Click Chemistry in Peptide-Based Drug Design

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
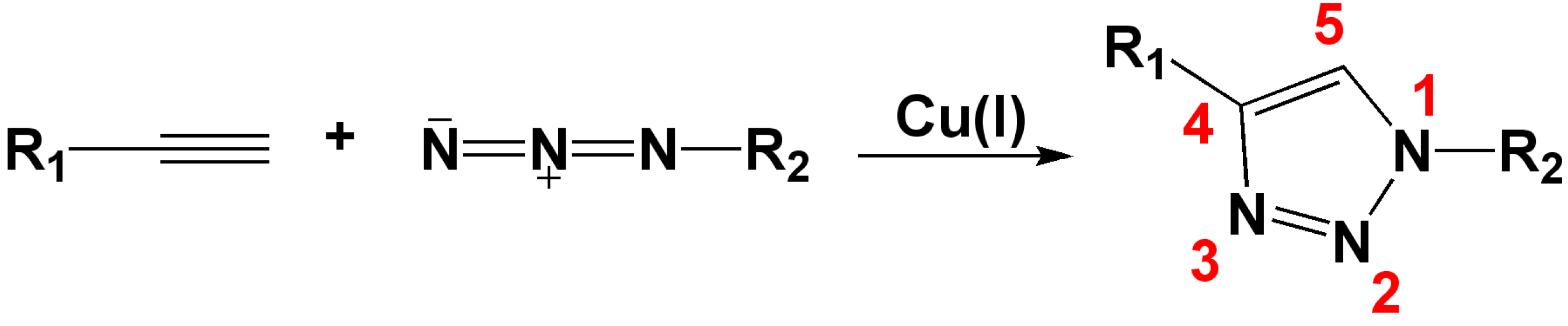{kind=link}
Abstract
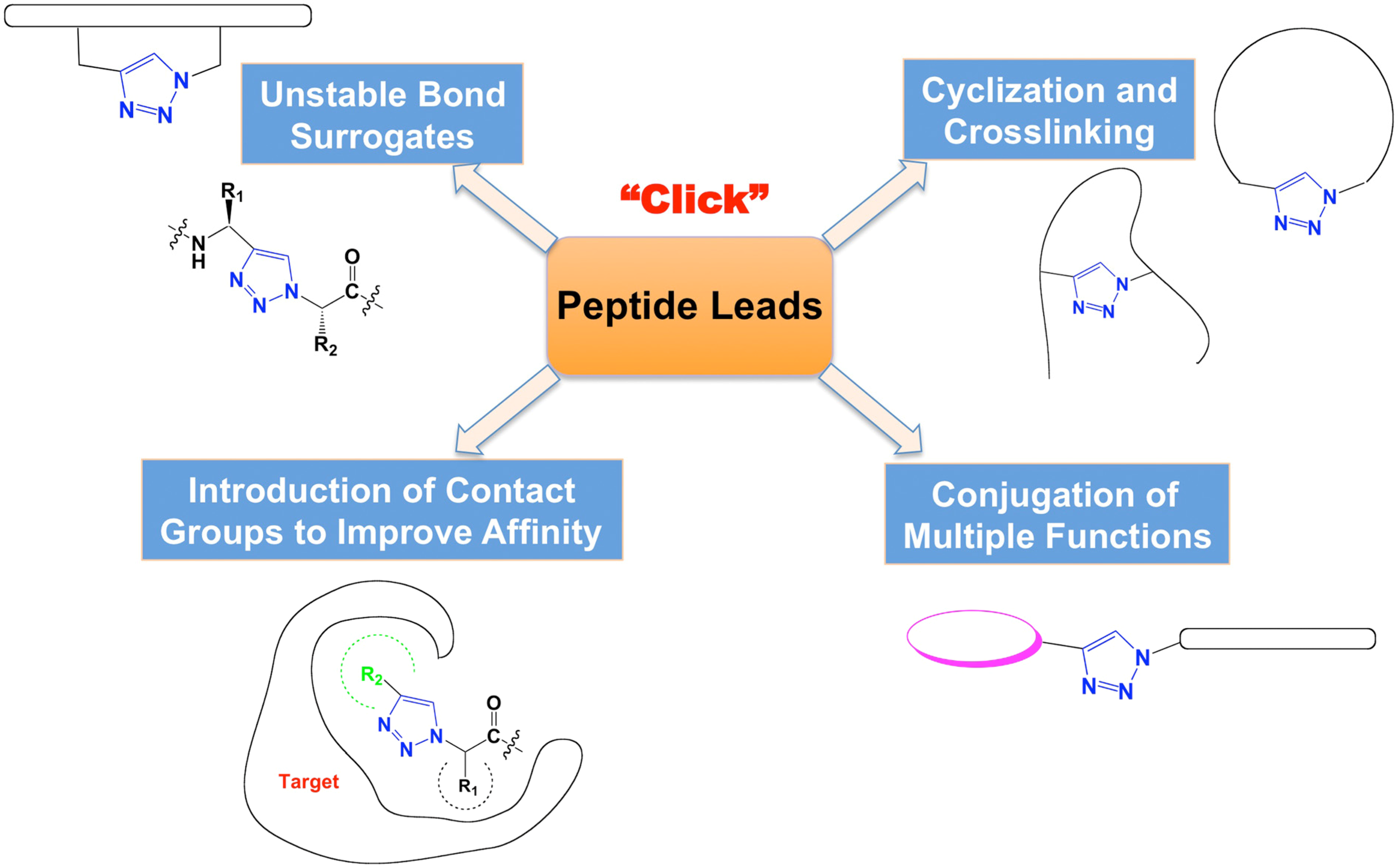
:1. Introduction
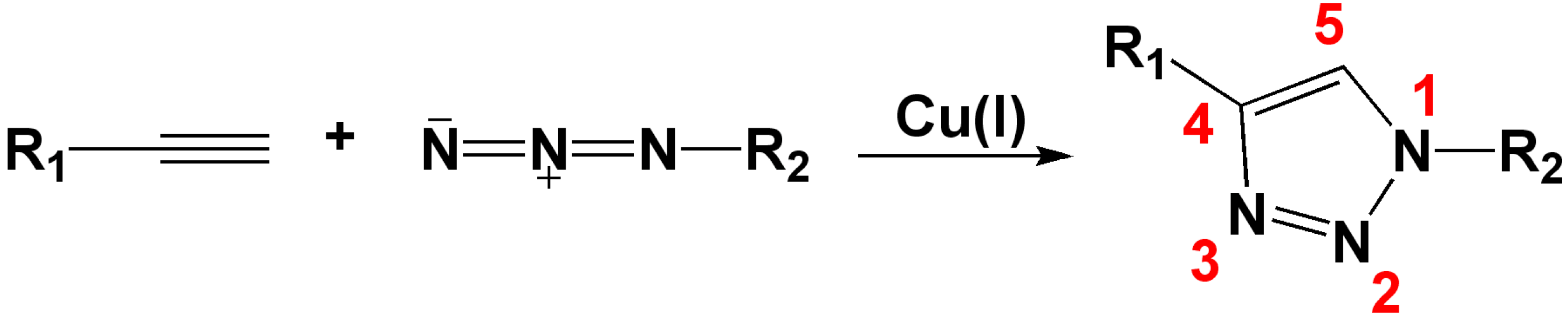
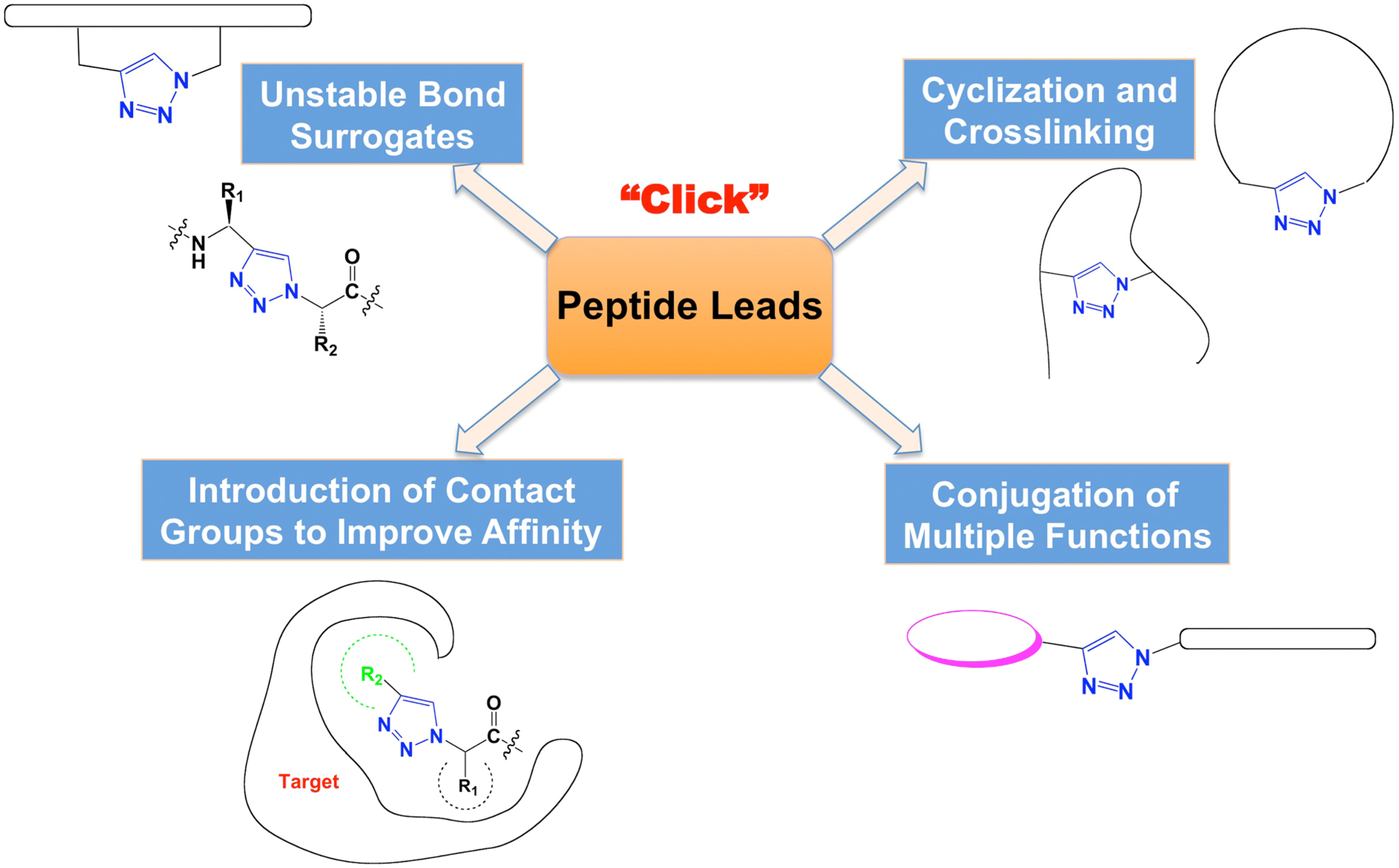
2. 1,2,3-Triazoles as Surrogates for Unstable Bonds
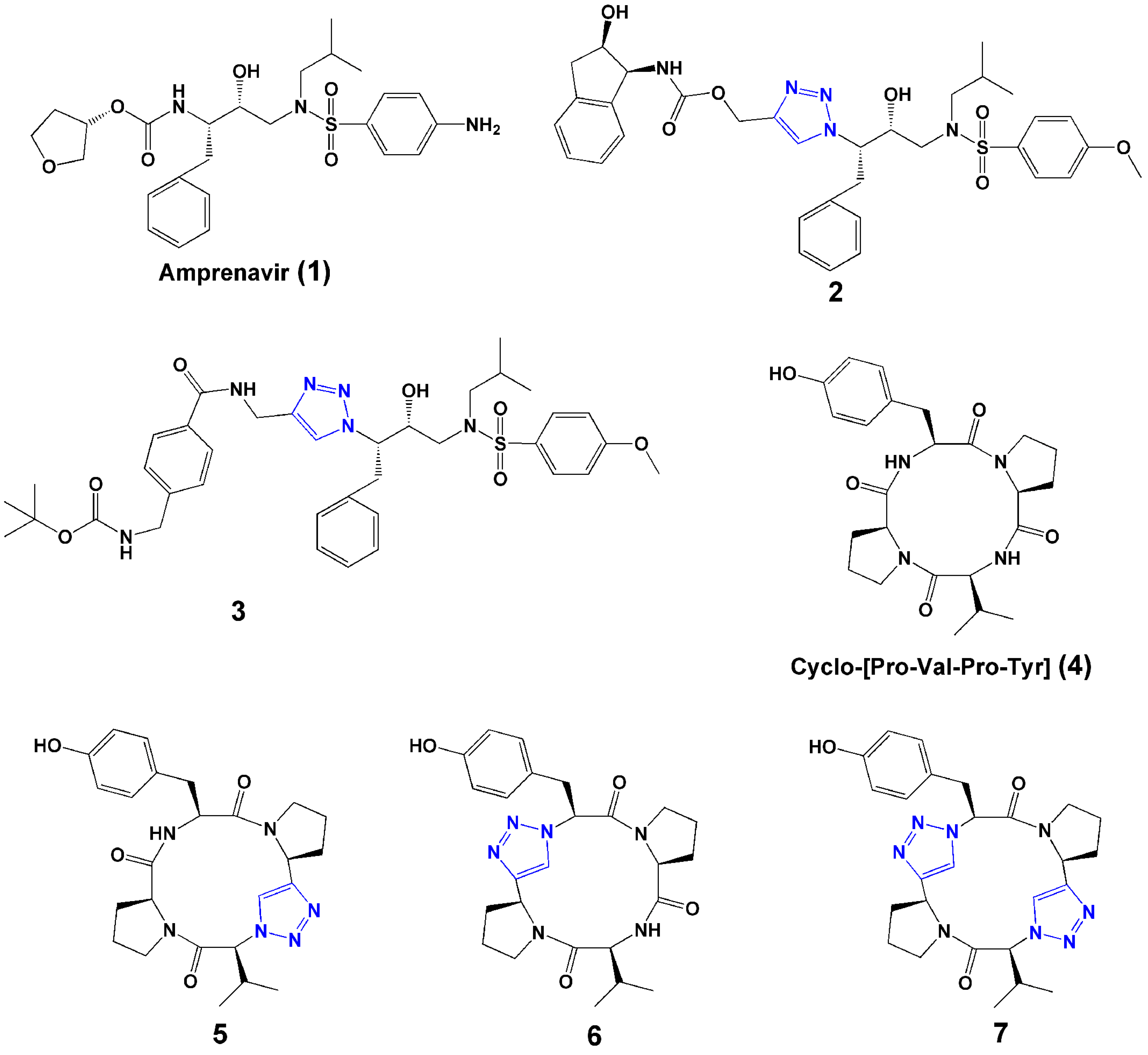
2.1. Bioisostere of Amide Bond
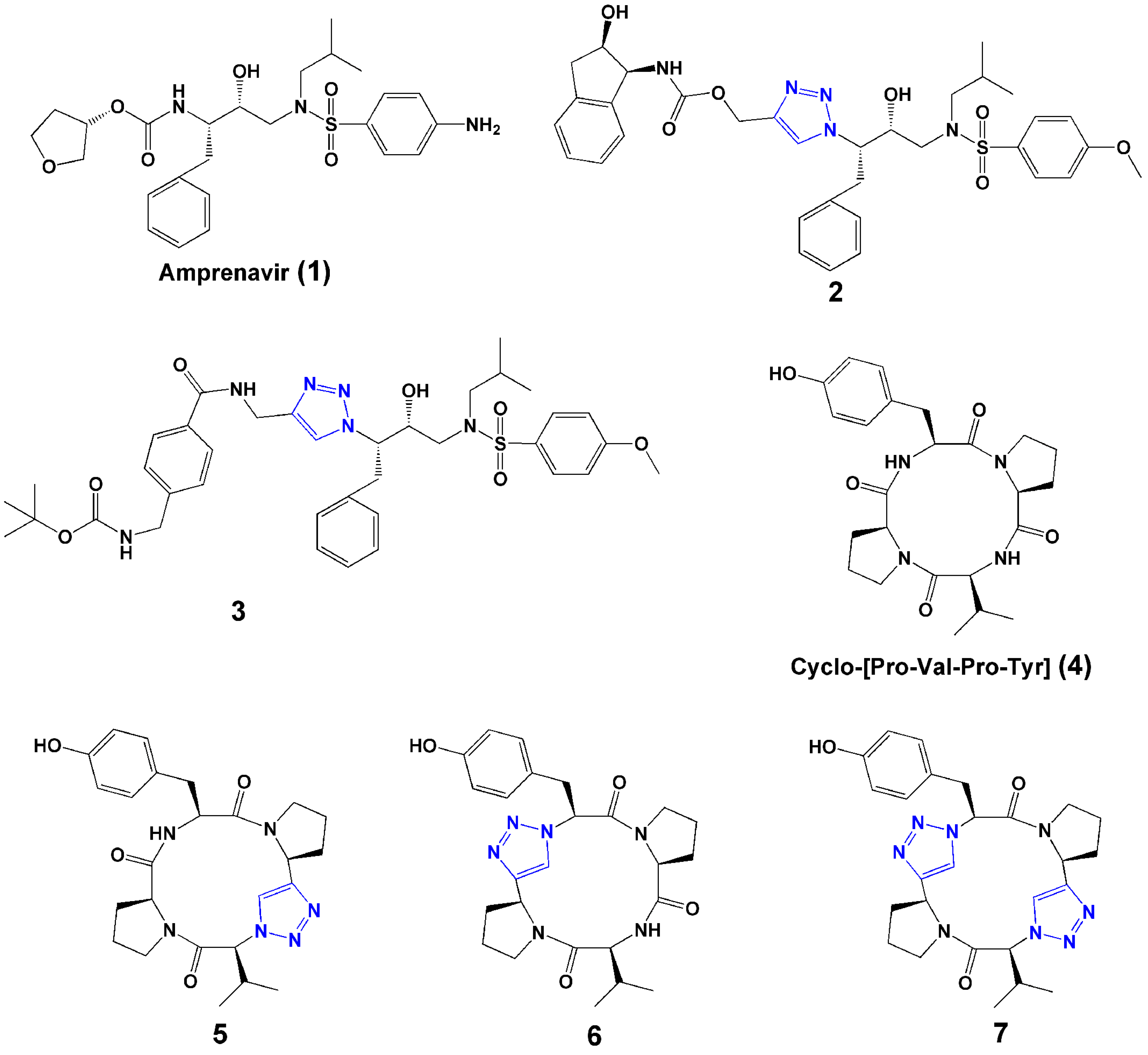
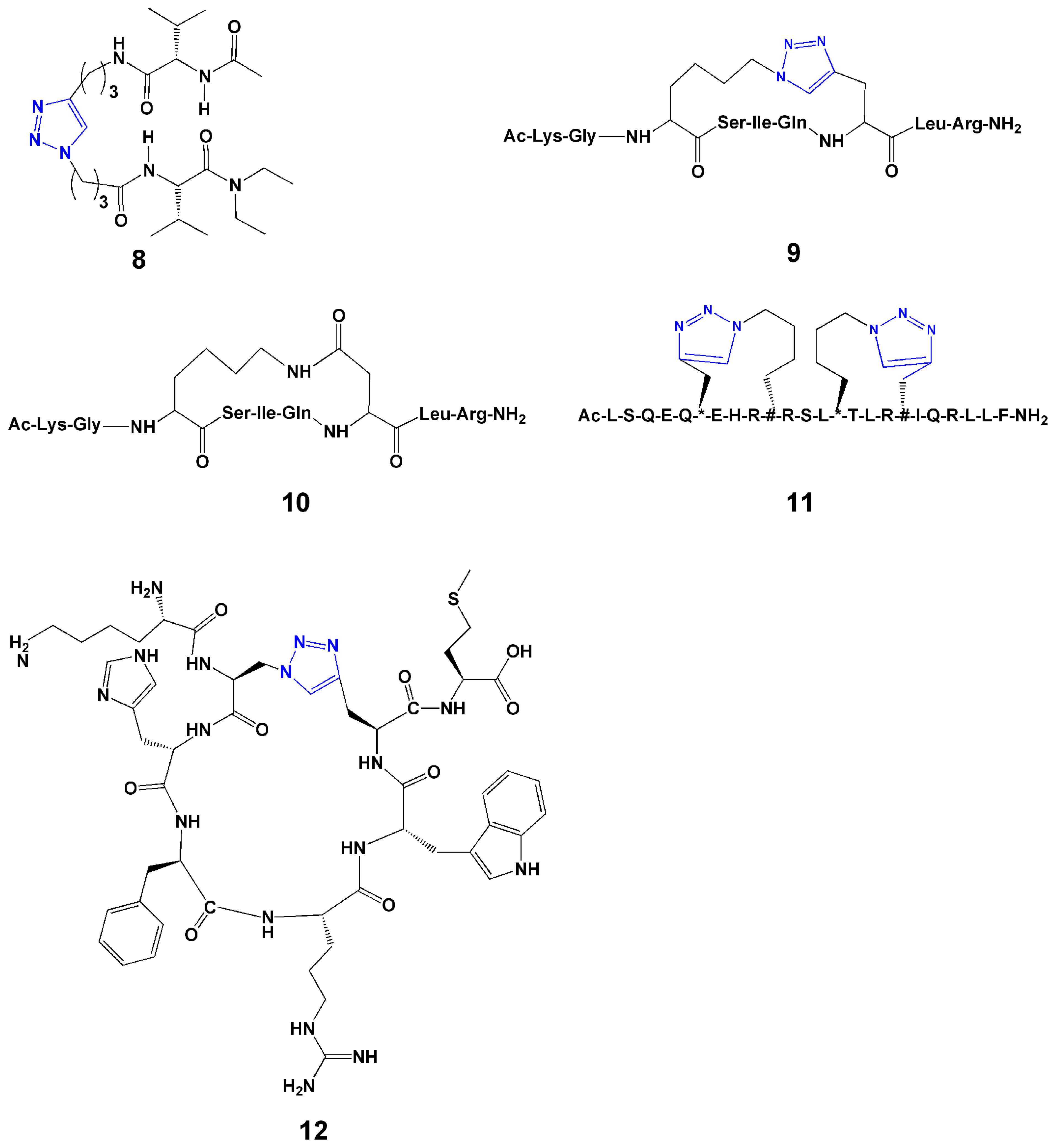
2.2. Replacement of Disulfide Bond
2.3. Substitution of Aromatic Rings and Double Bonds
3. Crosslinking and Cyclization to Stabilize Functional Structures
3.1. β-turn Surrogate
3.2. Cis/Trans Conformation
3.3. Stabilizing α-Helical Structure
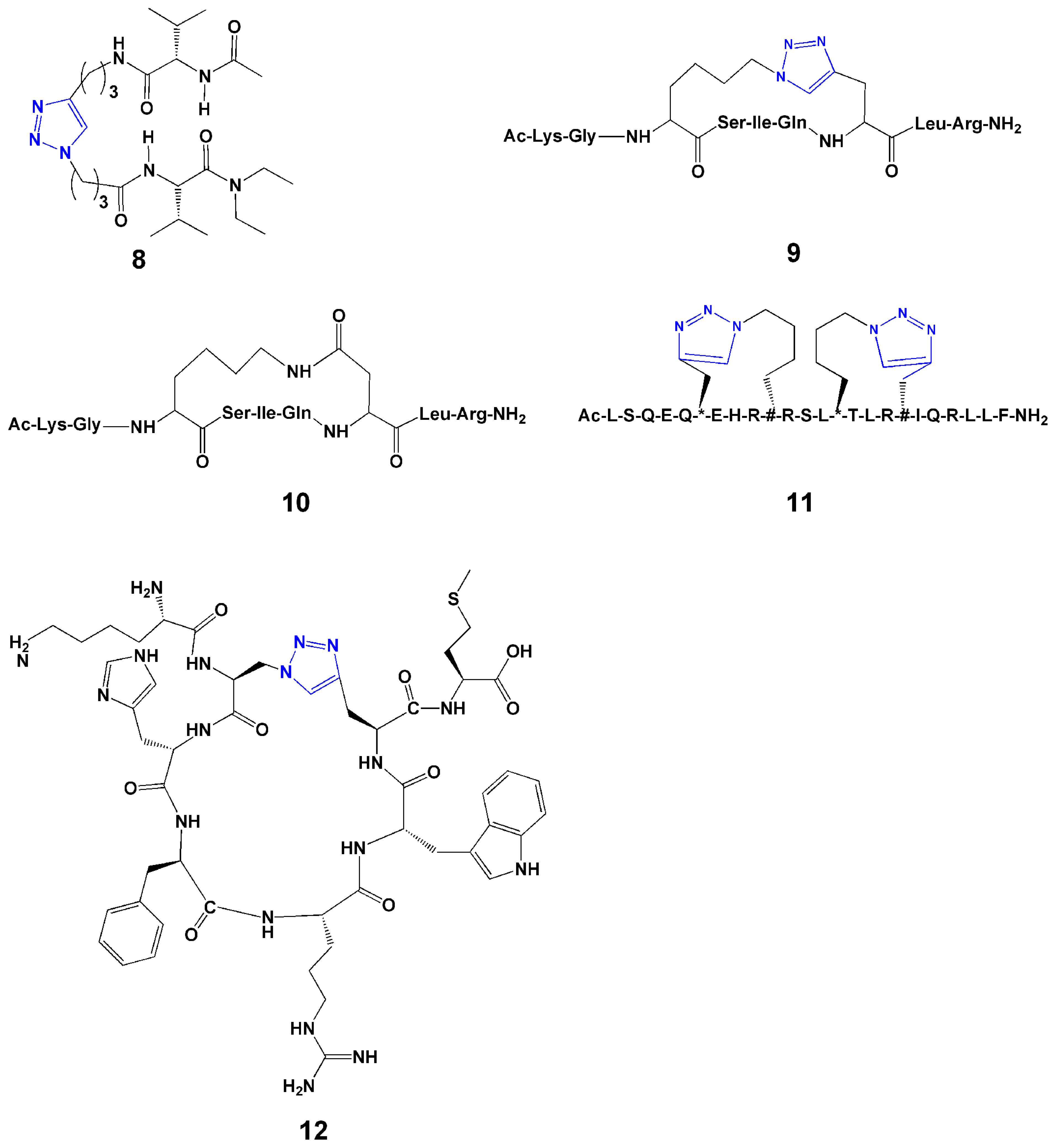
3.4. Peptide Cyclization
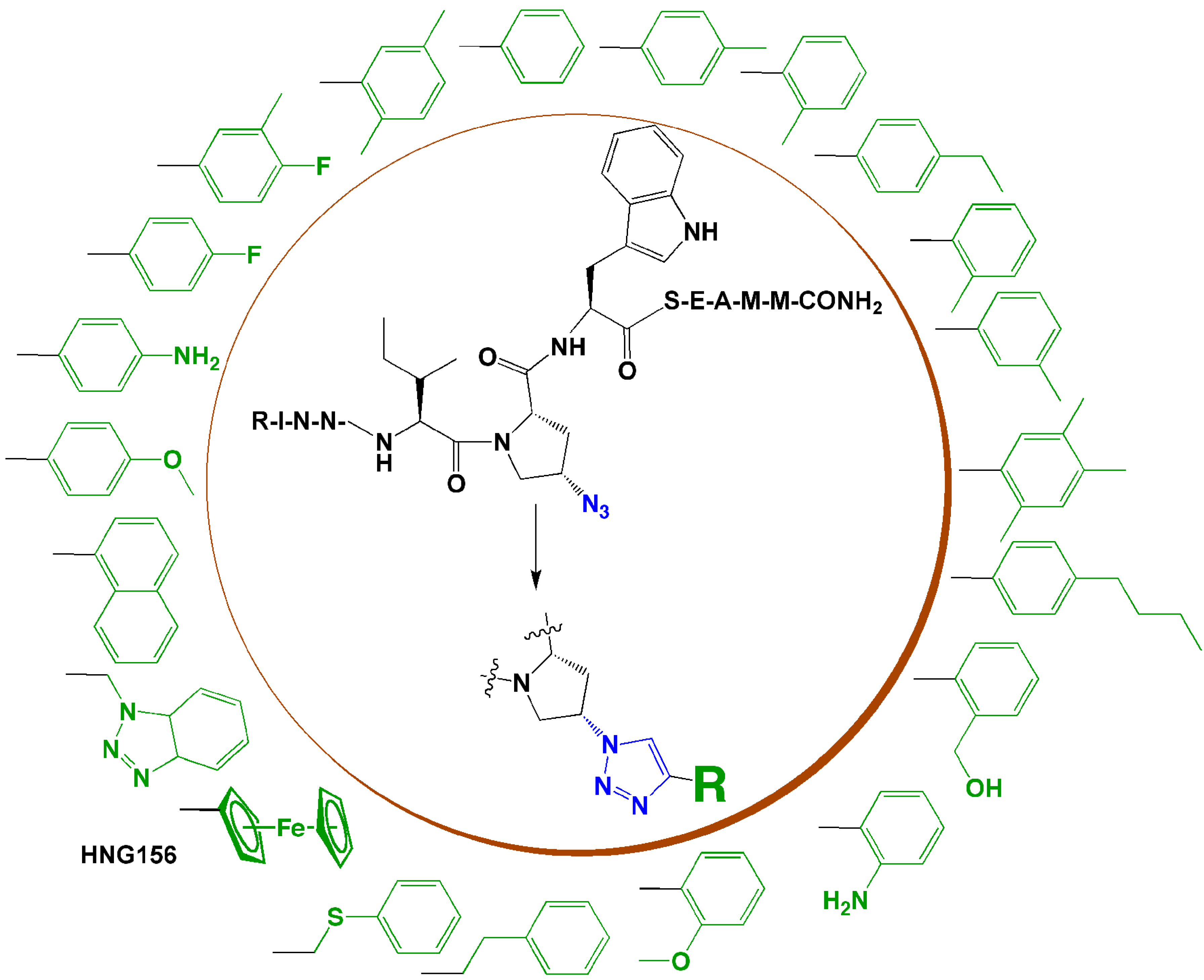
4. Introduction of Contact Groups to Enhance Affinity
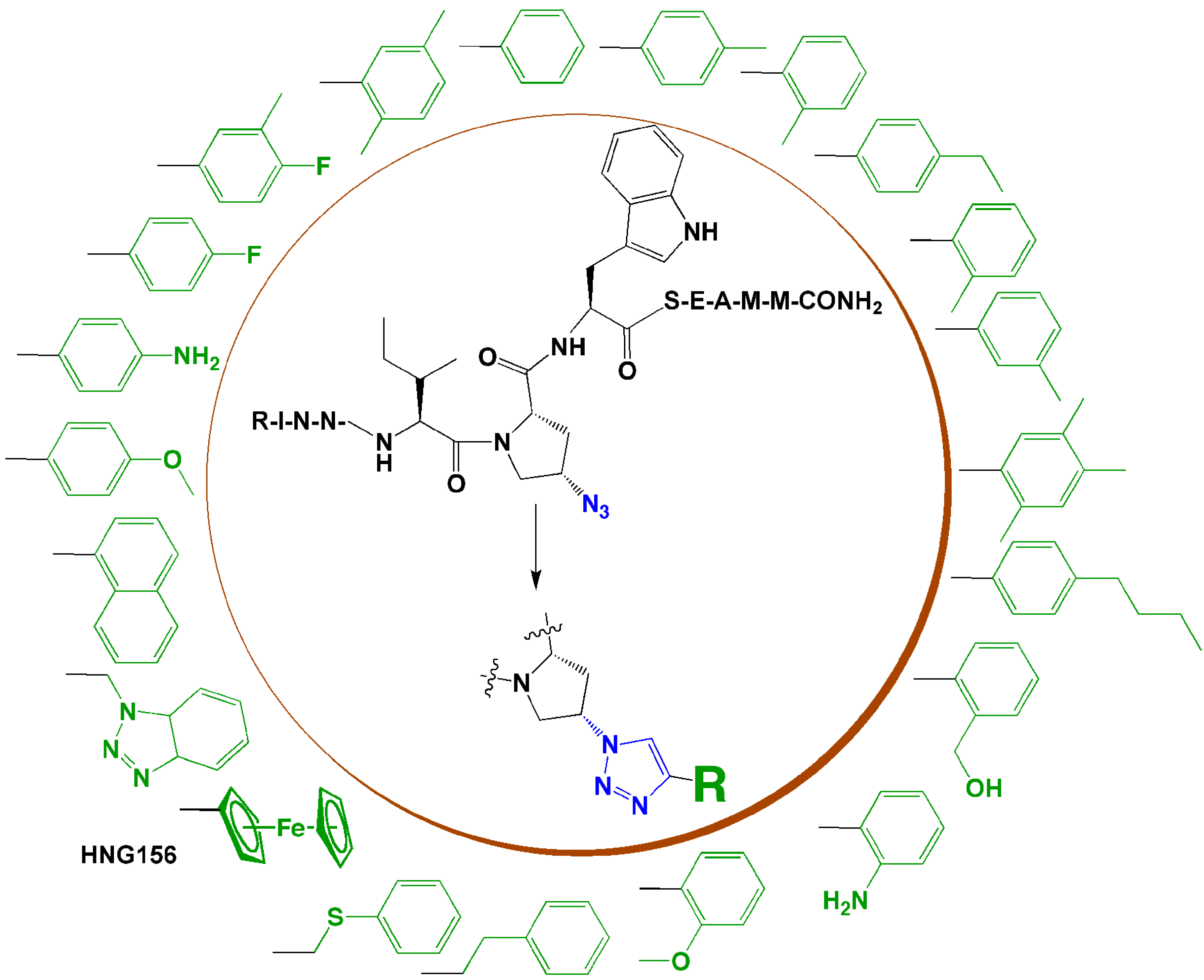
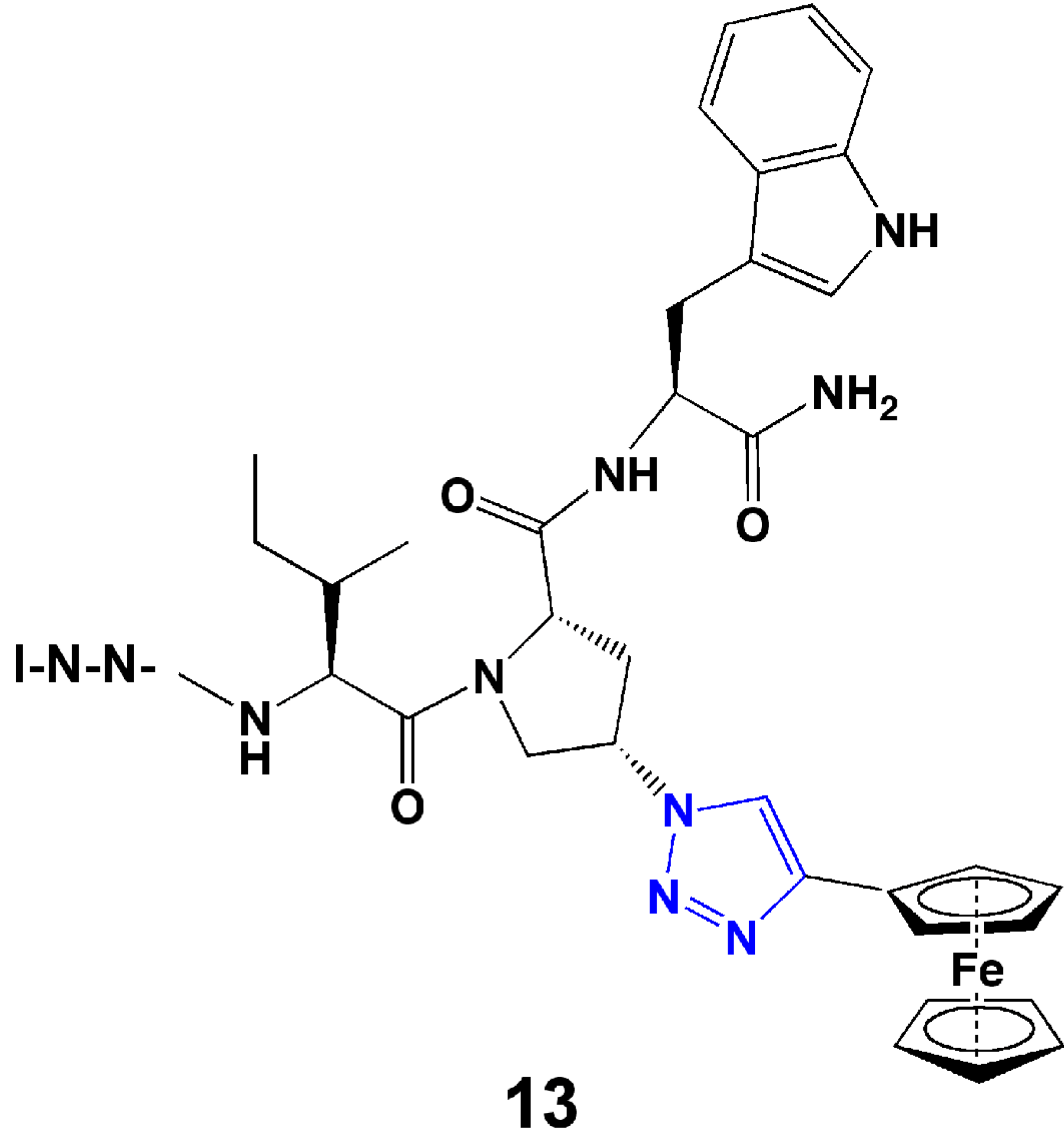
5. Bioconjugation to Combine Units with Different Functions
5.1. Peptide-Carbohydrate Conjugates
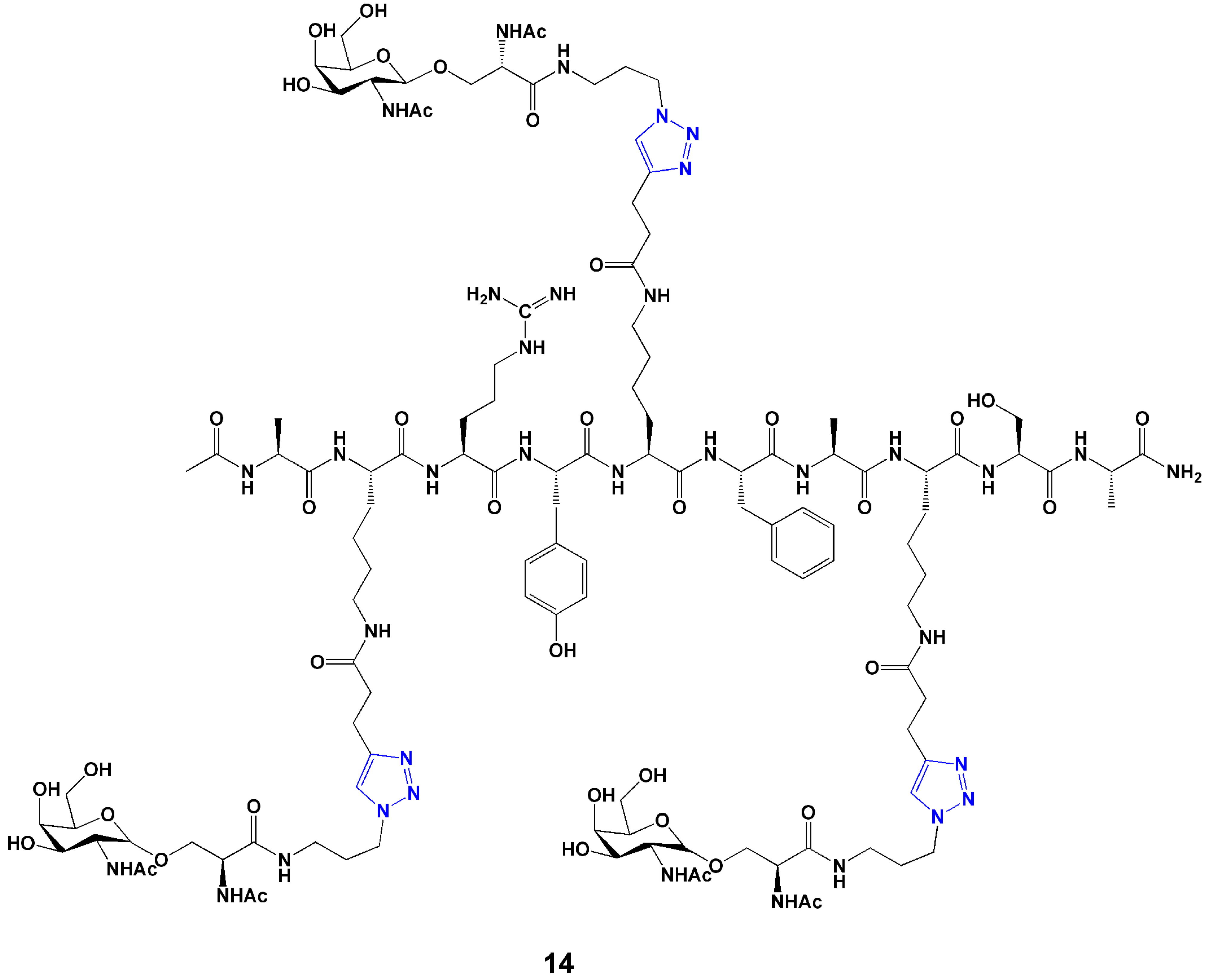
5.2. Conjugation for Drug Delivery
5.3. Radio- and Fluorophore-Labeling
5.4. Immobilization of Peptides for Separation and Imaging
6. Conclusions
Conflicts of Interest
References
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: science and market. Drug. Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. Engl. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Tornoe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar cycloadditions. Past and future. Angew. Chem. Int. Ed. Engl. 1963, 2, 565–632. [Google Scholar] [CrossRef]
- Huisgen, R. Kinetics and mechanism of 1,3-dipolar cycloadditions. Angew. Chem. Int. Ed. Engl. 1963, 2, 633–696. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Moorhouse, A.D.; Moses, J.E. Click chemistry and medicinal chemistry: a case of “cyclo-addiction”. ChemMedChem 2008, 3, 715–723. [Google Scholar] [CrossRef]
- Moses, J.E.; Moorhouse, A.D. The growing applications of click chemistry. Chem. Soc. Rev. 2007, 36, 1249–1262. [Google Scholar] [CrossRef]
- Lutz, J.F.; Zarafshani, Z. Efficient construction of therapeutics, bioconjugates, biomaterials and bioactive surfaces using azide-alkyne “click” chemistry. Adv. Drug Deliv. Rev. 2008, 60, 958–970. [Google Scholar] [CrossRef]
- Lallana, E.; Fernandez-Trillo, F.; Sousa-Herves, A.; Riguera, R.; Fernandez-Megia, E. Click chemistry with polymers, dendrimers, and hydrogels for drug delivery. Pharm. Res. 2012, 29, 902–921. [Google Scholar] [CrossRef]
- Boren, B.C.; Narayan, S.; Rasmussen, L.K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.; Fokin, V.V. Ruthenium-catalyzed azide-alkyne cycloaddition: scope and mechanism. J. Am. Chem. Soc. 2008, 130, 8923–8930. [Google Scholar]
