Quantum-Mechanical Calculations on Molecular Substructures Involved in Nanosystems

Abstract
:
1. Introduction
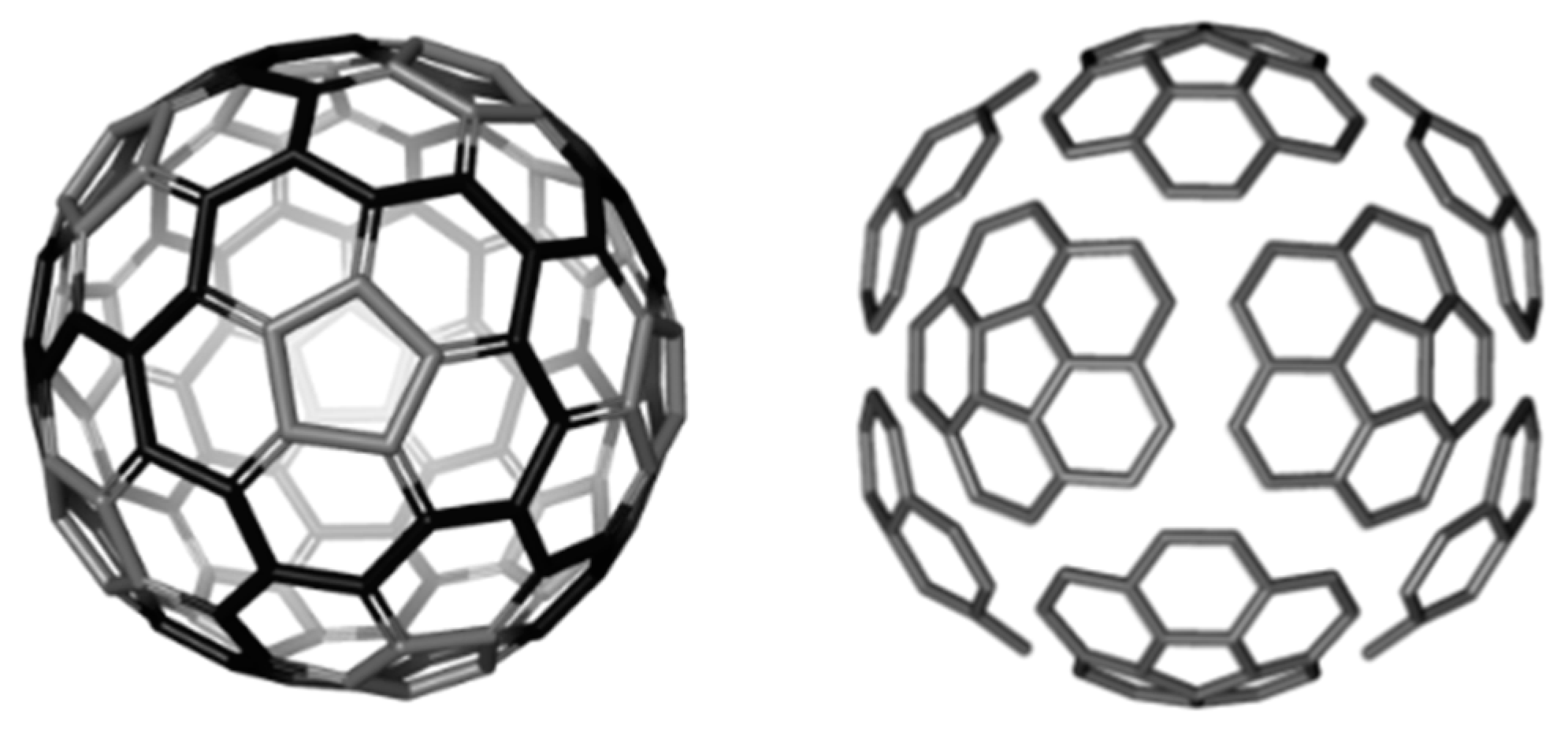
2. Circulene Patched Fullerenes

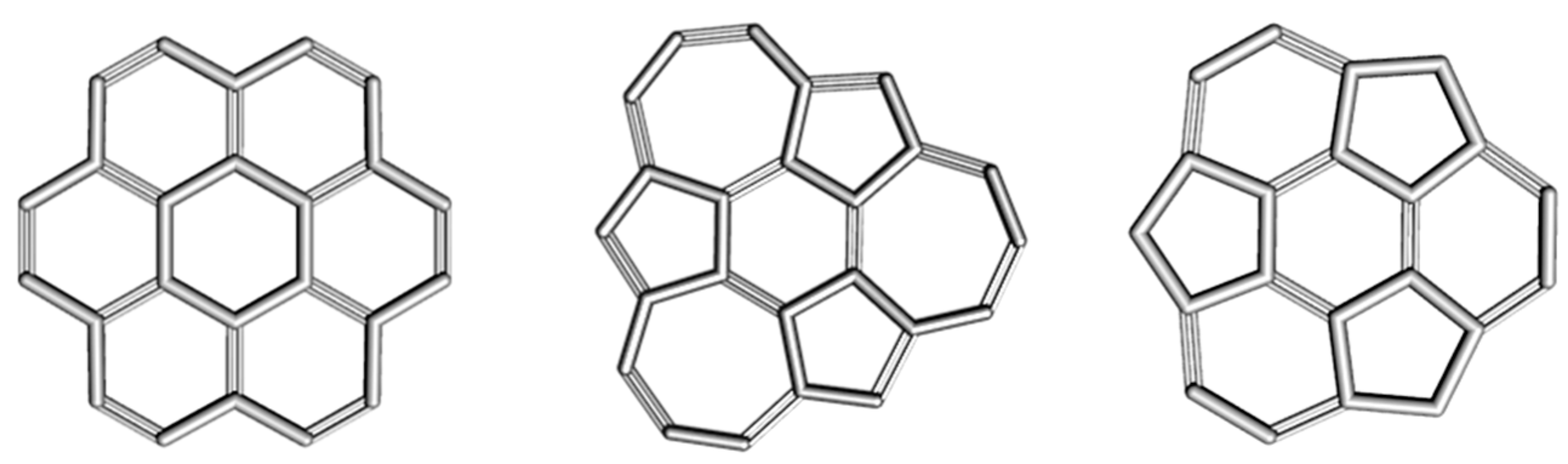
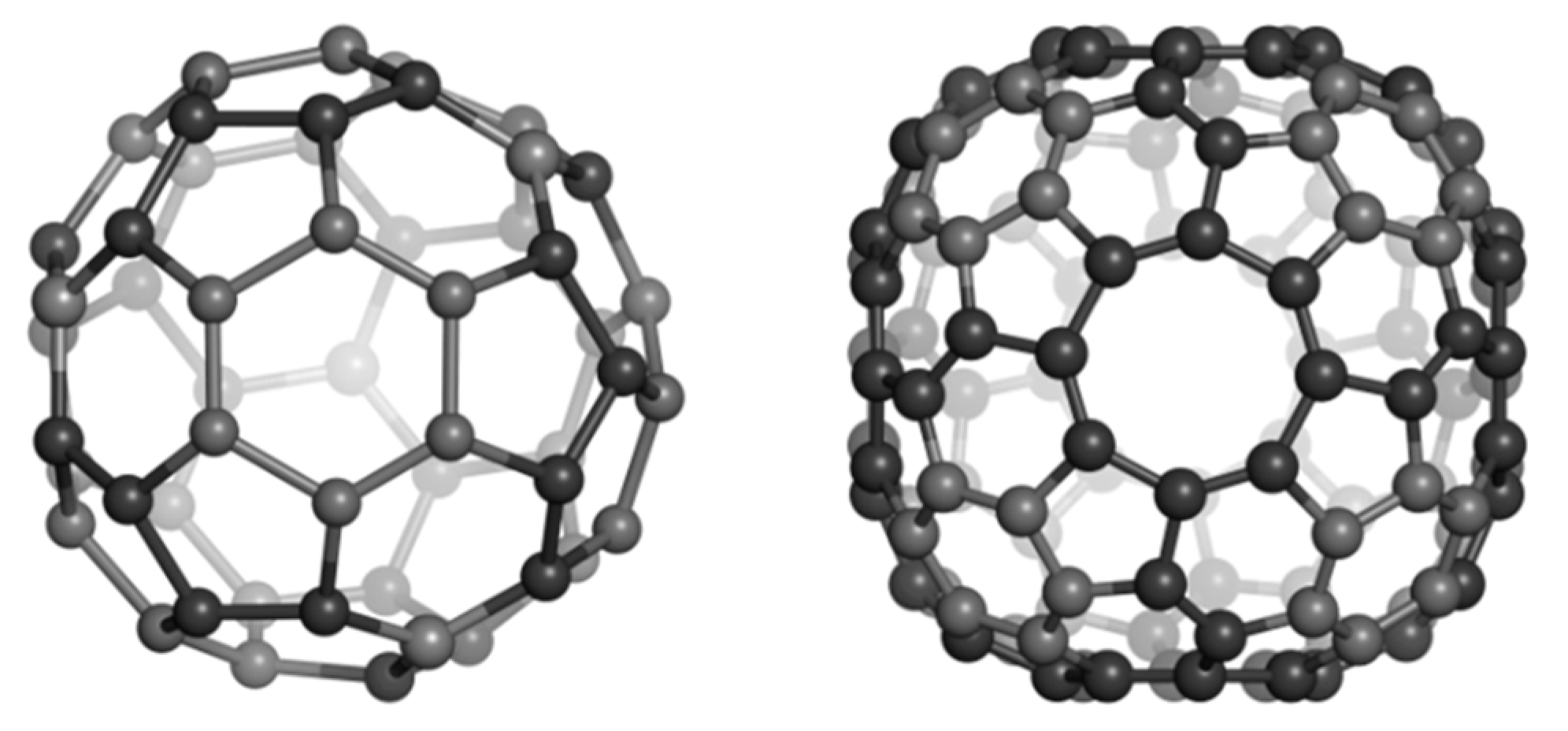
2.1. Circulenes with Hexagonal Core

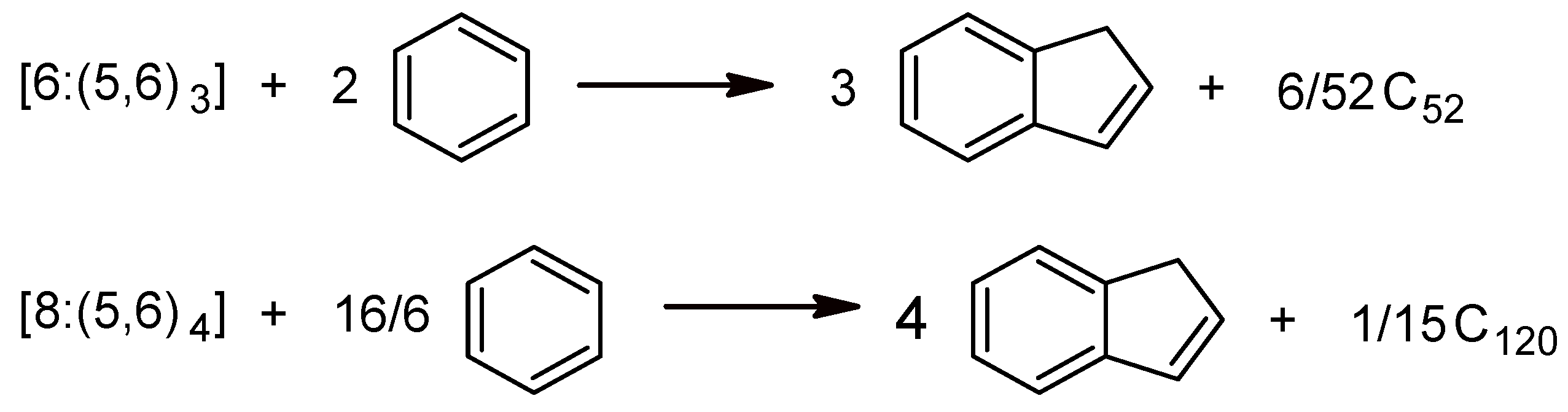
2.1.1. Global Stability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Symmetry | Etot | Etot/C | HL Gap |
|---|---|---|---|---|
| Coronene [6:66] | D6h | −915.953 | −38.164 | 8.960 |
| Isocoronene [6:(5,7)3] | Cs | −915.780 | −38.156 | 7.260 |
| Sumanene [6:(5,6)3] | C3v | −802.190 | −38.200 | 10.160 |
2.1.2. Aromaticity
| Coronene [6:66] | NICS(0) | NICS(1) | HOMA |
|---|---|---|---|
| R[6] core | −0.009 | −4.429 | 0.618 |
| R[6] petal | −10.406 | −12.453 | 0.764 |
| Isocoronene [6:(5,7)3] | NICS(0) | NICS(1) | HOMA |
|---|---|---|---|
| R[6] core | −2.908 | −5.156 | 0.867 |
| R[7] petal | 0.394 | −2.712 | 0.013 |
| R[5] petal | −3.136 | −5.637 | −0.036 |
| Sumanene [6:(5,6)3] | NICS(0) | NICS(1) | HOMA |
|---|---|---|---|
| R[6] core | −2.767 | −10.385 | 0.708 |
| R[6] petal | −10.080 | −16.892 | 0.925 |
| R[5] petal | 3.189 | −5.192 | −1.955 |
| Structure | Theory Level | Etot | Etot/C | HL Gap |
|---|---|---|---|---|
| Cor_T-84 | HF/6-31G(d,p) | −3194.384 | −33.275 | 7.347 |
| Sum_T_84 | −3155.466 | −38.028 | 7.562 | |
| C60(Ih) | −2271.830 | −37.864 | 7.418 | |
| Cor_T-84 | B3LYP/6-31G(d,p) | −3215.331 | −35.333 | 2.268 |
| Sum_T_84 | −3214.968 | −38.273 | 2.520 | |
| C60(Ih) | B3LYP/6311 + G(d,p) | −2286.610 | −38.110 | 2.724 |
| Structure | Substructure | HOMA | Strain | NICS(−1) | NICS(0) | NICS(+1) |
|---|---|---|---|---|---|---|
| Cor_T_84 | ||||||
| HF | R6,core | 0.525 | 0.387 | −5.258 | −0.789 | −5.257 |
| R6,plane | 0.908 | 0.709 | −16.429 | −11.928 | −11.342 | |
| R6,bound | 0.047 | 2.699 | −13.533 | −4.518 | −1.812 | |
| patch | 0.374 | 2.053 | ||||
| molecule | 0.348 | 1.477 | ||||
| B3LYP | R6,core | 0.529 | 0.474 | −9.184 | −1.749 | −2.808 |
| R6,plane | 0.804 | 0.765 | −16.555 | −12.123 | −11.443 | |
| R6,bound | 0.162 | 2.543 | −14.554 | −5.532 | −2.259 | |
| patch | 0.422 | 1.938 | ||||
| molecule | 0.392 | 1.435 | ||||
| Sum_T_84 | ||||||
| HF | R6,Core | 0.849 | 2.488 | −11.775 | −2.313 | −2.078 |
| R6 | 0.896 | 1.169 | −16.264 | −11.349 | −9.801 | |
| R5 | −1.685 | 2.357 | −7.314 | 1.773 | 2.203 | |
| patch | −0.548 | 1.684 | ||||
| molecule | −0.476 | 1.685 | ||||
| B3LYP | R6Core | 0.889 | 3.158 | −11.807 | −2.437 | −1.798 |
| R6 | 0.850 | 1.408 | −16.177 | −11.067 | −9.047 | |
| R5 | −1.162 | 2.568 | −7.006 | 2.779 | 2.117 | |
| patch | −0.296 | 1.832 | ||||
| molecule | −0.229 | 1.833 |

