scholar] [CrossRef]
- Daly, J.W.; Spande, T.F.; Garraffo, H.M. Alkaloids from amphibian skin: a tabulation of over eight-hundred compounds. J. Nat. Prod. 2005, 68, 1556–1575. [Google Scholar] [CrossRef]
- Airiau, E.; Girard, N.; Pizzeti, M.; Salvadori, J.; Taddei, M.; Mann, A. Hydroformylation of alkenylamines. Concise approaches toward piperidines, quinolizidines, and related alkaloids. J. Org. Chem. 2010, 75, 8670–8673. [Google Scholar]
- Chou, S.S.P.; Zhang, J.W.; Chen, K.H. Synthetic studies of quinolizidine 195C and derivatives. Tetrahedron 2013, 69, 1499–1508. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Sun, C.M. A facile synthesis of stable precursors to 2-alkylated and 2,3-dialkylated 1,3-butadienes. Tetrahedron Lett. 1990, 31, 1035–1038. [Google Scholar] [CrossRef]
- Chou, S.S.P.; Lee, B.H.; Ni, C.H.; Lin, Y.H. Synthesis and reactions of sulfur-substituted indolizidinones. Tetrahedron 2011, 67, 5395–5401. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chou, S.-S.P.; Huang, J.-L. Studies toward the First Stereoselective Total Synthesis of (±)-Quinolizidine 195C and Other Transformations. Molecules 2013, 18, 8243-8256. https://doi.org/10.3390/molecules18078243
Chou S-SP, Huang J-L. Studies toward the First Stereoselective Total Synthesis of (±)-Quinolizidine 195C and Other Transformations. Molecules. 2013; 18(7):8243-8256. https://doi.org/10.3390/molecules18078243
Chicago/Turabian StyleChou, Shang-Shing P., and Jhih-Liang Huang. 2013. "Studies toward the First Stereoselective Total Synthesis of (±)-Quinolizidine 195C and Other Transformations" Molecules 18, no. 7: 8243-8256. https://doi.org/10.3390/molecules18078243
APA StyleChou, S.-S. P., & Huang, J.-L. (2013). Studies toward the First Stereoselective Total Synthesis of (±)-Quinolizidine 195C and Other Transformations. Molecules, 18(7), 8243-8256. https://doi.org/10.3390/molecules18078243