- Baskin, J.M.; Prescher, J.A.; Laughlin, S.T.; Agard, N.J.; Chang, P.V.; Miller, I.A.; Lo, A.; Codelli, J.A.; Bertozzi, C.R. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797. [Google Scholar] [CrossRef]
- Campbell-Verduyn, L.; Elsinga, P.H.; Mirfeizi, L.; Dierckx, R.A.; Feringa, B.L. Copper-free 'click': 1,3-Dipolar Cycloaddition of azides and arynes. Org. Biomol. Chem. 2008, 6, 3461–3463. [Google Scholar] [CrossRef]
- Chang, P.V.; Prescher, J.A.; Sletten, E.M.; Baskin, J.M.; Miller, I.A.; Agard, N.J.; Lo, A.; Bertozzi, C.R. Copper-free click chemistry in living animals. Proc. Natl. Acad. Sci. USA 2010, 107, 1821–1826. [Google Scholar] [CrossRef]
- Jewett, J.C.; Bertozzi, C.R. Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev. 2010, 39, 1272–1279. [Google Scholar] [CrossRef]
- Tron, G.C.; Pirali, T.; Billington, R.A.; Canonico, P.L.; Sorba, G.; Genazzani, A.A. Click chemistry reactions in medicinal chemistry: applications of the 1,3-dipolar cycloaddition between azides and alkynes. Med. Res. Rev. 2008, 28, 278–308. [Google Scholar] [CrossRef]
- Hein, C.D.; Liu, X.M.; Wang, D. Click chemistry, a powerful tool for pharmaceutical sciences. Pharm. Res. 2008, 25, 2216–2230. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Manetsch, R. In situ click chemistry: A powerful means for lead discovery. Expert Opin. Drug Discov. 2006, 1, 525–538. [Google Scholar] [CrossRef]
- Hou, J.; Liu, X.; Shen, J.; Zhao, G.; Wang, P.G. The impact of click chemistry in medicinal chemistry. Expert Opin. Drug Discov. 2012, 7, 489–501. [Google Scholar] [CrossRef]
- Brik, A.; Alexandratos, J.; Lin, Y.C.; Elder, J.H.; Olson, A.J.; Wlodawer, A.; Goodsell, D.S.; Wong, C.H. 1,2,3-triazole as a peptide surrogate in the rapid synthesis of HIV-1 protease inhibitors. ChemBioChem 2005, 6, 1167–1169. [Google Scholar] [CrossRef]
- Bock, V.D.; Speijer, D.; Hiemstra, H.; van Maarseveen, J.H. 1,2,3-Triazoles as peptide bond isosteres: synthesis and biological evaluation of cyclotetrapeptide mimics. Org. Biomol. Chem. 2007, 5, 971–975. [Google Scholar] [CrossRef]
- Horne, W.S.; Yadav, M.K.; Stout, C.D.; Ghadiri, M.R. Heterocyclic peptide backbone modifications in an alpha-helical coiled coil. J. Am. Chem. Soc. 2004, 126, 15366–15367. [Google Scholar]
- Harbury, P.B.; Zhang, T.; Kim, P.S.; Alber, T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science 1993, 262, 1401–1407. [Google Scholar]
- Horne, W.S.; Olsen, C.A.; Beierle, J.M.; Montero, A.; Ghadiri, M.R. Probing the bioactive conformation of an archetypal natural product HDAC inhibitor with conformationally homogeneous triazole-modified cyclic tetrapeptides. Angew. Chem. Int. Ed. Engl. 2009, 48, 4718–4724. [Google Scholar] [CrossRef]