| Structure | Etot | Etot/C-atom | HL gap | Etot | Etot/C-atom | HL Gap |
|---|---|---|---|---|---|---|
| Theory level | HF | DFT | ||||
| [6:66] coronene | −915.640 | −38.151 | 8.956 | −922.071 | −38.420 | 4.026 |
| coronene_3 | −915.531 | −38.147 | 6.401 6.619 | −921.966 | −38.415 | 1.156 1.144 |
| [6:(5,7)3] isocoronene | −915.427 | −38.143 | 7.032 | −921.909 | −38.413 | 1.933 |
| isocoronene_3 | −915.416 | −38.142 | 6.598 7.151 | −921.878 | −25.608 | 1.073 1.061 |
| [6:(5,6)3] sumanene_2 | −800.033 | −38.097 | 8.378 8.347 | −805.640 | −38.364 | 1.619 2.838 |
| [6:(5,6)3] sumanene_4 | −805.588 | −38.361 | 3.520 1.408 | |||
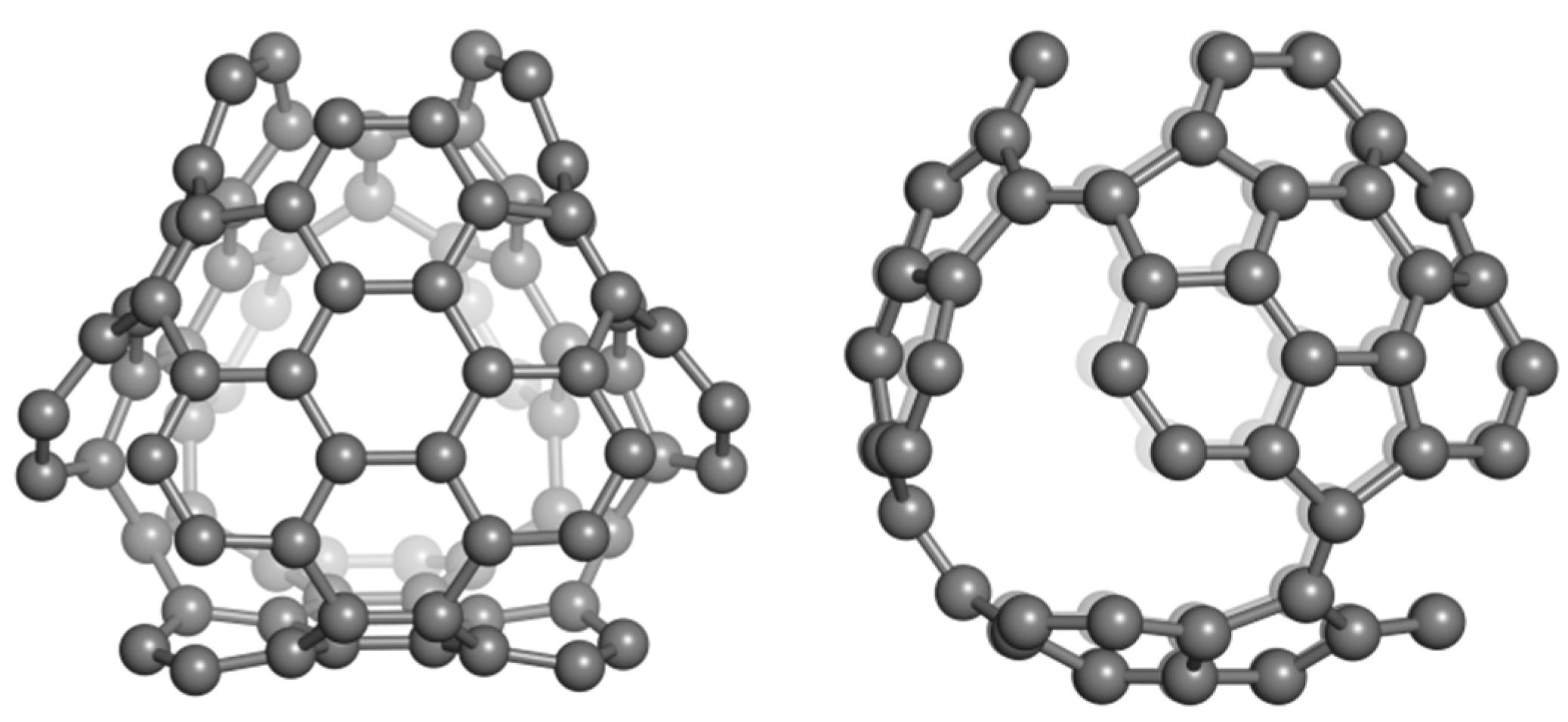
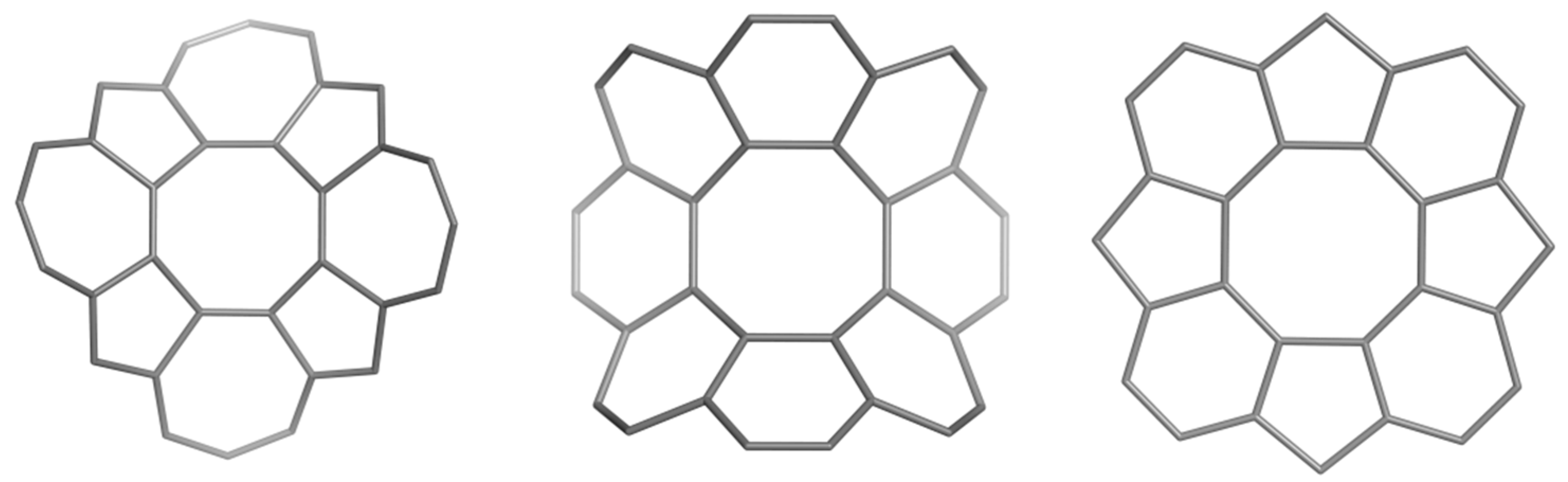
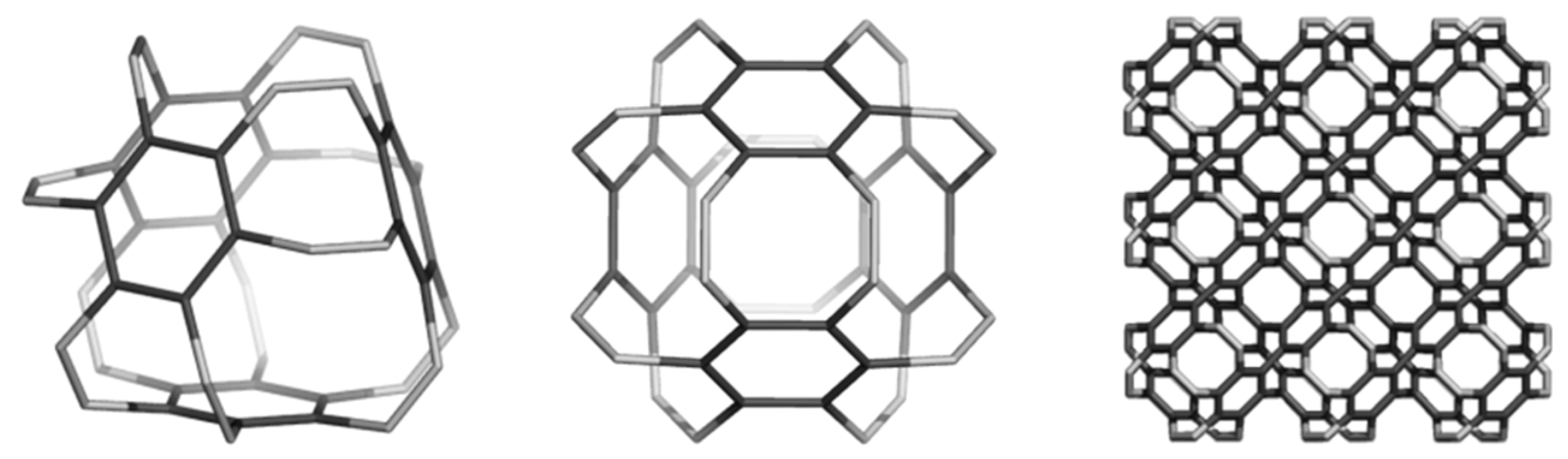
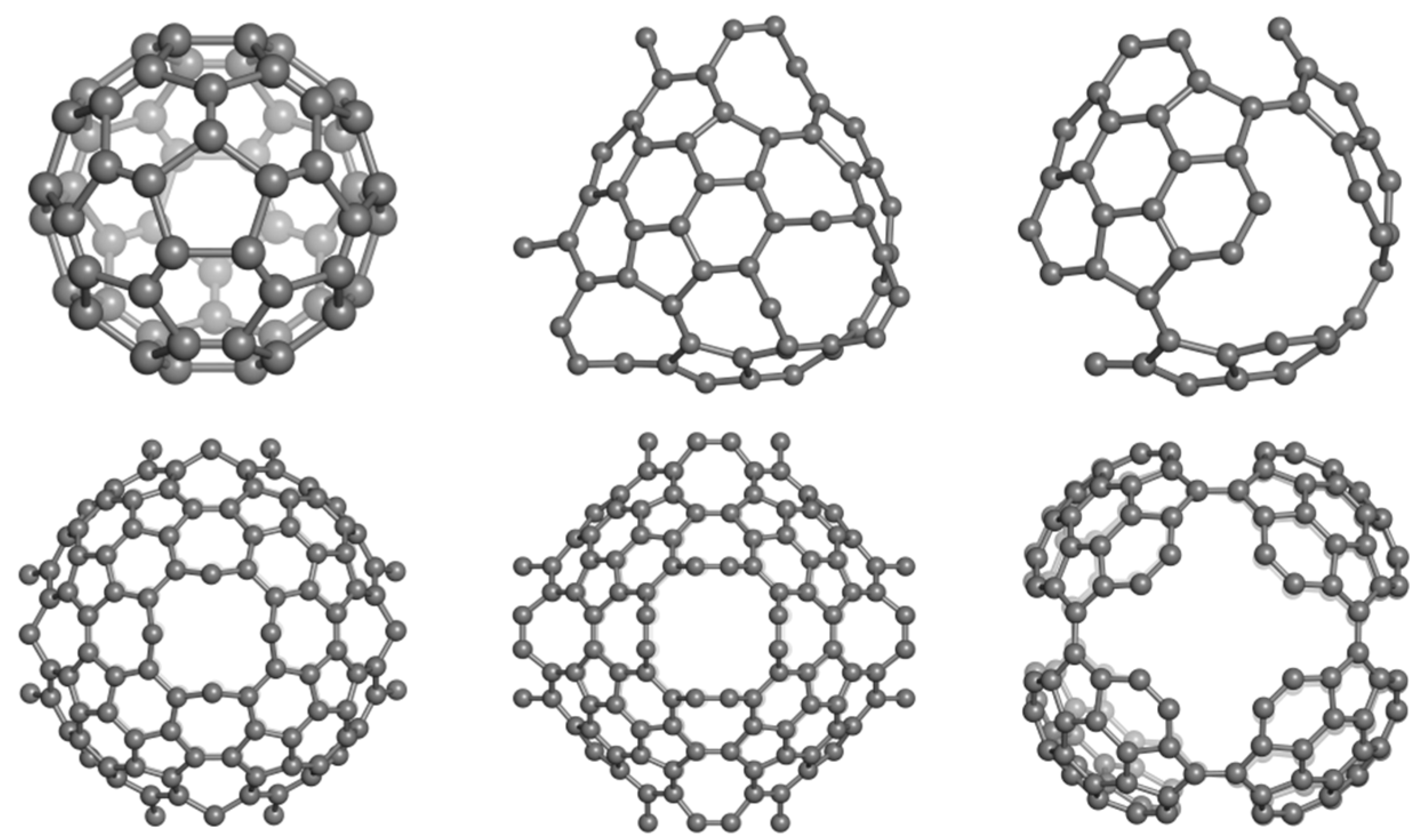
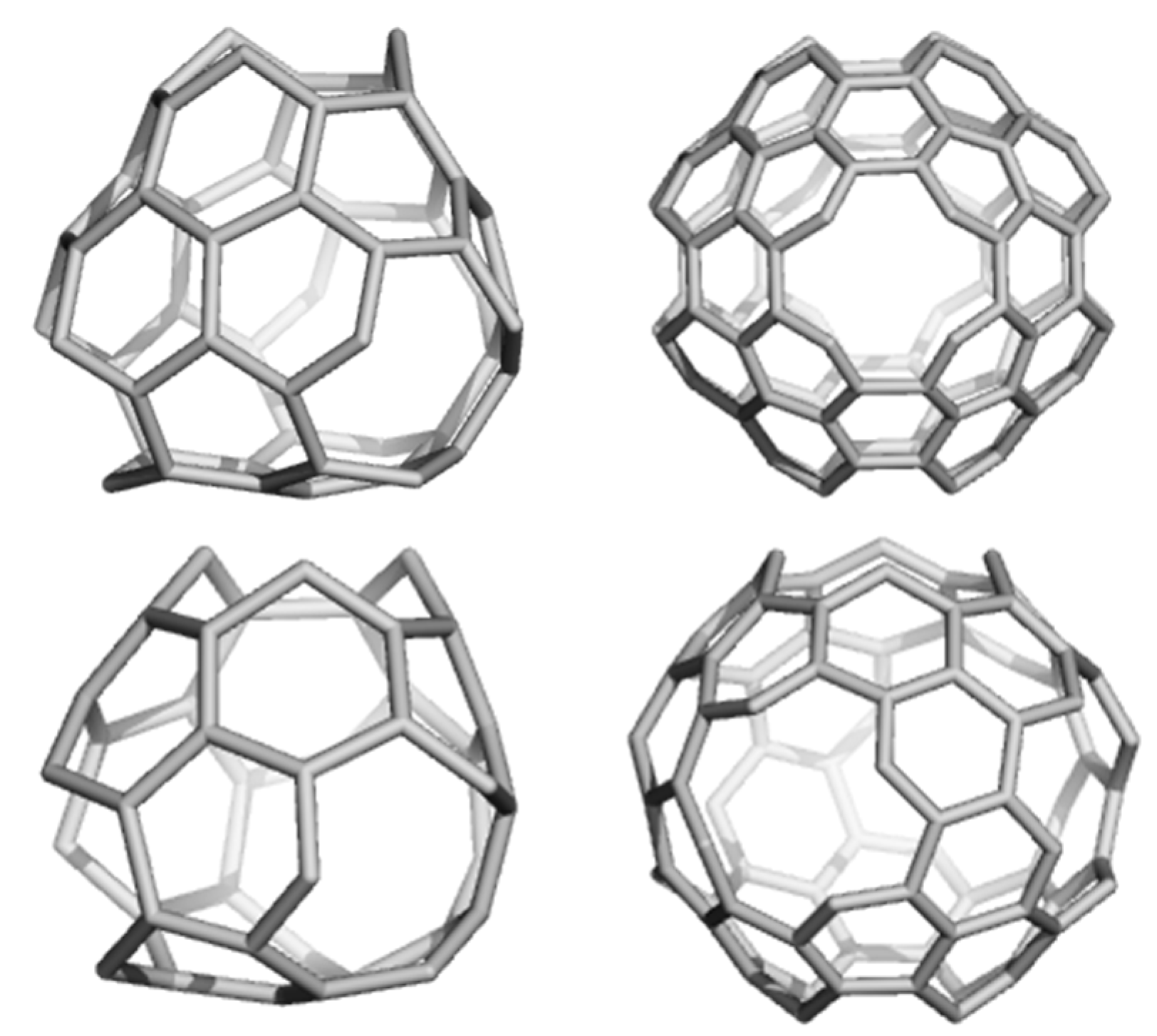
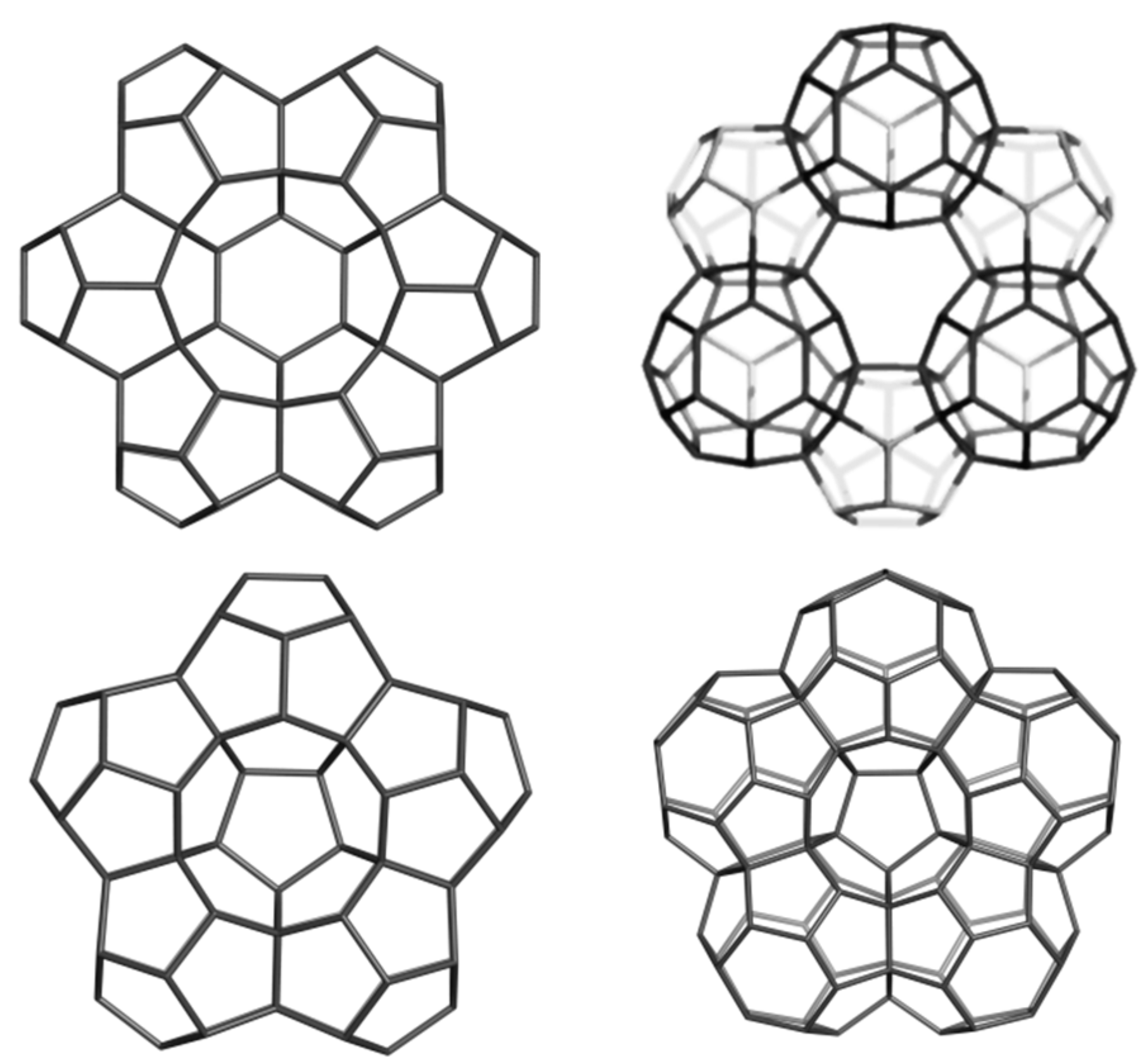
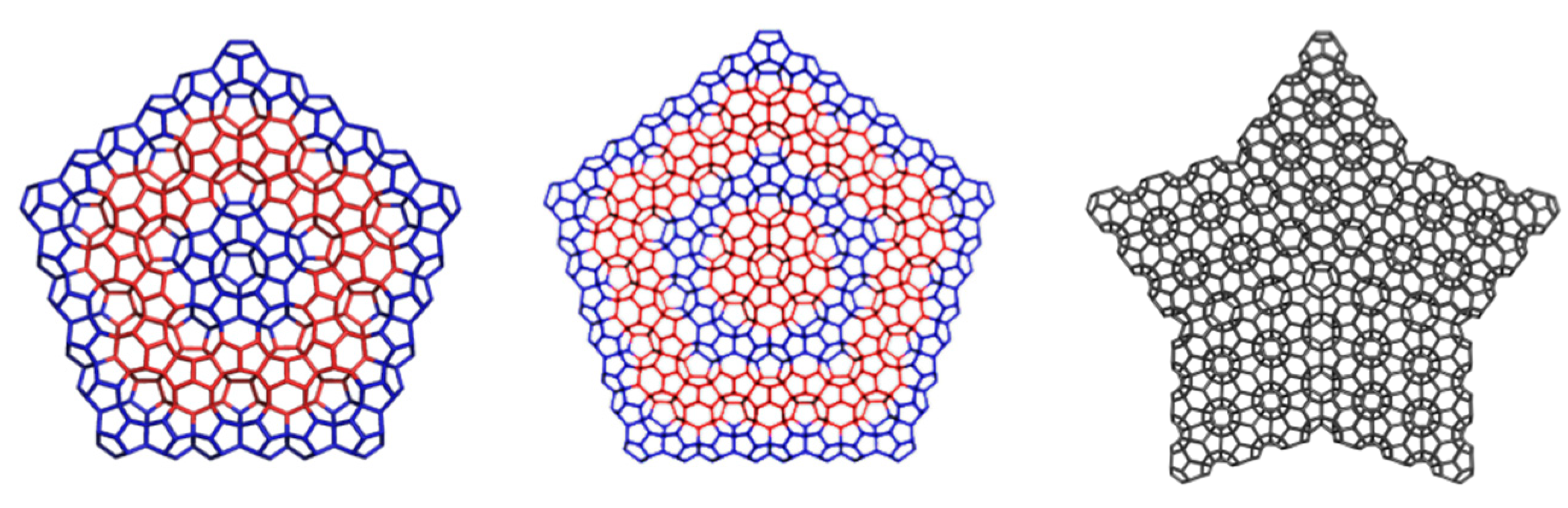
2.2. Circulenes with Octagonal Core

2.2.1. Enthalpy of Formation (Energetic Criterion)

2.2.2. 8-Flowers as Real Molecules
2.2.3. NICS(0) Index and Exaltation of Magnetic Susceptibility (Magnetic Criterion)
| Structure | Core | Petals (5 Atoms) | Petals (6 Atoms) | Petals (7 Atoms) |
|---|---|---|---|---|
| [8:(5,7)4] | −2.330 | −4.093 (−3.136) | - | 1.842 (0.393) |
| [8:68] | 9.465 | - | −7.788/−3.863 | - |
| (−10.406) | ||||
| [8:(5,6)4] | 8.172 | 0.686 (3.189) | −6.314 (−10.080) | - |
2.2.4. HOMA Index (Geometric Criterion)
| Structure | Core | Petals (5 Atoms) | Petals (6 Atoms) | Petals (7 Atoms) |
|---|---|---|---|---|
| [8:(5,7)4] | −0.811 | −0.983 | - | −0.414 |
| [8:68] | −0.432 | - | 0.703/0.335 | - |
| [8:(5,6)4] | −0.524 | −0.817 | 0.960 | - |
2.2.5. Reactivity Descriptors
| Circulene | η (eV) | ω (eV) |
|---|---|---|
| [8:(5,7)4] | 1.07 | 5.60 |
| [8:68] | 1.62 | 4.08 |
| [8:(5,6)4] | 1.93 | 3.38 |

- -
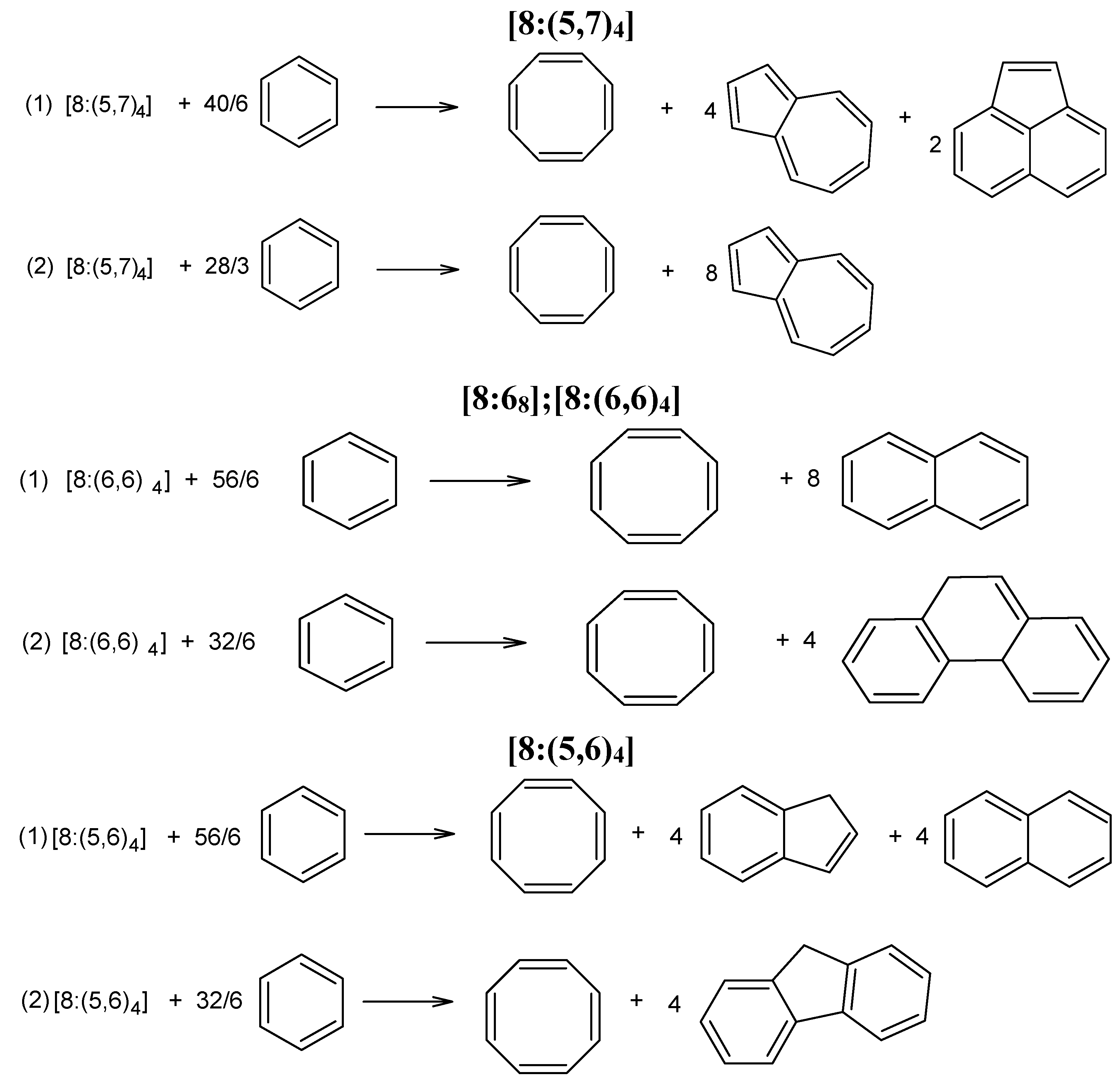
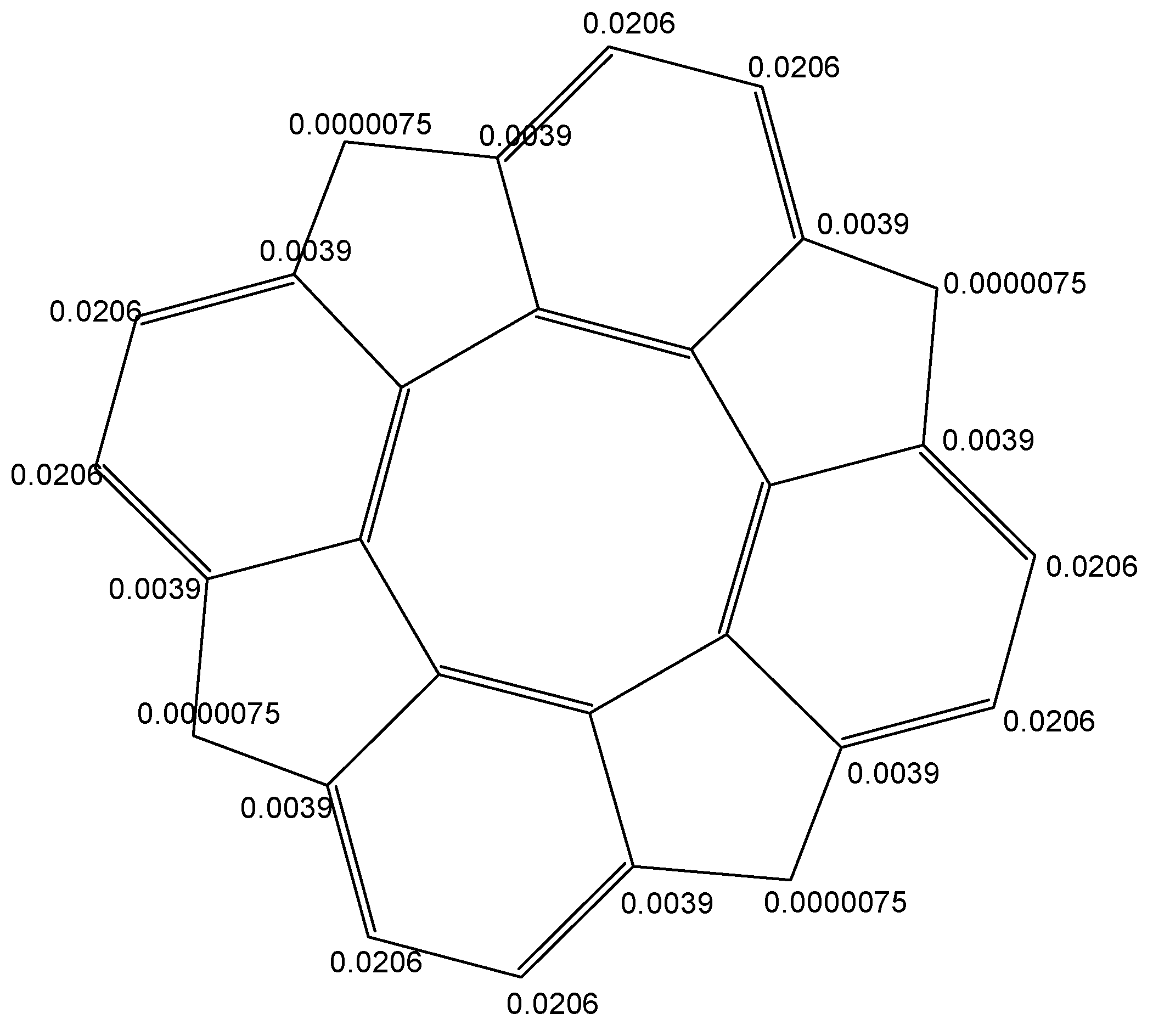
- in circulene [8:(5,7)4], the three carbon atoms in the 7-atom petals show different reactivity, suggesting a kind of circular polarization of pi-electrons;
- -
- in flower [8:68], the 6-atom petals show alternating properties, also shown by the values of local aromaticity indices (HOMA and NICS(0));
- -
- circulene [8:(5,6)4], the only planar structure, has two equivalents C-atoms on each benzene unit, with a uniform distribution around the molecule.