- Empting, M.; Avrutina, O.; Meusinger, R.; Fabritz, S.; Reinwarth, M.; Biesalski, M.; Voigt, S.; Buntkowsky, G.; Kolmar, H. “Triazole bridge”: disulfide-bond replacement by ruthenium-catalyzed formation of 1,5-disubstituted 1,2,3-triazoles. Angew. Chem. Int. Ed. Engl. 2011, 50, 5207–5211. [Google Scholar] [CrossRef]
- Holland-Nell, K.; Meldal, M. Maintaining biological activity by using triazoles as disulfide bond mimetics. Angew. Chem. Int. Ed. Engl. 2011, 50, 5204–5206. [Google Scholar] [CrossRef]
- Oh, K.; Guan, Z. A convergent synthesis of new beta-turn mimics by click chemistry. Chem. Commun. (Camb) 2006, 29, 3069–3071. [Google Scholar]
- Beierle, J.M.; Horne, W.S.; van Maarseveen, J.H.; Waser, B.; Reubi, J.C.; Ghadiri, M.R. Conformationally homogeneous heterocyclic pseudotetrapeptides as three-dimensional scaffolds for rational drug design: receptor-selective somatostatin analogues. Angew. Chem. Int. Ed. Engl. 2009, 48, 4725–4729. [Google Scholar] [CrossRef]
- Paul, A.; Bittermann, H.; Gmeiner, P. Triazolopeptides: chirospecific synthesis and cis/trans prolyl ratios of structural isomers. Tetrahedron 2006, 62, 8919–8927. [Google Scholar] [CrossRef]
- Cantel, S.; Isaad Ale, C.; Scrima, M.; Levy, J.J.; DiMarchi, R.D.; Rovero, P.; Halperin, J.A.; D'Ursi, A.M.; Papini, A.M.; Chorev, M. Synthesis and conformational analysis of a cyclic peptide obtained via i to i+4 intramolecular side-chain to side-chain azide-alkyne 1,3-dipolar cycloaddition. J. Org. Chem. 2008, 73, 5663–5674. [Google Scholar] [CrossRef]
- Scrima, M.; Chevalier-Isaad, A.L.; Rovero, P.; Papini, A.M.; Chorev, M.; D'Ursi, A.M. Cu(I)-catalyzed azide-alkyne intramolecular i-to-(i+4) side-chain-to-side-chain cyclization promotes the formation of helix-like secondary structures. Eur. J. Org. Chem. 2010, 3, 446–457. [Google Scholar]
- Jacobsen, O.; Maekawa, H.; Ge, N.H.; Gorbitz, C.H.; Rongved, P.; Ottersen, O.P.; Amiry-Moghaddam, M.; Klaveness, J. Stapling of a 3(10)-helix with click chemistry. J. Org. Chem. 2011, 76, 1228–1238. [Google Scholar]
- Kawamoto, S.A.; Coleska, A.; Ran, X.; Yi, H.; Yang, C.Y.; Wang, S. Design of triazole-stapled BCL9 alpha-helical peptides to target the beta-catenin/B-cell CLL/lymphoma 9 (BCL9) protein-protein interaction. J. Med. Chem. 2012, 55, 1137–1146. [Google Scholar] [CrossRef]
- Roice, M.; Johannsen, I.; Meldal, M. High capacity poly(ethylene glycol) based amino polymers for peptide and organic synthesis. Qsar Comb. Sci. 2004, 23, 662–673. [Google Scholar] [CrossRef]
- Bock, V.D.; Perciaccante, R.; Jansen, T.P.; Hiemstra, H.; van Maarseveen, J.H. Click chemistry as a route to cyclic tetrapeptide analogues: synthesis of cyclo-[Pro-Val-psi(triazole)-Pro-Tyr]. Org. Lett. 2006, 8, 919–922. [Google Scholar] [CrossRef]
- Jagasia, R.; Holub, J.M.; Bollinger, M.; Kirshenbaum, K.; Finn, M.G. Peptide cyclization and cyclodimerization by Cu(I)-mediated azide-alkyne cycloaddition. J. Org. Chem. 2009, 74, 2964–2974. [Google Scholar] [CrossRef]