| Structure/ Substructure | Etot/C (au) | HL Gap (eV) | Hf(kJ/mol) | NICS(−1) | NICS(0) | NICS(+1) | HOMA |
|---|---|---|---|---|---|---|---|
| C52 | −37.864 | 5.317 | 2095.9 | ||||
| R[5] petal | −17.783 | −2.292 | −0.275 | −0.093 | |||
| R[6] petal | −28.663 | −15.704 | −6.059 | 0.333 | |||
| R[6] core | −28.281 | −11.372 | −2.803 | 0.187 | |||
| C120 | −37.871 | 6.251 | 2181.8 | ||||
| R[5] petal | −12.058 | −0.242 | −0.711 | −0.553 | |||
| R[6] petal | −16.646 | −7.635 | −3.589 | 0.537 | |||
| R[6] core | −16.746 | −7.563 | −3.565 | 0.821 | |||
| R[8] core | −3.498 | 5.164 | 6.196 | −1.478 |

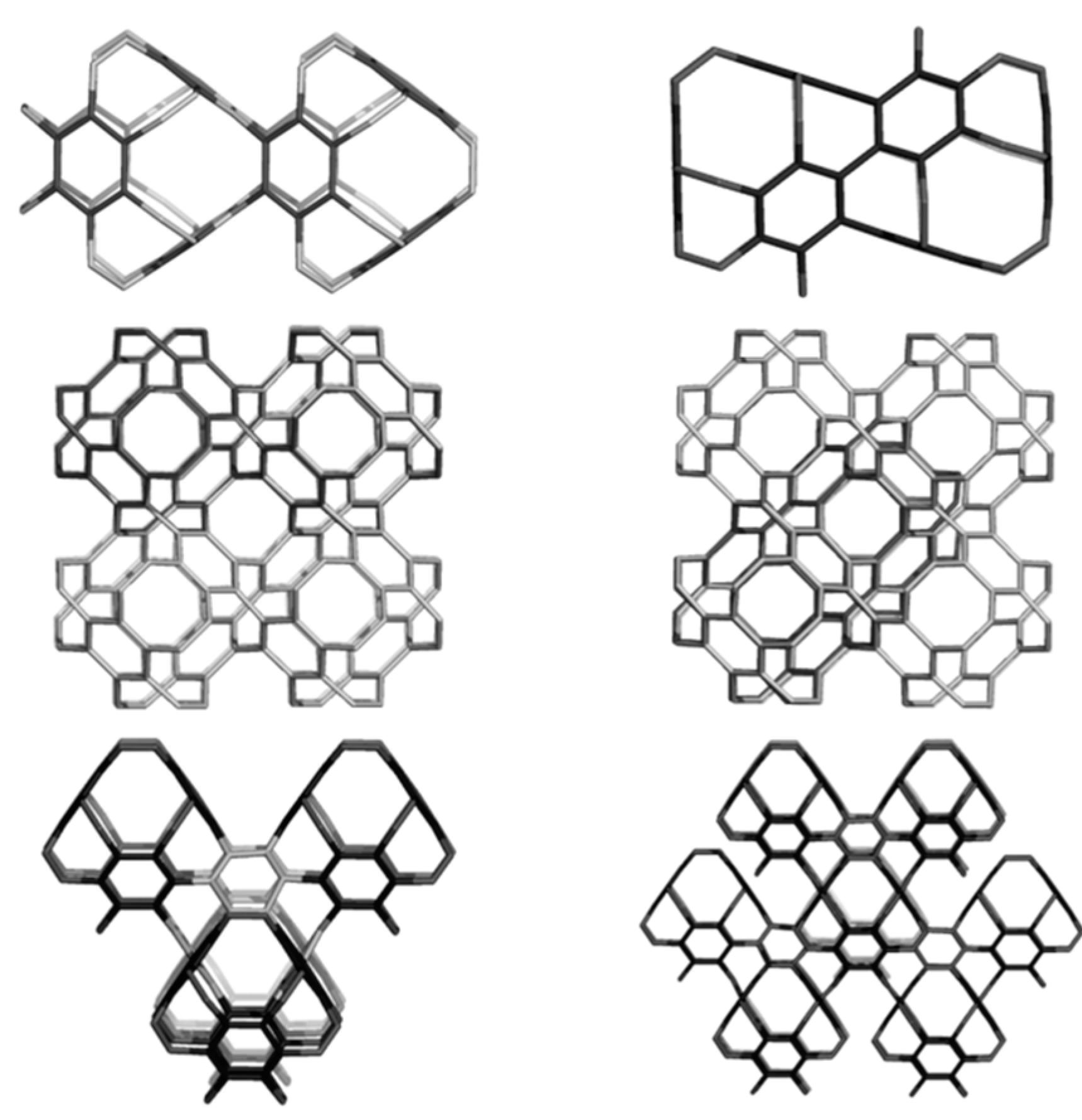
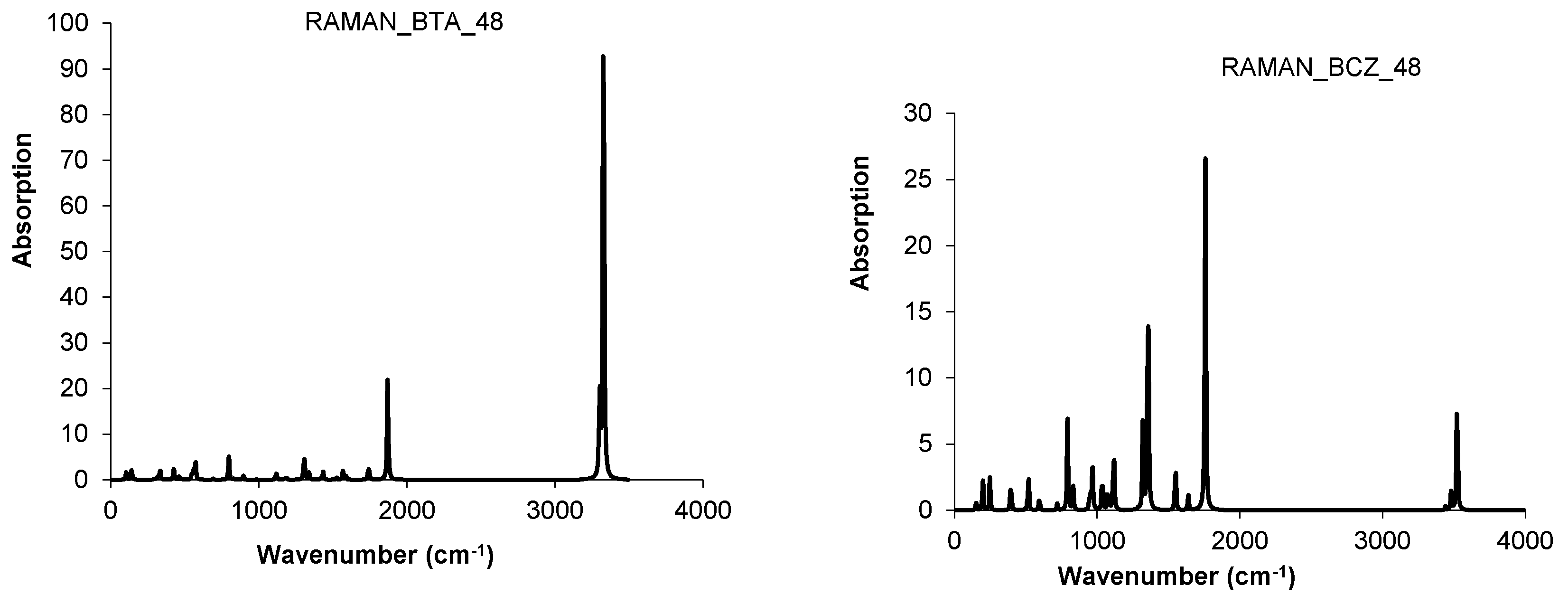
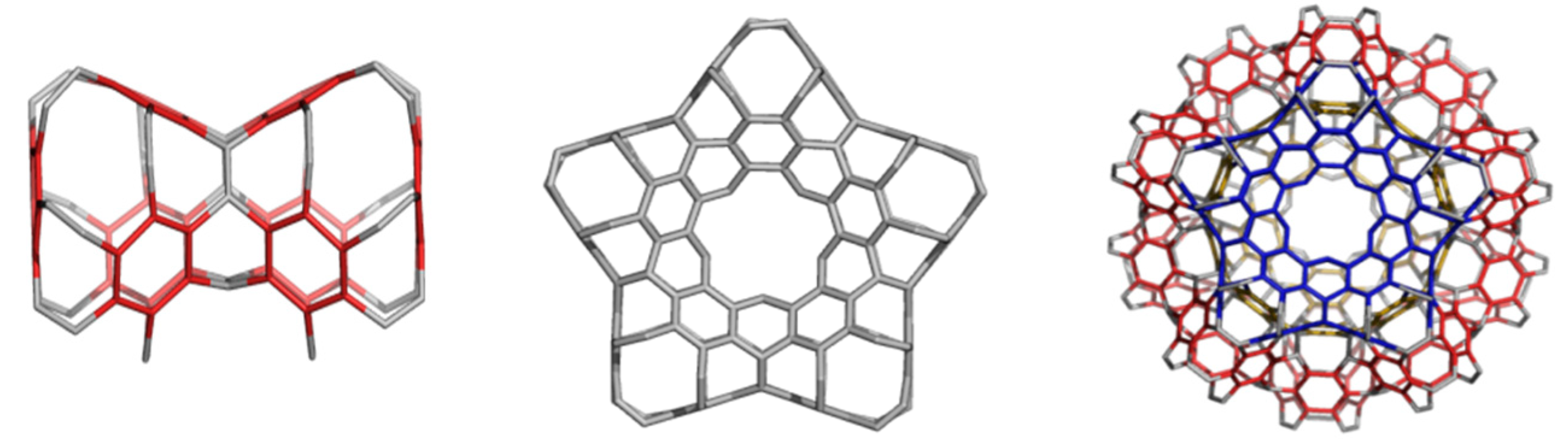
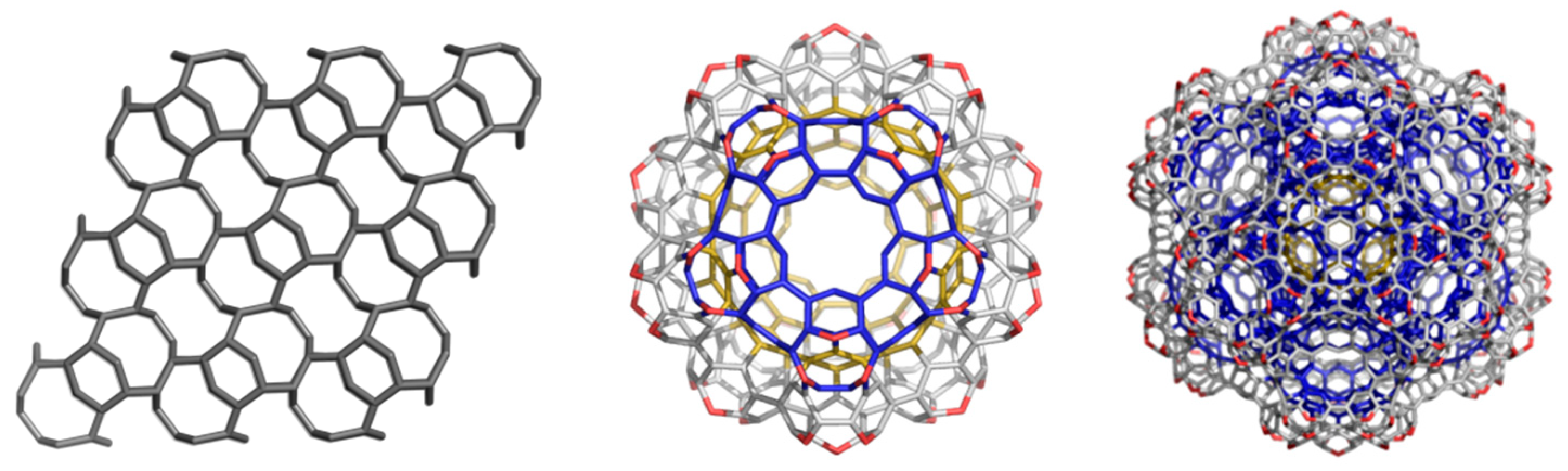
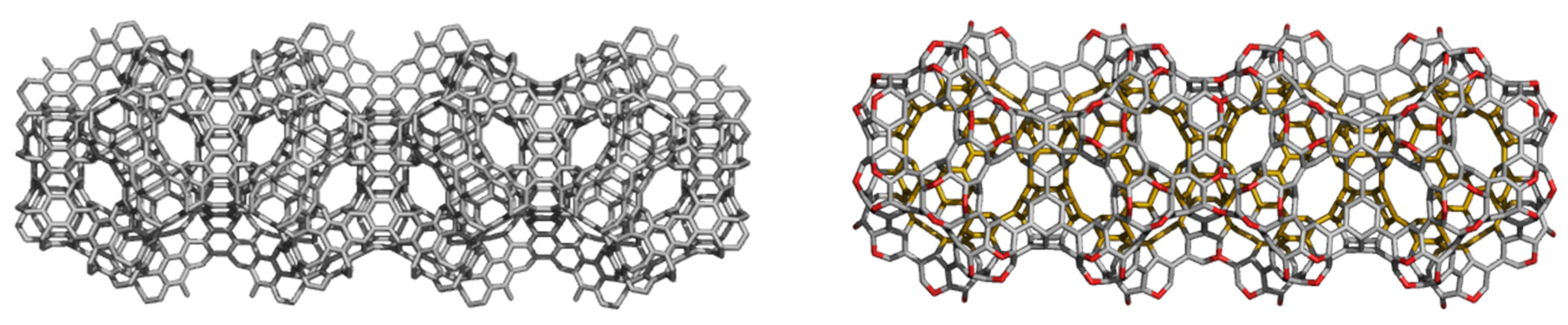
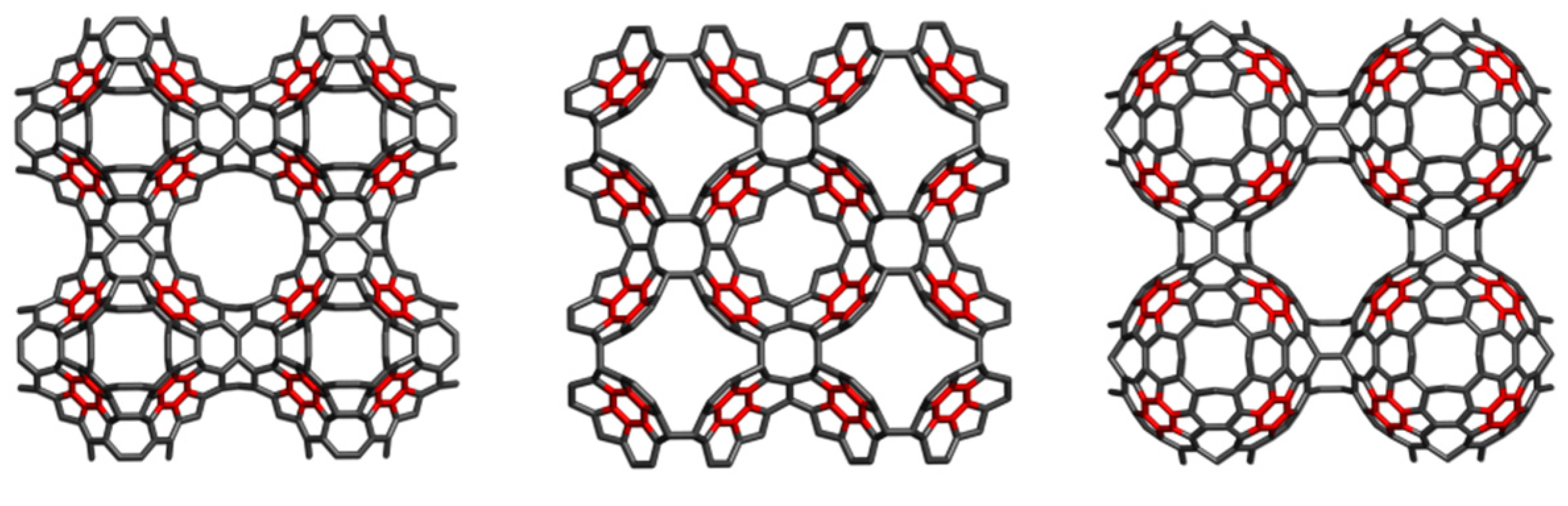
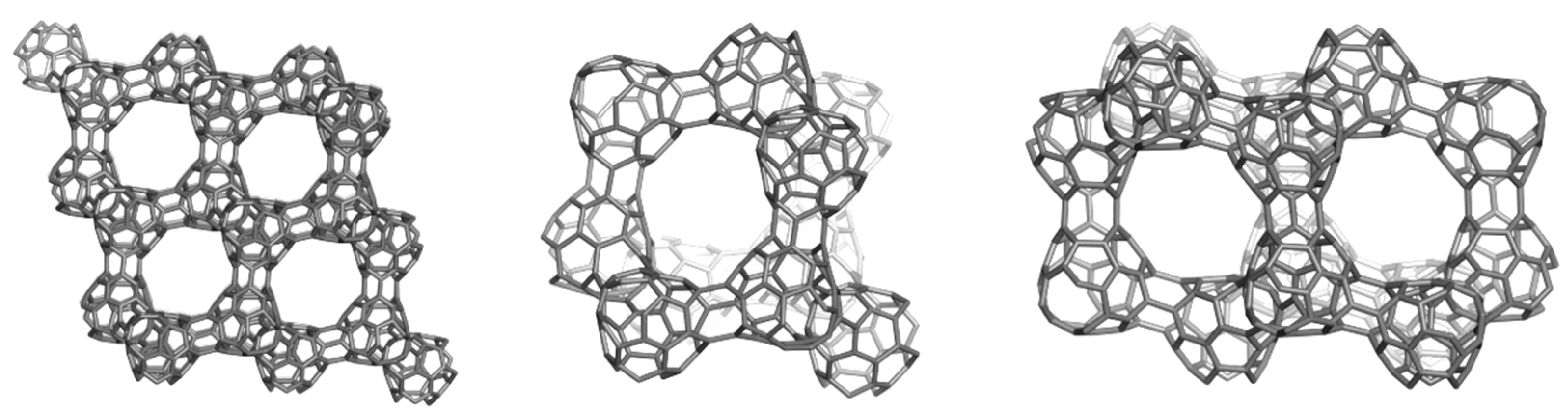
3. Polybenzenes



| Structure | No Units | Etot (au) | Etot/C (au) | HL Gap (eV) | Strain/C (kcal/mol) | HOMA R[6] | |
|---|---|---|---|---|---|---|---|
| 1 | BTA_48 | 1 | −1831.484 | −38.156 | 11.285 | 0.083 | 0.951 |
| 2 | BCZ_48 | 1 | −1831.097 | −38.148 | 8.134 | 3.395 | 0.989 |
| 3 | BTA2dia_88 | 2 | −3355.431 | −38.130 | 10.970 | 0.074 | 0.972 |
| 4 | BTA2dend_84 | 2 | −3201.679 | −38.115 | 10.895 | 0.061 | 0.975 |
| 5 | BTA3dend_120 | 3 | −4571.874 | −38.099 | 10.771 | 0.056 | 0.978 |
| 6 | BTA4dend_156 | 4 | −5942.070 | −38.090 | 10.684 | 0.054 | 0.978 |
| 7 | BTA5dend_192 | 5 | −7312.265 | −38.085 | 10.594 | 0.055 | 0.988 |
| 8 | C60(Ih) | 1 | −2271.830 | −37.864 | 7.418 | 8.256 | 0.493 |
| Structure | Etot | Etot/C | HL Gap | Etot | Etot/C | HL Gap |
|---|---|---|---|---|---|---|
| HF | DFT | |||||
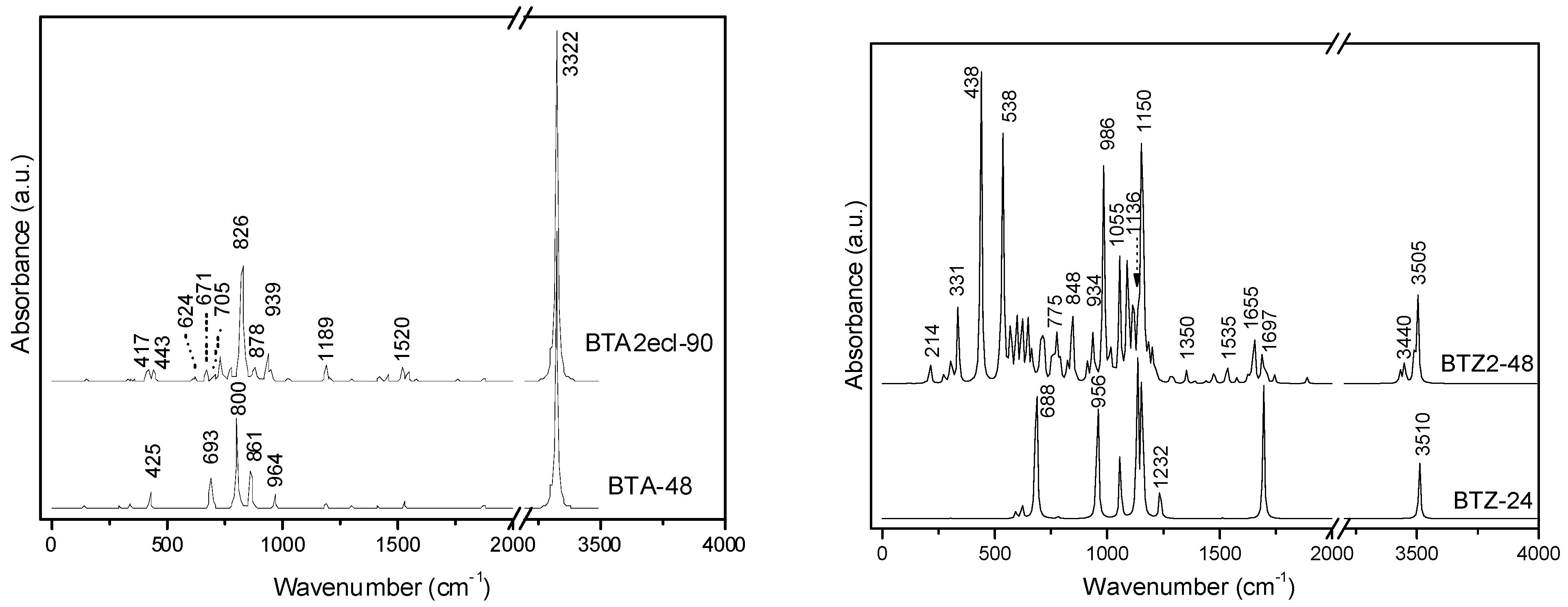
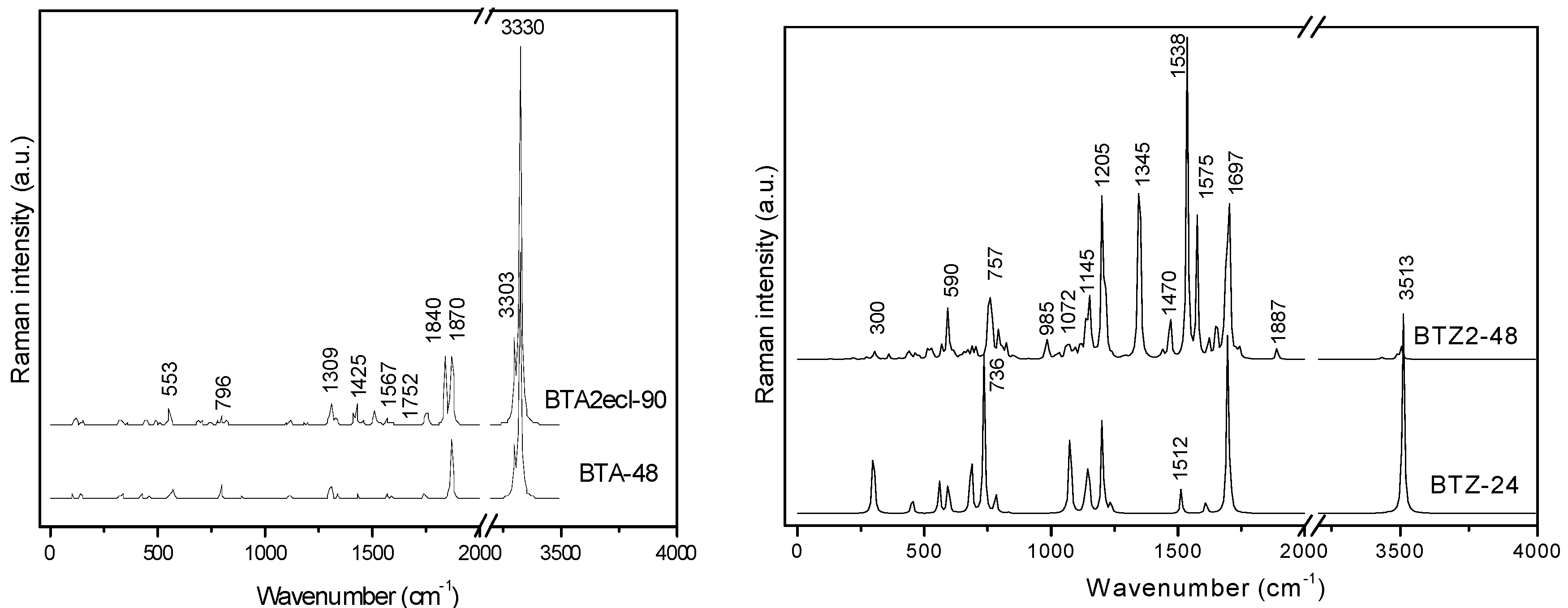
| BTA_48 | −1831.484 | −38.156 | 11.285 | −1843.743 | −38.411 | 5.052 |
| BTA2ecl_90 | −3428.847 | −38.098 | 10.085 | −3450.946 | −38.344 | 4.200 |
| BTA2dia_88 | −3355.431 | −38.130 | 10.970 | - | - | - |
| BTA2int_84 | −3201.679 | −38.115 | 10.895 | - | - | - |
| BTACy,5_210 | −7986.806 | −38.032 | 9.545 | - | - | - |
| BTZ_24 | −915.092 | −38.129 | 8.221 | −921.359 | −38.390 | 2.753 |
| BTZ2ecl_48 | −1826.768 | −38.058 | 6.194 | −1839.181 | −38.316 | 1.124 |
| BTZCy,5_120 | −4558.826 | −37.990 | 7.178 | - | - | - |
| C60(Ih) | −2271.830 | −37.864 | 7.418 | −2286.610 | −38.110 | 2.724 |








4. P-Type Surface Coverings
4.1. Sumanene Including Structures


| Structure | No. C | Etot | Etot/C | HL Gap |
|---|---|---|---|---|
| C60(Ih) | 60 | −2271.830 | −37.864 | 7.418 |
| Sum_TA_108 | 108 | −4103.136 | −37.992 | 7.259 |
| Sum_T_84 | 84 | −3194.384 | −38.028 | 7.562 |
| Sum_CZ_192 | 192 | −7298.367 | −38.012 | 6.044 |
| Sum_CA_216 | 216 | −8206.401 | −37.993 | 6.442 |
| Sum_S2LeX_168 | 168 | −6389.018 | −38.030 | 6.637 |
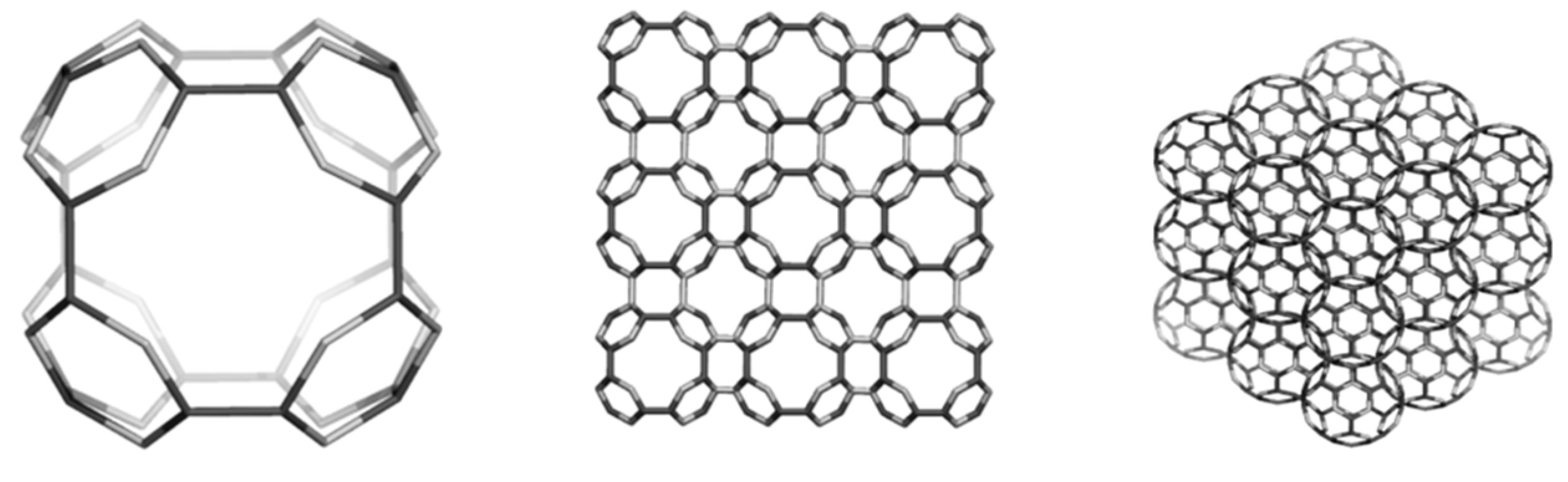
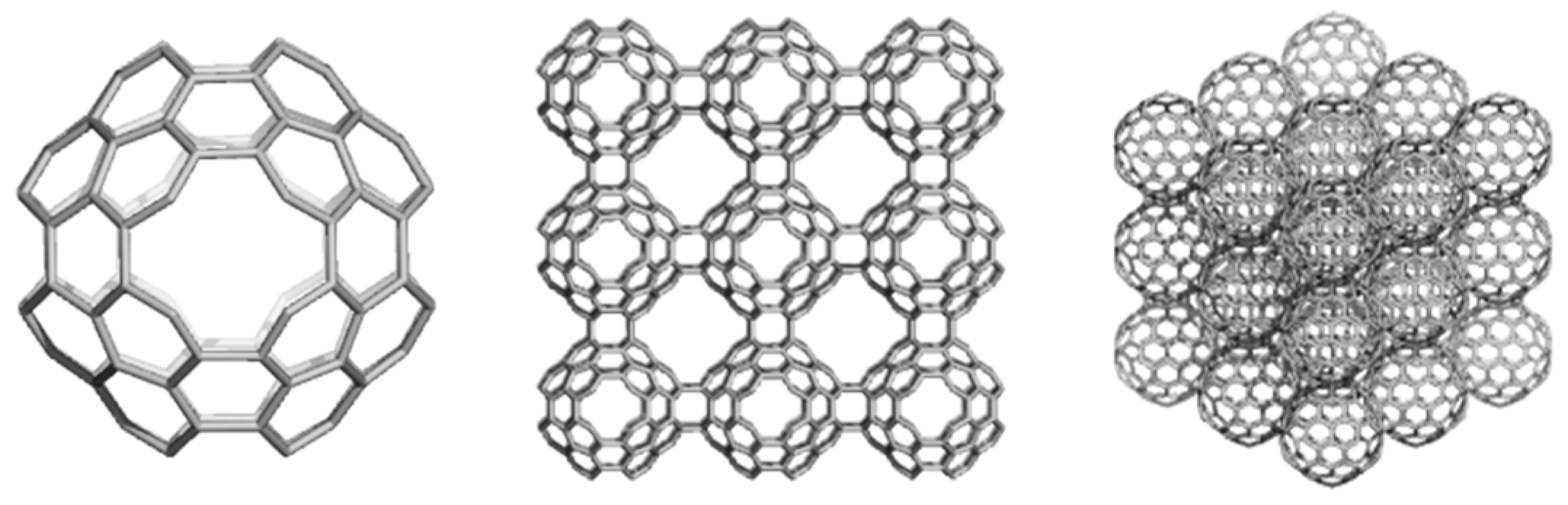
4.2. Spanned Cages Patched by Hexagons Only



| Structure | Etot/C (au) | HL Gap (eV) | Strain/C (kcal/mol) | HOMA Patch | Kekulé Count | |
|---|---|---|---|---|---|---|
| 1 | T_3HexZ_52 | −37.986 | 6.140 | 5.435 | −0.131 | 972 |
| 2 | C_3HexZ_104 | −37.999 | 5.342 | 2.329 | 0.258 | 944784 |
| 3 | T_3HextwZ_40 | −38.021 | 6.681 | 5.799 | −0.583 | 72 |
| 4 | C_3HextwZ_80 | −38.036 | 6.050 | 2.551 | −0.020 | 11025 |
| 5 | C60(Ih) | −37.864 | 7.418 | 8.256 | 0.493 | 12500 |