- Punna, S.; Kuzelka, J.; Wang, Q.; Finn, M.G. Head-to-tail peptide cyclodimerization by copper-catalyzed azide-alkyne cycloaddition. Angew. Chem. Int. Ed. Engl. 2005, 44, 2215–2220. [Google Scholar] [CrossRef]
- Ingale, S.; Dawson, P.E. On resin side-chain cyclization of complex peptides using CuAAC. Org. Lett. 2011, 13, 2822–2825. [Google Scholar] [CrossRef]
- Angell, Y.; Burgess, K. Ring closure to beta-turn mimics via copper-catalyzed azide/alkyne cycloadditions. J. Org. Chem. 2005, 70, 9595–9598. [Google Scholar] [CrossRef]
- van Maarseveen, J.H.; Horne, W.S.; Ghadiri, M.R. Efficient route to C2 symmetric heterocyclic backbone modified cyclic peptides. Org. Lett. 2005, 7, 4503–4506. [Google Scholar] [CrossRef]
- Li, X. Click to join peptides/proteins together. Asian J. Chem. 2011, 6, 2606–2616. [Google Scholar] [CrossRef]
- Ferrer, M.; Harrison, S.C. Peptide ligands to human immunodeficiency virus type 1 gp120 identified from phage display libraries. J. Virol. 1999, 73, 5795–802. [Google Scholar]
- Gopi, H.N.; Tirupula, K.C.; Baxter, S.; Ajith, S.; Chaiken, I.M. Click chemistry on azidoproline: High-affinity dual antagonist for HIV-1 envelope glycoprotein gp120. ChemMedChem 2006, 1, 54–57. [Google Scholar] [CrossRef]
- Gopi, H.; Umashankara, M.; Pirrone, V.; LaLonde, J.; Madani, N.; Tuzer, F.; Baxter, S.; Zentner, I.; Cocklin, S.; Jawanda, N.; et al. Structural determinants for affinity enhancement of a dual antagonist peptide entry inhibitor of human immunodeficiency virus type-1. J. Med. Chem. 2008, 51, 2638–2647. [Google Scholar] [CrossRef]
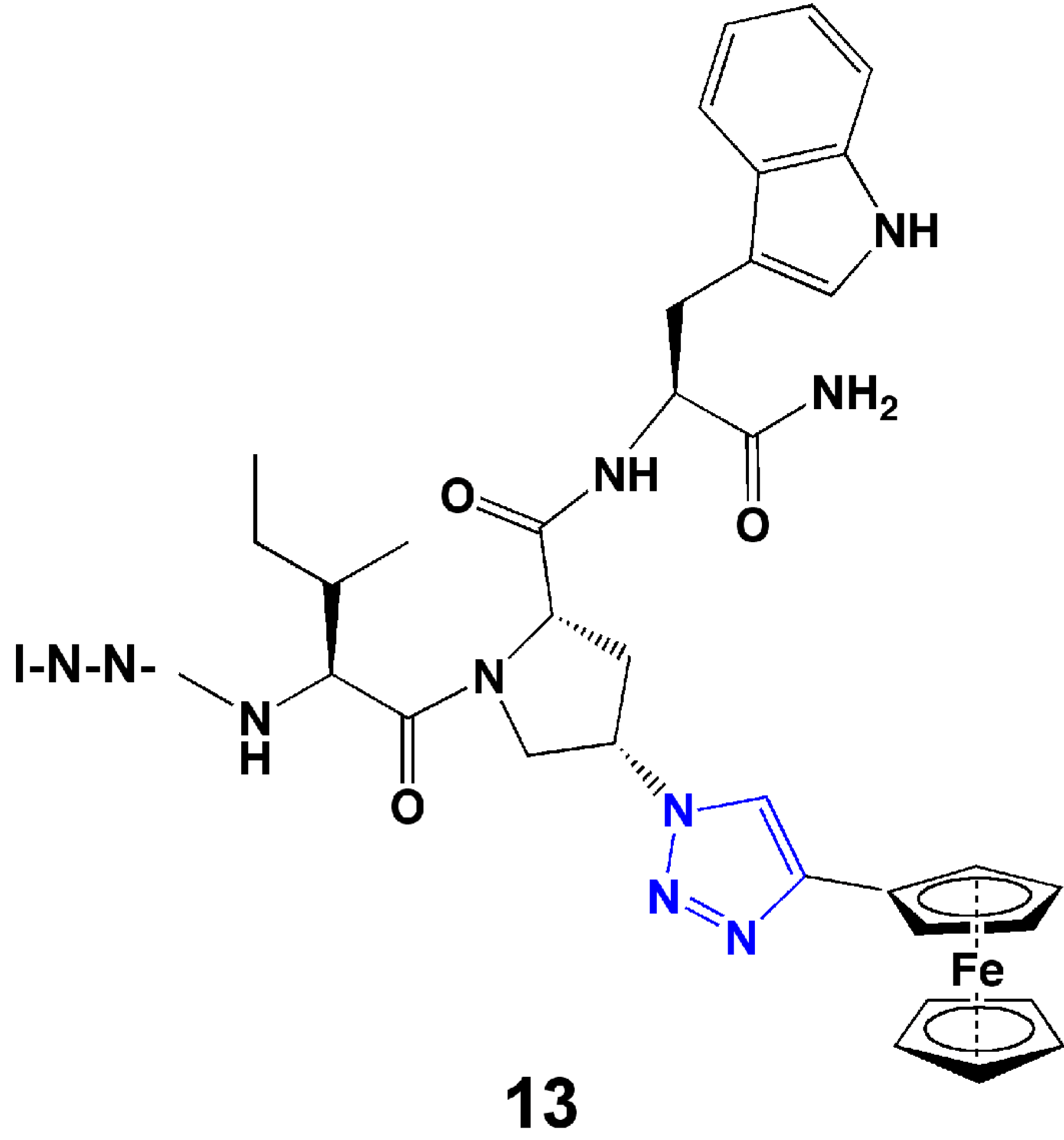
- Gopi, H.; Cocklin, S.; Pirrone, V.; McFadden, K.; Tuzer, F.; Zentner, I.; Ajith, S.; Baxter, S.; Jawanda, N.; Krebs, F.C.; et al. Introducing metallocene into a triazole peptide conjugate reduces its off-rate and enhances its affinity and antiviral potency for HIV-1 gp120. J. Mol. Recognit. 2009, 22, 169–174. [Google Scholar] [CrossRef]
- Umashankara, M.; McFadden, K.; Zentner, I.; Schon, A.; Rajagopal, S.; Tuzer, F.; Kuriakose, S.A.; Contarino, M.; Lalonde, J.; Freire, E.; et al. The active core in a triazole peptide dual-site antagonist of HIV-1 gp120. ChemMedChem 2010, 5, 1871–1879. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, L.; Luo, T.; Zhou, J.; Sun, L.; Xu, Y. Synthesis and evaluation of a dipeptide-drug conjugate library as substrates for PEPT1. ACS Comb. Sci. 2012, 14, 108–114. [Google Scholar] [CrossRef]
- Congreve, M.; Chessari, G.; Tisi, D.; Woodhead, A.J. Recent developments in fragment-based drug discovery. J. Med. Chem. 2008, 51, 3661–3680. [Google Scholar] [CrossRef]
- de Kloe, G.E.; Bailey, D.; Leurs, R.; de Esch, I.J. Transforming fragments into candidates: small becomes big in medicinal chemistry. Drug Discov. Today 2009, 14, 630–646. [Google Scholar] [CrossRef]
- Irie, T.; Fujii, I.; Sawa, M. Design and combinatorial synthesis of a novel kinase-focused library using click chemistry-based fragment assembly. Bioorg. Med. Chem. Lett. 2012, 22, 591–596. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, X.; Xue, P.; Sun, H.H.; Williams, I.D.; Sharpless, K.B.; Fokin, V.V.; Jia, G. Ruthenium-catalyzed cycloaddition of alkynes and organic azides. J. Am. Chem. Soc. 2005, 127, 15998–15999. [Google Scholar]
- Fittler, H.; Avrutina, O.; Glotzbach, B.; Empting, M.; Kolmar, H. Combinatorial tuning of peptidic drug candidates: High-affinity matriptase inhibitors through incremental structure-guided optimization. Org. Biomol. Chem. 2013, 11, 1848–1857. [Google Scholar] [CrossRef]
- Wan, Q.; Chen, J.; Chen, G.; Danishefsky, S.J. A potentially valuable advance in the synthesis of carbohydrate-based anticancer vaccines through extended cycloaddition chemistry. J. Org. Chem. 2006, 71, 8244–8249. [Google Scholar]
- Varki, A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology 1993, 3, 97–130. [Google Scholar] [CrossRef]
- Dwek, R.A. Glycobiology: Toward Understanding the Function of Sugars. Chem. Rev. 1996, 96, 683–720. [Google Scholar] [CrossRef]
- Dondoni, A.; Giovannini, P.P.; Massi, A. Assembling heterocycle-tethered C-glycosyl and alpha-amino acid residues via 1,3-dipolar cycloaddition reactions. Org. Lett. 2004, 6, 2929–2932. [Google Scholar] [CrossRef]
- Kuijpers, B.H.; Groothuys, S.; Keereweer, A.B.; Quaedflieg, P.J.; Blaauw, R.H.; van Delft, F.L.; Rutjes, F.P. Expedient synthesis of triazole-linked glycosyl amino acids and peptides. Org. Lett. 2004, 6, 3123–3126. [Google Scholar] [CrossRef]
- Palomo, C.; Aizpurua, J.M.; Balentova, E.; Azcune, I.; Santos, J.I.; Jimenez-Barbero, J.; Canada, J.; Miranda, J.I. “Click” saccharide/beta-lactam hybrids for lectin inhibition. Org. Lett. 2008, 10, 2227–2230. [Google Scholar] [CrossRef]
- Chakrabarty, S.P.; Ramapanicker, R.; Mishra, R.; Chandrasekaran, S.; Balaram, H. Development and characterization of lysine based tripeptide analogues as inhibitors of Sir2 activity. Bioorg. Med. Chem. 2009, 17, 8060–8072. [Google Scholar] [CrossRef]
- Miller, N.; Williams, G.M.; Brimble, M.A. Synthesis of fish antifreeze neoglycopeptides using microwave-assisted “click chemistry”. Org. Lett. 2009, 11, 2409–2412. [Google Scholar] [CrossRef]
- Yang, J.W.; He, X.P.; Li, C.; Gao, L.X.; Sheng, L.; Xie, J.; Shi, X.X.; Tang, Y.; Li, J.; Chen, G.R. A unique and rapid approach toward the efficient development of novel protein tyrosine phosphatase (PTP) inhibitors based on 'clicked' pseudo-glycopeptides. Bioorg. Med. Chem. Lett. 2011, 21, 1092–1096. [Google Scholar]
- Zhong, J.; Chau, Y. Synthesis, Characterization, and Thermodynamic study of a polyvalent lytic peptide-polymer conjugate as novel anticancer agent. Bioconjugate Chem. 2010, 21, 2055–2064. [Google Scholar] [CrossRef]