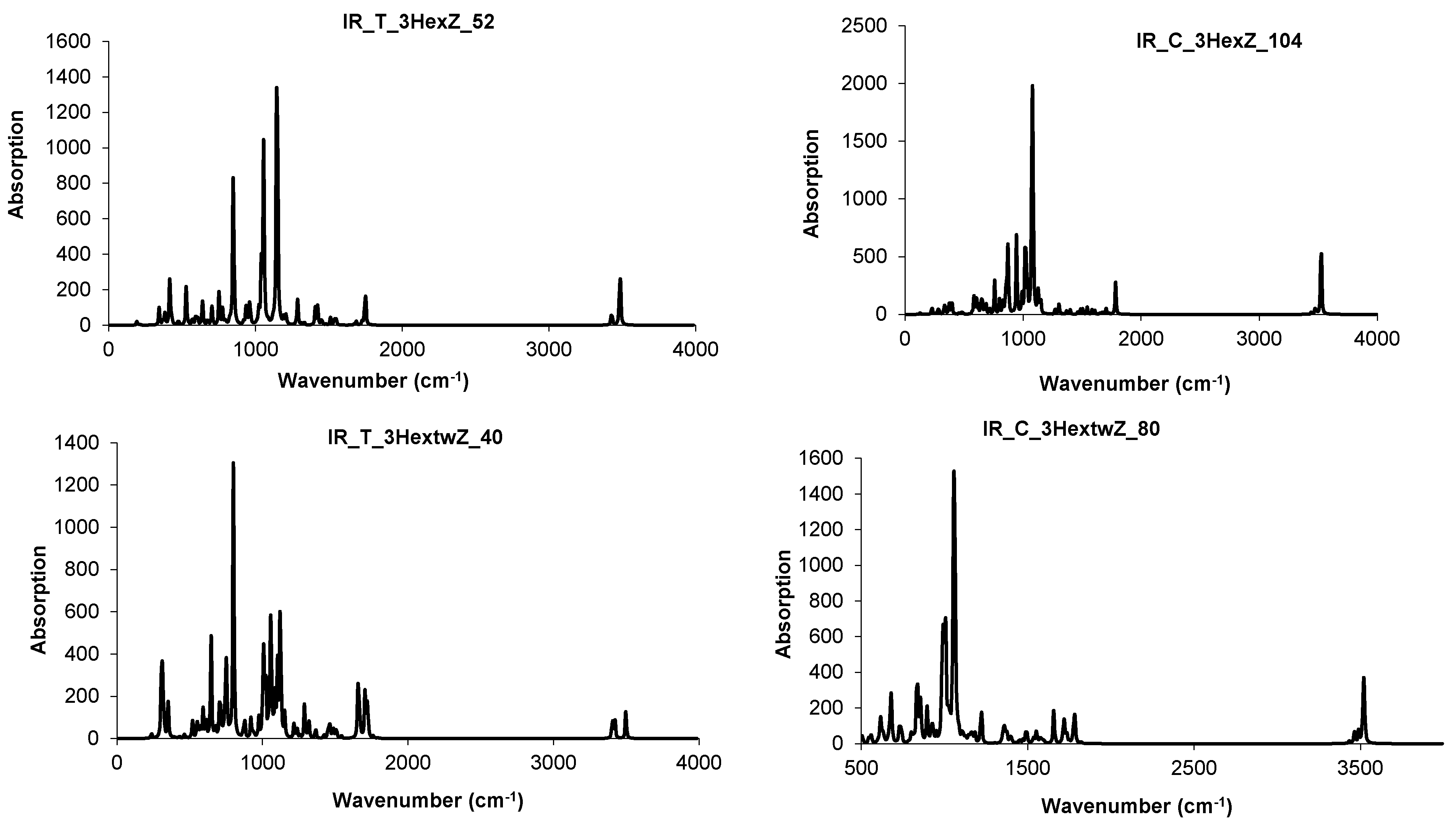
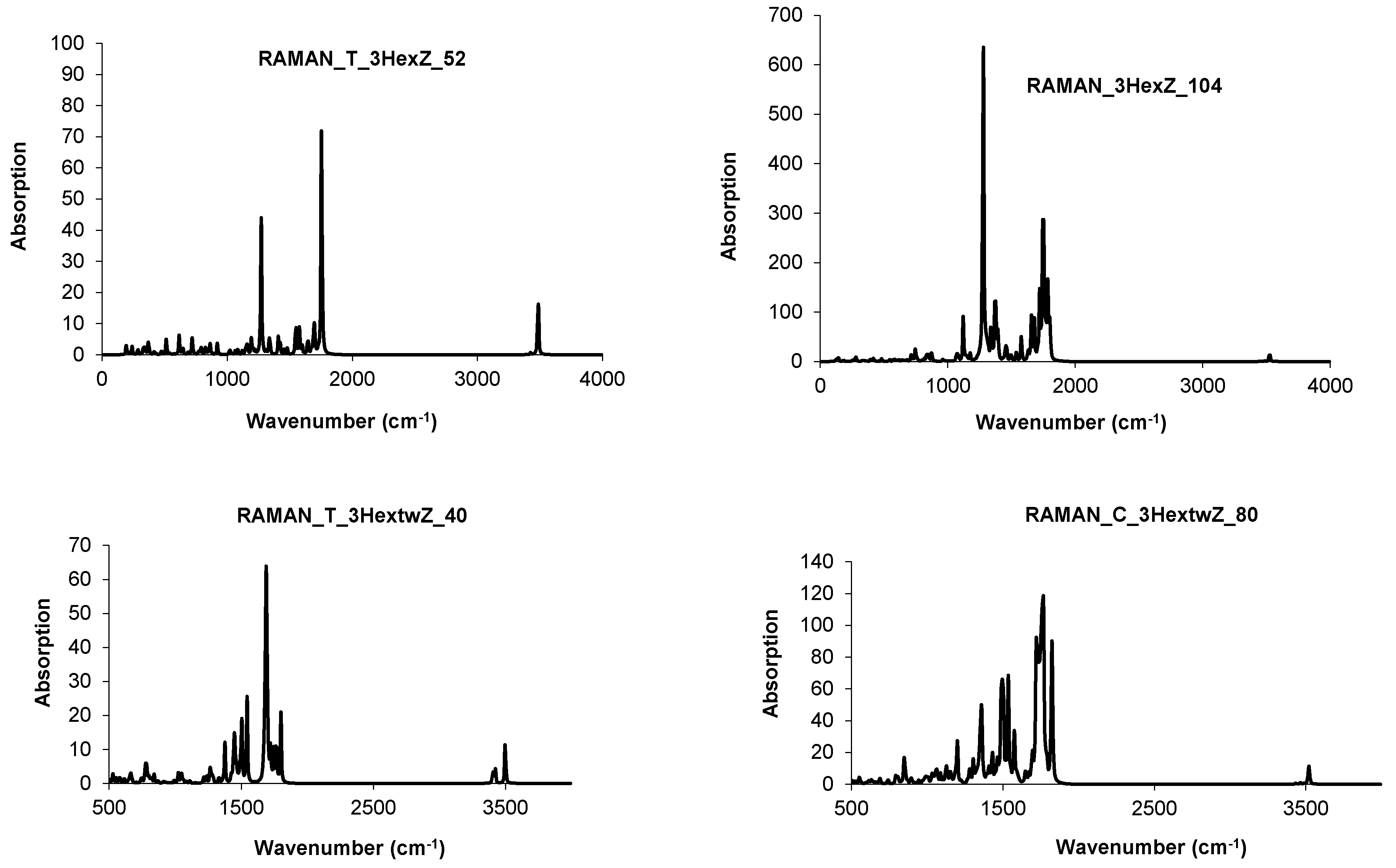
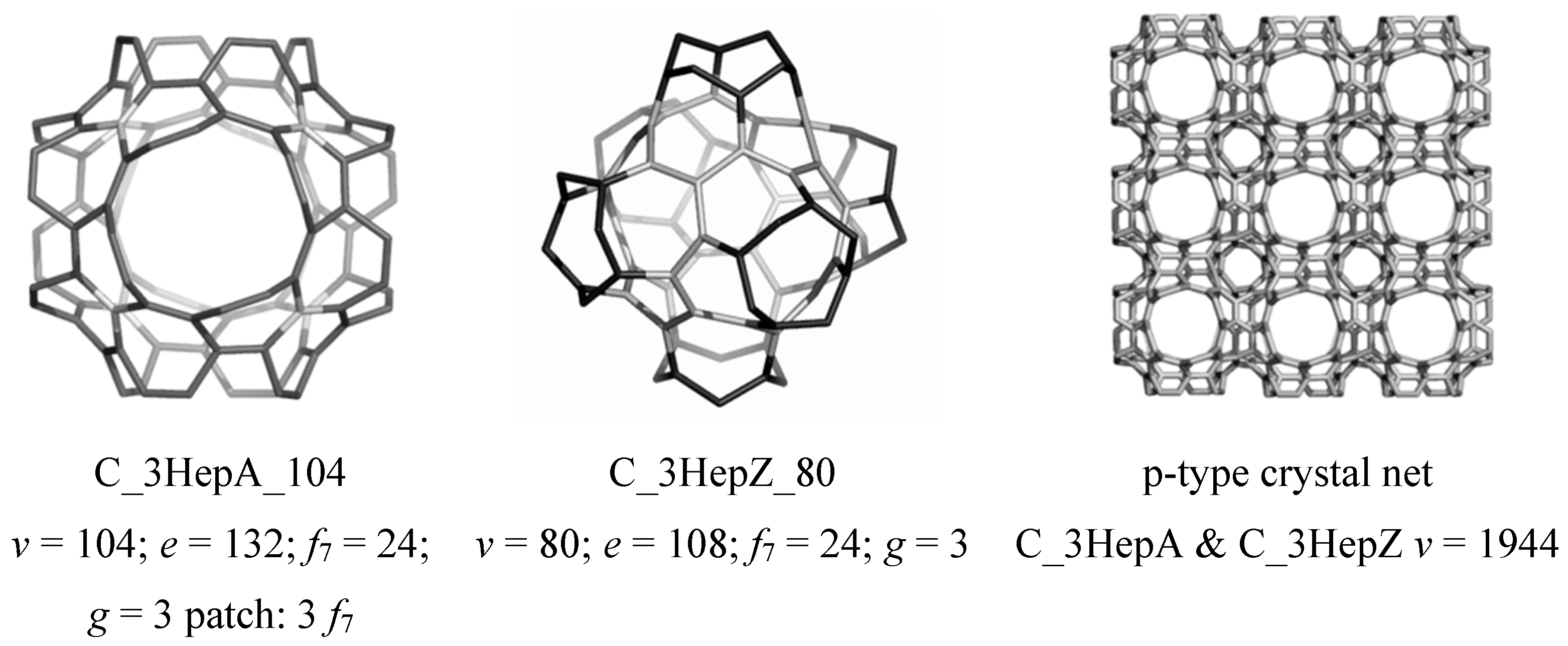
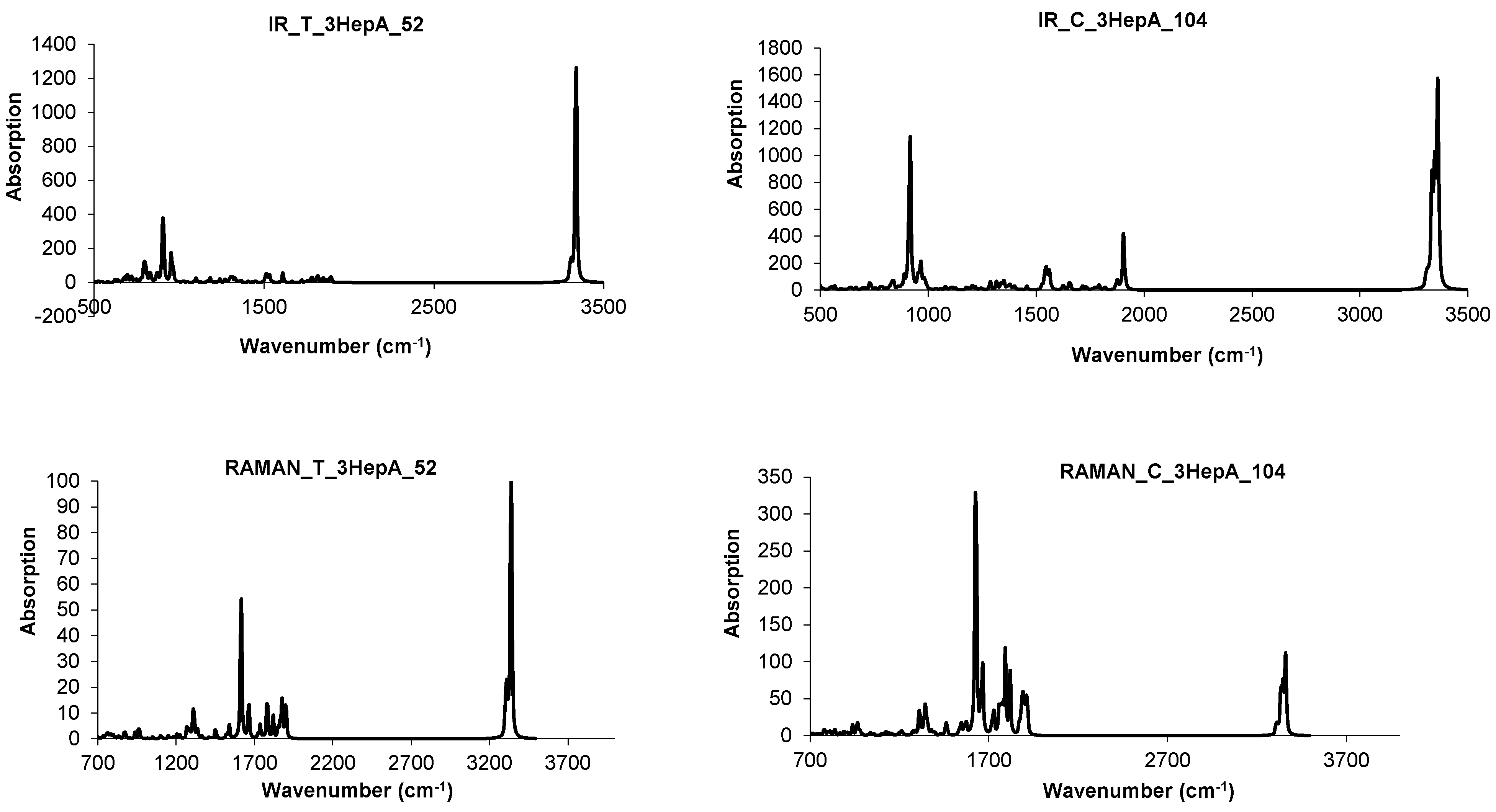
4.3. Nanotube Junctions Patched by Heptagons Only