- Maeda, H. Tumor-selective delivery of macromolecular drugs via the EPR effect: Background and future prospects. Bioconjugate Chem. 2010, 21, 797–802. [Google Scholar] [CrossRef]
- Dutot, L.; Lecorche, P.; Burlina, F.; Marquant, R.; Point, V.; Sagan, S.; Chassaing, G.; Mallet, J.M.; Lavielle, S. Glycosylated cell-penetrating peptides and their conjugates to a proapoptotic peptide: preparation by click chemistry and cell viability studies. J. Chem. Biol. 2009, 3, 51–65. [Google Scholar]
- Papst, S.; Noisier, A.F.; Brimble, M.A.; Yang, Y.; Krissansen, G.W. Synthesis and biological evaluation of tyrosine modified analogues of the alpha4beta7 integrin inhibitor biotin-R(8)ERY. Bioorg. Med. Chem. 2012, 20, 5139–5149. [Google Scholar] [CrossRef]
- Papst, S.; Noisier, A.; Brimble, M.A.; Yang, Y.; Krissansen, G.W. Tyrosine modified analogues of the alpha4beta7 integrin inhibitor biotin-R(8)ERY prepared via Click Chemistry: Synthesis and biological evaluation. Bioorg. Med. Chem. 2012, 20, 2638–2644. [Google Scholar] [CrossRef]
- van Dijk, M.; van Nostrum, C.F.; Hennink, W.E.; Rijkers, D.T.; Liskamp, R.M. Synthesis and characterization of enzymatically biodegradable PEG and peptide-based hydrogels prepared by click chemistry. Biomacromolecules 2010, 11, 1608–1614. [Google Scholar] [CrossRef]
- De Groot, C.J.; Cadee, J.A.; Koten, J.W.; Hennink, W.E.; Den Otter, W. Therapeutic efficacy of IL-2-loaded hydrogels in a mouse tumor model. Int. J. Cancer 2002, 98, 134–140. [Google Scholar] [CrossRef]
- Parrish, B.; Breitenkamp, R.B.; Emrick, T. PEG- and peptide-grafted aliphatic polyesters by click chemistry. J. Am. Chem. Soc. 2005, 127, 7404–7410. [Google Scholar] [CrossRef]
- Carberry, T.P.; Tarallo, R.; Falanga, A.; Finamore, E.; Galdiero, M.; Weck, M.; Galdiero, S. Dendrimer functionalization with a membrane-interacting domain of herpes simplex virus type 1: Towards intracellular delivery. Chemistry 2012, 18, 13678–13685. [Google Scholar] [CrossRef]
- Jolck, R.I.; Berg, R.H.; Andresen, T.L. Solid-phase synthesis of PEGylated lipopeptides using click chemistry. Bioconjugate Chem. 2010, 21, 807–810. [Google Scholar] [CrossRef]
- Terada, T.; Iwai, M.; Kawakami, S.; Yamashita, F.; Hashida, M. Novel PEG-matrix metalloproteinase-2 cleavable peptide-lipid containing galactosylated liposomes for hepatocellular carcinoma-selective targeting. J. Control. Release. 2006, 111, 333–342. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Ito, E.; Akita, H.; Oishi, M.; Nagasaki, Y.; Futaki, S.; Harashima, H. A pH-sensitive fusogenic peptide facilitates endosomal escape and greatly enhances the gene silencing of siRNA-containing nanoparticles in vitro and in vivo. J. Control. Release. 2009, 139, 127–132. [Google Scholar] [CrossRef]