| No. | Structure | Etot/C
(au) | HLGap
(eV) | Strain
(kcal/mol) | C60(Ih) Relat. Strain | HOMA | Kekulé
Count | C-C
Min/Max | C-C
Average |
|---|---|---|---|---|---|---|---|---|---|
| HF | |||||||||
| 1 | C20 | −37.828 | 6.819 | 27.250 | 3.301 | 0.194 | 36 | 1.405 1.472 | 1.439 |
| 2 | T_3HepA_52 | −38.126 | 7.057 | 0.410 | 0.050 | −0.951 | 75 | 1.312 1.500 | 1.406 |
| 3 | C_3HepA_104 | −38.127 | 6.942 | 0.240 | 0.029 | −0.842 | 3600 | 1.313 1.503 | 1.408 |
| 4 | C60(Ih) | −37.864 | 7.418 | 8.256 | 1.000 | 0.343 | 12500 | 1.373 1.449 | 1.411 |
| DFT | |||||||||
| 5 | C20 | −38.080 | 1.912 | 26.730 | 3.238 | −0.361 | 36 | 1.398 1.537 | 1.467 |
| 6 | T_3HepA_52 | −38.382 | 1.413 | 0.410 | 0.050 | −0.951 | 75 | 1.312 1.500 | 1.406 |
| 7 | C_3HepA_104 | −38.376 | 1.354 | 0.192 | 0.023 | −0.244 | 3600 | 1.334 1.482 | 1.408 |
| 8 | C60(Ih) | −38.110 | 2.724 | 8.256 | 1.000 | 0.299 | 12,500 | 1.392 1.452 | 1.422 |



| Structure | T-3HepA_52 | C-3HepA_104 | ||
|---|---|---|---|---|
| spectrum | IR | Raman | IR | Raman |
| cm−1 | 903 | 1617 | 917 | 1617 |
| 3332 | 3332 | 3332 | 1624 | |
| 3339 | 3339 | 3339 | 1631 | |
| 3346 | 3346 | 3346 | ||
| 3360 | 3360 |
5. Allotropes of D5 as Hyper-Graphenes



| C20_Hyper-Graphene | C atoms | Etot (au) | Etot/C-atom (au) | HL gap (eV) |
|---|---|---|---|---|
| C20HG_11_90H60 = (C20)6_90H60 | 90 | −178.393 | −1.982 | 8.992 |
| C20HG_22_252H136 | 252 | −487.798 | −1.936 | 8.447 |
| C20HG_44_780H360 | 780 | −1487.55 | −1.907 | 8.191 |
| C20HGCor_621_384H192 | 384 | −737.736 | −1.921 | 8.307 |
| C20HGCor_631_882H396 | 882 | −1678.02 | −1.903 | 8.155 |
| C60H60 | 60 | −125.584 | −2.093 | 10.412 |
| Structure | C Atoms | Etot (au) | Etot/C-Atom (au) | HL Gap (eV) |
|---|---|---|---|---|
| C60(Ih) | 60 | −102.185 | −1.703 | 1.930 |
| C20 | 20 | −33.429 | −1.671 | 0.731 |
| C24 | 24 | −40.142 | −1.673 | 1.667 |
| C28 | 28 | −47.101 | −1.682 | 0.351 |
| (C24)5_90 | 90 | −152.998 | −1.700 | 1.634 |
| (C20C28)3_114 | 114 | −192.488 | −1.688 | 0.166 |
| (C20)5_75H50 | 75 | −146.956 | −1.959 | 9.969 |
| (C24)5_90H60 | 90 | −175.282 | −1.948 | 9.103 |
| (C28)5_110H80 | 110 | −220.185 | −2.002 | 9.270 |
| (C20)6_90H60 | 90 | −178.393 | −1.982 | 8.992 |
| (C20C28)3_114H84 | 114 | −226.346 | −1.985 | 10.278 |
| C60H60 | 60 | −125.584 | −2.093 | 10.412 |
| C20H20 | 20 | −41.659 | −2.083 | 12.295 |
| C24H24 | 24 | −49.752 | −2.073 | 12.247 |
| C28H28 | 28 | −58.301 | −2.082 | 12.384 |