- Meers, P. Enzyme-activated targeting of liposomes. Adv. Drug Delivery Rev. 2001, 53, 265–272. [Google Scholar] [CrossRef]
- Zhang, J.X.; Zalipsky, S.; Mullah, N.; Pechar, M.; Allen, T.M. Pharmaco attributes of dioleoylphosphatidylethanolamine/cholesterylhemisuccinate liposomes containing different types of cleavable lipopolymers. Pharmacol. Res. 2004, 49, 185–198. [Google Scholar] [CrossRef]
- Arnusch, C.J.; Pieters, R.J.; Breukink, E. Enhanced membrane pore formation through high-affinity targeted antimicrobial peptides. PLoS One 2012, 7, e39768. [Google Scholar] [CrossRef]
- Stewart, K.M.; Horton, K.L.; Kelley, S.O. Cell-penetrating peptides as delivery vehicles for biology and medicine. Org. Biomol. Chem. 2008, 6, 2242–2255. [Google Scholar] [CrossRef]
- Takeuchi, T.; Kosuge, M.; Tadokoro, A.; Sugiura, Y.; Nishi, M.; Kawata, M.; Sakai, N.; Matile, S.; Futaki, S. Direct and rapid cytosolic delivery using cell-penetrating peptides mediated by pyrenebutyrate. ACS Chem. Biol. 2006, 1, 299–303. [Google Scholar] [CrossRef]
- Loh, Y.; Shi, H.; Hu, M.; Yao, S.Q. “Click”synthesis of small molecule-peptide conjugates for organelle-specific delivery and inhibition of lysosomal cysteine proteases. Chem. Commun. (Camb) 2010, 46, 8407–8409. [Google Scholar] [CrossRef]
- Brooks, H.; Lebleu, B.; Vives, E. Tat peptide-mediated cellular delivery: back to basics. Adv. Drug Deliv. Rev. 2005, 57, 559–577. [Google Scholar] [CrossRef]
- Pouton, C.W.; Wagstaff, K.M.; Roth, D.M.; Moseley, G.W.; Jans, D.A. Targeted delivery to the nucleus. Adv. Drug Delivery Rev. 2007, 59, 698–717. [Google Scholar] [CrossRef]
- Resh, M.D. Regulation of cellular signalling by fatty acid acylation and prenylation of signal transduction proteins. Cell. Signal. 1996, 8, 403–412. [Google Scholar] [CrossRef]
- Marik, J.; Sutcliffe, J.L. Click for PET: Rapid preparation of [18F] fluoropeptides using Cu(I) catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006, 47, 6681–6684. [Google Scholar] [CrossRef]
- Glaser, M.; Arstad, E. “Click labeling” with 2-[18f]fluoroethylazide for positron emission tomography. Bioconjugate Chem. 2007, 18, 989–993. [Google Scholar] [CrossRef]
- Hausner, S.H.; Marik, J.; Gagnon, M.K.; Sutcliffe, J.L. In vivo positron emission tomography (PET) imaging with an alphavbeta6 specific peptide radiolabeled using 18F-"click" chemistry: evaluation and comparison with the corresponding 4-[18F]fluorobenzoyl- and 2-[18F]fluoropropionyl-peptides. J. Med. Chem. 2008, 51, 5901–5904. [Google Scholar] [CrossRef]
- Li, Z.B.; Wu, Z.; Chen, K.; Chin, F.T.; Chen, X. Click chemistry for (18)F-labeling of RGD peptides and microPET imaging of tumor integrin alphavbeta3 expression. Bioconjugate Chem. 2007, 18, 1987–1994. [Google Scholar] [CrossRef]
- Glaser, M.; Solbakken, M.; Turton, D.R.; Pettitt, R.; Barnett, J.; Arukwe, J.; Karlsen, H.; Cuthbertson, A.; Luthra, S.K.; Arstad, E. Methods for 18F-labeling of RGD peptides: comparison of aminooxy [18F]fluorobenzaldehyde condensation with 'click labeling' using 2-[18F]fluoroethylazide, and S-alkylation with [18F]fluoropropanethiol. Amino Acids 2009, 37, 717–724. [Google Scholar] [CrossRef]
- Maschauer, S.; Einsiedel, J.; Haubner, R.; Hocke, C.; Ocker, M.; Hubner, H.; Kuwert, T.; Gmeiner, P.; Prante, O. Labeling and glycosylation of peptides using click chemistry: a general approach to (18)F-glycopeptides as effective imaging probes for positron emission tomography. Angew. Chem. Int. Ed. Engl. 2010, 49, 976–979. [Google Scholar] [CrossRef]
- Mindt, T.L.; Struthers, H.; Brans, L.; Anguelov, T.; Schweinsberg, C.; Maes, V.; Tourwe, D.; Schibli, R. “Click to chelate”: Synthesis and installation of metal chelates into biomolecules in a single step. J. Am. Chem. Soc. 2006, 128, 15096–15097. [Google Scholar] [CrossRef]
- Struthers, H.; Spingler, B.; Mindt, T.L.; Schibli, R. “Click-to-chelate”: Design and incorporation of triazole-containing metal-chelating systems into biomolecules of diagnostic and therapeutic interest. Chemistry 2008, 14, 6173–6183. [Google Scholar] [CrossRef]
- Kim, E. M.; Joung, M.H.; Lee, C.M.; Jeong, H.J.; Lim, S.T.; Sohn, M.H.; Kim, D.W. Synthesis of Tc-99m labeled 1,2,3-triazole-4-yl c-met binding peptide as a potential c-met receptor kinase positive tumor imaging agent. Bioorg. Med. Chem. Lett. 2010, 20, 4240–4243. [Google Scholar] [CrossRef]
- Kele, P.; Mezo, G.; Achatz, D.; Wolfbeis, O.S. Dual labeling of biomolecules by using click chemistry: A sequential approach. Angew. Chem. Int. Ed. Engl. 2009, 48, 344–347. [Google Scholar] [CrossRef]
- Punna, S.; Kaltgrad, E.; Finn, M.G. “Clickable”agarose for affinity chromatography. Bioconjugate Chem. 2005, 16, 1536–1541. [Google Scholar] [CrossRef]
- Wang, Q.; Chan, T.R.; Hilgraf, R.; Fokin, V.V.; Sharpless, K.B.; Finn, M.G. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 3192–3193. [Google Scholar] [CrossRef]
- Roughley, S.D.; Jordan, A.M. The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, H.; Aneja, R.; Chaiken, I. Click Chemistry in Peptide-Based Drug Design. Molecules 2013, 18, 9797-9817. https://doi.org/10.3390/molecules18089797
Li H, Aneja R, Chaiken I. Click Chemistry in Peptide-Based Drug Design. Molecules. 2013; 18(8):9797-9817. https://doi.org/10.3390/molecules18089797
Chicago/Turabian StyleLi, Huiyuan, Rachna Aneja, and Irwin Chaiken. 2013. "Click Chemistry in Peptide-Based Drug Design" Molecules 18, no. 8: 9797-9817. https://doi.org/10.3390/molecules18089797
APA StyleLi, H., Aneja, R., & Chaiken, I. (2013). Click Chemistry in Peptide-Based Drug Design. Molecules, 18(8), 9797-9817. https://doi.org/10.3390/molecules18089797