| Structure | C Atoms | Etot (au) | Etot/C | HL Gap(eV) |
|---|---|---|---|---|
| C60(Ih) | 60 | −102.185 | −1.703 | 1.930 |
| C60P2J5_115 | 115 | −195.708 | −1.702 | 2.044 |
| C60P2J6_114 | 114 | −194.183 | −1.703 | 1.444 |
| C60P3J555_165 | 165 | −280.787 | −1.702 | 0.608 |
| C60P3J556_164 | 164 | −281.658 | −1.717 | 0.333 |
| C60P3_J566_163 | 163 | −280.238 | −1.719 | 0.391 |
| C60P3J666_162 | 163 | −278.935 | −1.722 | 1.481 |
| Hex(C60J5)_330 | 330 | −567.506 | −1.710 | 0.179 |
| Hex(C60J6)_324 | 324 | −557.737 | −1.721 | 1.255 |
| Le(Cor(C20))J5_165_1560 | 1560 | −2652.462 | −1.700 | 0.021 |
| Cor(C60)J6_162_1512 | 1512 | −2603.270 | −1.722 | 1.095 |
| Structure | C Atoms | Theory | Etot (au) | Etot/C (au) | Gap(eV) |
|---|---|---|---|---|---|
| C20_Cy5J5_75 | 75 | HF | −2838.062 | −37.841 | 4.158 |
| C20_Cy6J5_90 | 90 | −3405.751 | −37.842 | 5.990 | |
| C28_Cy5J6_110 | 110 | −4163.361 | −37.849 | 5.533 | |
| C28_Cy6J6_132 | 132 | −4996.056 | −37.849 | 5.421 | |
| C60(Ih)_HF | 60 | −2271.830 | −37.864 | 7.418 | |
| C60P2J5_115 | 115 | −4354.333 | −37.864 | 7.597 | |
| C60P2J6_114 | 114 | −4316.491 | −37.864 | 6.270 | |
| C20_Cy5J5_75 | 75 | DFT | −2856.161 | −38.082 | 0.600 |
| C20_Cy6J5_90 | 90 | −3427.462 | −38.083 | 0.900 | |
| C28_Cy5J6_110 | 110 | −4189.837 | −38.089 | 1.072 | |
| C28_Cy6J6_132 | 132 | −5027.845 | −38.090 | 1.059 | |
| C60(Ih)_DFT | 60 | −2286.174 | −38.103 | 2.760 | |
| C60P2J5_115 | 115 | −4381.797 | −38.103 | 2.907 | |
| C60P2J6_114 | 114 | −4343.730 | −38.103 | 1.908 | |
| C20_Cy5J5_75 | 75 | DFTB | −126.324 | −1.684 | 0.113 |
| C20_Cy6J5_90 | 90 | −151.694 | −1.684 | 0.195 | |
| C28_Cy5J6_110 | 110 | −185.928 | −1.690 | 0.006 | |
| C28_Cy6J6_132 | 132 | −223.166 | −1.691 | 0.035 | |
| C60(Ih)_DFTB | 60 | −102.185 | −1.703 | 1.930 | |
| C60P2J5_115 | 115 | −195.708 | −1.702 | 2.044 | |
| C60P2J6_114 | 114 | −194.183 | −1.703 | 1.444 |
6. Computational Details
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Diudea, M.V.; Nagy, C.L. Periodic Nanostructures; Springer: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Diudea, M.V. Nanostructures, Novel Architecture; Diudea, M.V., Ed.; Nova: New York, NY, USA, 2005. [Google Scholar]
- Diamond and Related Nanostructures; Diudea, M.V.; Nagy, C.L. (Eds.) Springer: Dordrecht, The Netherlands, 2013.
- Patzke, G.R.; Krumeich, F.; Nesper, R. Oxidic nanotubes and nanorods-anisotropic modules for a future nanotechnology. Angew. Chem. Int. Ed. 2002, 41, 2447–2461. [Google Scholar]
- Tenne, R. Inorganic nanotubes and fullerene-like materials. Chem. Eur. J. 2002, 8, 5296–5304. [Google Scholar]
- Imai, H.; Matsuta, M.; Shimizu, K.; Hirashima, H.; Negishi, N. Morphology transcription with TiO2 using chemical solution growth and its application for photocatalysts. Solid State Ion. 2002, 151, 183–187. [Google Scholar]
- Abouimrane, A.; Compton, O.C.; Amine, K.; Nguyen, S.T. Non-annealed graphene paper as a Binder-free anode for Lithium-ion batteries. J. Phys. Chem. C 2010, 114, 12800–12804. [Google Scholar]
- Yu, D.S.; Dai, L.M. Self-assembled graphene/carbon nanotube hybrid films for supercapacitors. J. Phys. Chem. Lett. 2010, 1, 467–470. [Google Scholar]
- Wang, Y.; Shi, Z.Q.; Huang, Y.; Ma, Y.F.; Wang, C.Y.; Chen, M.M.; Chen, Y.S. Supercapacitor devices based on graphene materials. J. Phys. Chem. C 2009, 113, 13103–13107. [Google Scholar]
- Ng, Y.H.; Lightcap, I.V.; Goodwin, K.; Matsumura, M.; Kamat, P.V. To what extent do graphene scaffolds improve the photovoltaic and photocatalytic response of TiO2 nanostructured films? J. Phys. Chem. Lett. 2010, 1, 2222–2227. [Google Scholar] [CrossRef]
- Randić, M. Aromaticity of polycyclic conjugated hydrocarbons. Chem. Rev. 2003, 103, 3449–3605. [Google Scholar] [CrossRef]
- Kirby, E.C. Fully arenoid toroidal fullerenes, both benzenoid and non-benzenoid. MATCH Commun. Math. Comput. Chem. 1996, 33, 147–156. [Google Scholar]
- Bühl, M.; Hirsch, A. Spherical aromaticity of fullerenes. Chem. Rev. 2001, 101, 1153–1183. [Google Scholar] [CrossRef]
- Gutman, I.; Milun, M.; Trinajstić, N. Topological definition of delocalisation energy. MATCH Commun. Math. Comput. Chem. 1975, 1, 171–175. [Google Scholar]
- Gutman, I.; Milun, M.; Trinajstić, N. Graph theory and molecular orbitals. 19. Nonparametric resonance energies of arbitrary conjugated systems. J. Am. Chem. Soc. 1977, 99, 1692–1704. [Google Scholar]
- Aihara, J. A New Definition of Dewar-type resonance energies. J. Am. Chem. Soc. 1976, 98, 2750–2758. [Google Scholar]
- Randić, M. Algebraic Kekulé formulas for benzenoid hydrocarbons. J. Chem. Inf. Comput. Sci. 2004, 44, 365–372. [Google Scholar]
- Gutman, I.; Vukičević, D.; Graovac, A.; Randić, M. Algebraic Kekulé structures of benzenoid hydrocarbons. J. Chem. Inf. Comput. Sci. 2004, 44, 296–299. [Google Scholar]
- Miličević, A.; Nikolić, S.; Trinajstić, N. Coding and ordering Kekulé structures. J. Chem. Inf. Comput. Sci. 2004, 44, 415–421. [Google Scholar]
- Randić, M.; Balaban, A.T. Partitioning of π-electrons in rings of polycyclic conjugated hydrocarbons. Part 1. Catacondensed benzenoids. Polycycl. Aromat. Compd. 2004, 24, 173–193. [Google Scholar] [CrossRef]
- Balaban, A.T.; Randić, M. Partitioning of π-electrons in rings of polycyclic conjugated hydrocarbons. Part 3. Perifusene. New J. Chem. 2004, 28, 800–806. [Google Scholar]
- Vukicevic, D.; Randić, M.; Balaban, A.T. Partitioning of π- electrons in rings of polycyclic conjugated hydrocarbons. Part 4. Benzenoids with more than one geometric Kekule structure corresponding to the same algebraic Kekulé structure. J. Math. Chem. 2004, 36, 271–279. [Google Scholar]
- Balaban, A.T.; Randić, M. Partitioning of π- electrons in rings of polycyclic conjugated hydrocarbons. Part 5. Nonalternant Compounds. J. Chem. Inf. Comput. Sci. 2004, 44, 1701–1707. [Google Scholar]
- Balaban, A.T.; Randić, M. Partitioning of π- electrons in rings of polycyclic conjugated hydrocarbons. Part 6. Comparison with other methods for estimating the local aromaticity of rings in polycyclic benzenoids. J. Math. Chem. 2005, 37, 443–453. [Google Scholar]
- Clar, E. Polycyclic Hydrocarbons; Acad. Press: London, UK, 1964. [Google Scholar]
- Clar, E. The Aromatic Sextet; Wiley: New York, NY, USA, 1972. [Google Scholar]
- Trinajstić, N. Chemical Graph Theory; CRC Press: Boca Raton, FL, USA, 1992; pp. 161–187. [Google Scholar]
- Fowler, P.W.; Collins, D.J.; Austin, S.J. Is aromaticity a useful concept for C60 and its derivatives? Aromatisation of C60 by regioselective addition. J. Chem. Soc. Perkin Trans. 1993, 2, 275–277. [Google Scholar]
- Austin, S.J.; Fowler, P.W.; Hansen, P.; Manolopoulos, D.E.; Zheng, M. Fullerene isomers of C60. Kekulé counts versus stability. Chem. Phys. Lett. 1994, 228, 478–484. [Google Scholar] [CrossRef]
- Liu, X.; Klein, D.J.; Seitz, W.A.; Schmalz, T.G. Sixty-atom carbon cages. J. Comp. Chem. 1991, 12, 1265–1269. [Google Scholar]
- Randić, M. Conjugated circuits and resonance energies of benzenoid hydrocarbons. Chem. Phys. Lett. 1976, 38, 68–70. [Google Scholar]
- Randić, M. Aromaticity and conjugation. J. Am. Chem. Soc. 1977, 99, 444–450. [Google Scholar]
- Randić, M. A graph theoretical approach to conjugation and resonance energies of hydrocarbons. Tetrahedron 1977, 33, 1905–1920. [Google Scholar]
- Ozaki, M.; Takahashi, A. On Electronic States and Bond Lengths of Truncated Icosahedral C60 Molecule. Chem. Phys. Lett. 1986, 127, 242–244. [Google Scholar] [CrossRef]
- Disch, R.L.; Schulman, J.M. On symmetrical clusters of carbon atoms: C60. Chem. Phys. Lett. 1986, 125, 465–466. [Google Scholar] [CrossRef]
- Bird, C.W. Heteroaromaticity.13. Bond alternation and its consequences for conjugation energies and other criteria of aromaticity. Tetrahedron 1985, 41, 1409–1414. [Google Scholar]
- Julg, A.; Francois, P. Structural studies of some non-altemant hydrocarbons; a new definition of aromaticity. Theor. Chim. Acta 1967, 7, 249–261. [Google Scholar]
- Cyranski, M.K.; Howard, S.T.; Chodkiewicz, M.L. Bond energy, aromatic stabilization energy and strain in IPR fullerenes. Chem. Commun. 2004, 2458–2459. [Google Scholar]
- Krygowski, T.M.; Ciesielski, A. Aromatic character in the benzene ring present in various topological environments in benzenoid hydrocarbons. Nonequivalence of indices of aromaticity. J. Chem. Inf. Comput. Sci. 1995, 35, 203–210. [Google Scholar]
- Krygowski, T.M.; Ciesielski, A. Local aromatic character of C60 and C70 and their derivatives. J. Chem. Inf. Comput. Sci. 1995, 35, 1001–1003. [Google Scholar]
- Krygowski, T.M.; Cyranski, M. Separation of the energetic and geometric contributions to the aromaticity. Part IV. A general model for the pi-electron systems. Tetrahedron 1996, 52, 10255–10264. [Google Scholar]
- Dewar, M.J.S. Aromaticity and pericyclic reactions. Angew. Chem. Int. Ed. Engl. 1971, 10, 761–870. [Google Scholar]
- Jug, K.; Koester, A.M. Aromaticity as a multi-dimensional phenomenon. J. Phys. Org. Chem. 1991, 4, 163–169. [Google Scholar]
- Balaban, A.T.; Oniciu, D.C.; Katritzky, A.R. Aromaticity as a cornerstone in heterocyclic chemistry. Chem. Rev. 2004, 104, 2777–2812. [Google Scholar]
- Diudea, M.V. Nanomolecules and Nanostructures-Polynomials and Indices; MCM Series 10; MCM Series 10; University of Kragujevac Press: Kragujevac, Serbia, 2010. [Google Scholar]
- Amsharov, K.Y.; Jansen, M.A. C78 Fullerene precursor: Toward the direct synthesis of higher fullerenes. J. Org. Chem. 2008, 73, 2931–2934. [Google Scholar]
- Amsharov, K.Y.; Jansen, M. Synthesis of a higher fullerene precursor-an “unrolled” C84 fullerene. Chem. Commun. 2009, 2691–2693. [Google Scholar]
- Barborini, E.; Piseri, P.; Milani, P.; Benedek, G.; Ducati, C. Robertson Negatively curved spongy carbon. J. Appl. Phys. Lett. 2002, 81, 3359–3361. [Google Scholar]
- Benedek, G.; Vahedi-Tafreshi, H.; Barborini, E.; Piseri, P.; Milani, P.; Ducati, C.; Robertson, J. The structure of negatively curved spongy carbon. Diam. Relat. Mater. 2003, 12, 768–773. [Google Scholar]
- Fowler, P.W.; Pisanski, T. Leapfrog transformations and polyhedra of Clar type. J. Chem. Soc. Faraday Trans. 1994, 90, 2865–2871. [Google Scholar]
- Schleyer, P.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Pop, R.; Medeleanu, M.; Diudea, M.V.; Szefler, B.; Cioslowski, J. Fullerenes patched by flowers. Cent. Eur. J. Chem. 2013, 11, 527–534. [Google Scholar]
- Diudea, M.V. Nanostructures-Novel Architecture; Diudea, M.V., Ed.; Nova: New York, NY, USA, 2005; pp. 203–242. [Google Scholar]
- Ciesielski, A.; Cyrański, M.K.; Krygowski, T.M.; Fowler, P.W.; Lillington, M. Super-Delocalized Valence Isomer of Coronene. J. Org. Chem. 2006, 71, 6840–6845. [Google Scholar] [CrossRef]
- Ciesielski, A.; Krygowski, T.M.; Cyrański, M.K.; Balaban, A.T. Defining rules of aromaticity: A unified approach to the Hückel, Clar and Randić concepts. Phys. Chem. Chem. Phys. 2011, 13, 3737–3747. [Google Scholar]
- Löffler, A.; Bajales, N.; Cudaj, M.; Weis, P.; Lebedkin, S.; Bihlmeier, A.; Tew, D.P.; Klopper, W.; Böttcher, A.; Kappes, M.M. Non-IPR C60 solids. J. Chem. Phys. 2009, 130, 164705. [Google Scholar] [CrossRef]
- Haddon, R.C. Rehybridization and pi.-orbital overlap in nonplanar conjugated organic molecules: pi.-Orbital axis vector (POAV) analysis and three-dimensional Hueckel molecular orbital (3D-HMO) theory. J. Am. Chem. Soc. 1987, 109, 1676–1685. [Google Scholar]
- Pop, R.; Medeleanu, M.; Diudea, M.V.; Szefler, B.; Cioslowski, J. Fullerenes patched by flowers with octagonal core. Cent. Eur. J. Chem. 2014, 12, 90–97. [Google Scholar]
- Parthasarathi, R.; Padmanabhan, J.; Elango, M.; Subramanian, V.; Chatarraj, P.K. Intermolecular reactivity through the generalized philicity concept. Chem. Phys. Lett. 2004, 394, 225–230. [Google Scholar] [CrossRef]
- Contreras, R.; Fuentealba, P.; Galvan, M.; Perez, P. A direct evaluation of regional Fukui functions in molecules. Chem. Phys. Lett. 1999, 304, 405–413. [Google Scholar] [CrossRef]
- Roux, M.V.; Temprado, M.; Chickos, J.S.; Nagano, Y. Critically evaluated thermochemical properties of polycyclic aromatic hydrocarbons. J. Phys. Chem. 2008, 37, 1855–1996. [Google Scholar]
- Torres, L.A.; Campos, M.; Martinez, M.; Rojas, A. The thermochemistry of coronene revisited. J. Chem. Thermodyn. 2009, 41, 957–965. [Google Scholar]
- Nielsen, C.B.; Brock-Nannestad, T.; Reenberg, T.K.; Hammershøj, P.; Christensen, J.B.; Stouwdam, J.W.; Pittelkow, M. Organic light-emitting diodes from symmetrical and unsymmetrical π-extended tetraoxa[8]circulenes. Chem. Eur. J. 2010, 16, 13030–13034. [Google Scholar] [CrossRef]
- Eskildsen, J.; Reenberg, T.; Christensen, J.B. Substituted Tetraoxa[8]circulenes—New members of the liquid crystal family. Eur. J. Org. Chem. 2000, 1637–1640. [Google Scholar]
- Minaev, B.F.; Baryshnikov, G.V.; Minaeva, V.A. Density functional theory study of electronic structure and spectra of tetraoxa[8]circulenes. Comput. Theor. Chem. 2011, 972, 68–74. [Google Scholar]
- Eskildsen, J.; Hammershшj, P.; Reenberg, T.K.; Larsen, U.; Pittelkow, M.; Leth, S.M.; Peck, R.A.; Christensen, J.B. Substituted Tetraoxa[8]circulenes. Asian Chem. Lett. 2007, 11, 211–218. [Google Scholar]
- Chernichenko, K.Y.; Balenkova, E.S.; Nenajdenko, V.G. From thiophene to sulflower. Mendeleev Commun. 2008, 18, 171–179. [Google Scholar]
- Brock-Nannestad, T.; Nielsen, C.B.; Schau-Magnussen, M.; Hammershøj, P.; Reenberg, T.K.; Petersen, A.B.; Trpcevski, D.; Pittelkow, M. Tetra-tert-butyltetraoxa[8]circulene and Its Unusual Aggregation Behaviour. Eur. J. Org. Chem. 2011, 2011, 6320–6325. [Google Scholar]
- Perepichka, I.; Perepichka, D.; Meng, H.; Wudlt, F. Light-Emitting Polythiophenes. Adv. Mater. 2005, 17, 2281–2305. [Google Scholar] [CrossRef]
- Rathore, R.; Abdelwahed, S.H. Soluble cycloannulated tetroxa[8]circulane derivatives: Synthesis, optical and electrochemical properties, and generation of their robust cation–radical salts. Tetrahedron Lett. 2004, 45, 5267–5270. [Google Scholar]
- Baryshnikov, G.V.; Minaev, B.F.; Pittelkow, M.; Nielsen, C.B.; Salcedo, R. Nucleus-independent chemical shift criterion for aromaticity in π-extended tetraoxa[8]circulenes. J. Mol. Model. 2013, 19, 847–850. [Google Scholar]
- Minaeva, V.A.; Minaev, B.F.; Baryshnikov, G.V.; Agren, H.; Pittelkowc, M. Experimental and theoretical study of IR and Raman spectra of tetraoxa[8]circulenes. Vib. Spectrosc. 2012, 61, 156–166. [Google Scholar]
- Baryshnikov, G.V.; Minaev, B.F.; Minaeva, V.A.; Baryshnikova, A.T.; Pittelkow, M. DFT and QTAIM study of the tetra-tert-butyltetraoxa[8]circulene regioisomers structure. J. Mol. Struct. 2012, 1026, 127–132. [Google Scholar]
- Minaeva, V.A.; Minaev, B.F.; Baryshnikov, G.V.; Romeyko, O.M.; Pittelkow, M. The FTIR spectra of substituted tetraoxa[8]circulenes and their assignments based on DFT calculations. Vib. Spectrosc. 2013, 65, 147–158. [Google Scholar]
- Liljefors, T.; Wennerstrom, O. Molecular mechanics calculations on the structures and conformational properties of [8]circulene and some related phane compounds. Tetrahedron 1977, 33, 2999–3003. [Google Scholar]
- Salcedo, R.; Sansores, L.E.; Picazo, A.; Sansón, L. [8]Circulene. Theoretical approach. J. Mol. Struct. Theochem. 2004, 678, 211–215. [Google Scholar]
- Solà, M.; Feixas, F.; Jiménez-Halla, J.O.C.; Matito, E.; Poater, J. A critical assessment of the performance of magnetic and electronic indices of aromaticity. Symmetry 2010, 2, 1156–1179. [Google Scholar]
- Roy, D.R.; Bultinck, P.; Subramanian, V.; Chattaraj, P.K. Bonding, reactivity and aromaticity in the light of the multicenter indices. J. Mol. Struct. Theochem. 2008, 854, 35–39. [Google Scholar]
- O’Keeffe, M.; Adams, G.B.; Sankey, O.F. Predicted new low energy forms of carbon. Phys. Rev. Lett. 1992, 68, 2325–2328. [Google Scholar]
- Harary, F. Graph Theory; Addison-Wesley: Reading, MA, USA, 1969. [Google Scholar]
- Schwarz, H.A. Über Minimalflächen; Monatsber: Berlin Akad, Germany, 1865. (In German) [Google Scholar]
- Schwarz, H.A. Gesammelte Matematische Abhandlungen; Springer: Berlin, Germany, 1980. (In German) [Google Scholar]
- Meier, W.M.; Olson, D.H. Atlas of Zeolite Structure Types, 3rd ed.; Butterworth-Heineman: London, UK, 1992. [Google Scholar]
- Szefler, B.; Diudea, M.V. Polybenzene revisited. Acta Chim. Slov. 2012, 59, 795–802. [Google Scholar]
- Szefler, B.; Ponta, O.; Diudea, M.V. Energetics of polybenzene multi tori. J. Mol. Struct. 2012, 1022, 89–93. [Google Scholar]
- Szefler, B.; Diudea, M.V. Polybenzene multitori. Cent. Eur. J. Chem. 2012, 10, 1779–1785. [Google Scholar]
- Silverstein, R.; Webster, F. Spectrometric Identification of Organic Compounds, 6th ed.; John Wiley & Sons: New York, NY, USA, 1997; Chapter 3; p. 5. [Google Scholar]
- Keogh, S.M.; Hedderman, T.G.; Rüther, M.G.; Lyng, F.M.; Gregan, E.; Farrell, G.F.; Chambers, G.; Byrne, H.J. Temperature-Induced Nucleation of Poly(p-phenylene vinylene-co-2,5-dioctyloxy-m-phenylene vinylene) Crystallization by HiPco Single-Walled Carbon Nanotubes. J. Phys. Chem. B 2005, 109, 5600–5607. [Google Scholar] [CrossRef]
- Wondmagegn, W.T.; Curran, S.A. A study of C60–poly(m-phenylenevinylene-co-2,5-dioctoxy-p-phenylenevinylene) nanocomposite. Thin Solid Films 2006, 515, 2393–2397. [Google Scholar]
- Moret, R.; Wagberg, T.; Sundqvist, B. Influence of the pressure—Temperature treatment on the polymerization of C60 single crystals at 2 GPa–700 K. Carbon 2005, 43, 709–716. [Google Scholar]
- Diudea, M.V.; Petitjean, M. Symmetry in multi tori. Symmetry Cult. Sci. 2008, 19, 285–305. [Google Scholar]
- Szefler, B.; Saheli, M.; Diudea, M.V. Sumanene units in P-type surface networks. Acta Chim. Slov. 2012, 59, 177–182. [Google Scholar]
- Nagy, C.L.; Diudea, M.V. Nano Studio Software; “Babes-Bolyai” University: Cluj, Romania, 2009. [Google Scholar]
- Szefler, B.; Diudea, M.V. Strain in Platonic fullerenes. Struct. Chem. 2014, 25, 319–325. [Google Scholar]
- Diudea, M.V.; Szefler, B. Nanotube junctions and the genus of multi-tori. Phys. Chem. Chem. Phys. 2012, 14, 8111–8115. [Google Scholar]
- Diudea, M.V.; Ştefu, M.; John, P.E.; Graovac, A. Generalized operations on maps. Croat. Chem. Acta 2006, 79, 355–362. [Google Scholar]
- tefu, M.; Diudea, M.V.; John, P.E. Composite operations on maps. Stud. Univ. Babes-Bolyai Chem. 2005, 50, 165–174. [Google Scholar]
- Diudea, M.V. Manoporous carbon alloptropes by septupling map operations. J. Chem. Inf. Model. 2005, 45, 1002–1009. [Google Scholar]
- Böhme, B.; Guloy, A.; Tang, Z.; Schnelle, W.; Burkhardt, U.; Baitinger, M.; Grin, Yu. Oxidation of M4Si4 (M=Na, K) to clathrates by HCl or H2O. J. Am. Chem. Soc. 2007, 129, 5348–5349. [Google Scholar] [CrossRef]
- Adams, G.B.; O’Keeffe, M.; Demkov, A.A.; Sankey, O.F.; Huang, Y.-M. Wide-band-gap Si in open fourfold-coordinated clathrate structures. Phys. Rev. B 1994, 49, 8048–8053. [Google Scholar]
- Guloy, A.; Ramlau, R.; Tang, Z.; Schnelle, W.; Baitinger, M.; Grin, Yu. A quest-free germanium chlatrate. Nature 2006, 443, 320–323. [Google Scholar]
- Schwarz, U.; Wosylus, A.; Böhme, B.; Baitinger, M.; Hanfland, M.; Grin, Yu. A 3D network of four-bonded germanium: A link between open and dense. Angew. Chem. Int. Ed. 2008, 47, 6790–6793. [Google Scholar] [CrossRef]
- Diudea, M.V. Hyper-graphenes. Int. J. Chem. Model. 2013, 5, 211–220. [Google Scholar]
- Diudea, M.V.; Szefler, B.; Nagy, C.L.; Bende, A. Exotic allotropes of carbon. In Carbon Materials: Chemistry and Physics Exotic Properties of Carbon; Putz, M.V., Ori, O., Eds.; Springer: Dordrecht, The Netherlands, 2014; in press. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dauben, H.J.; Wilson, J.D.; Laity, J.L. Diamagnetic susceptibility exaltation as a criterion of aromaticity. J. Am. Chem. Soc. 1968, 90, 811–813. [Google Scholar]
- Chesnut, D.B.; Moore, K.D. Locally dense basis sets for chemical shift calculations. J. Comput. Chem. 1985, 10, 648–659. [Google Scholar]
- Krygowski, T.M.; Szatylowicz, H.; Stasyuk, O.A.; Dominikowska, J.; Palusiak, M. Aromaticity from the Viewpoint of Molecular Geometry: Application to Planar Systems. Chem. Rev. 2014, 114, 6383–6422. [Google Scholar] [CrossRef]
- Nagy, C.L.; Diudea, M.V. JSCHEM Software Program; “Babes-Bolyai” University: Cluj, Romania, 2004. [Google Scholar]
- Stefu, M.; Diudea, M.V. CVNET Software Program; “Babes-Bolyai” University: Cluj, Romania, 2005. [Google Scholar]
- Nagy, C.L.; Diudea, M.V. Nano-Studio Software Program; “Babes-Bolyai” University: Cluj, Romania, 2005. [Google Scholar]
- Chandra, A.; Nguyen, M. A density functional study of weakly bound hydrogen bonded complexes. Chem. Phys. 1998, 232, 299–306. [Google Scholar] [CrossRef]
- Pei, K.; Li, Y.; Li, H. Hydrogen-bonding interaction between acetic acid and pyridine 1:1 complex. J. Mol. Struct. 2003, 660, 113–118. [Google Scholar] [CrossRef]
- Chaundhari, A.; Lee, S. Computational study of glycine-(water) complex by density functional study. Chem. Phys. 2005, 310, 281–285. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Szatyłowicz, H.; Zachara, J.E. Molecular geometry as a source of chemical information. Part V. Substituent effect on proton transfer in para-substitued phenol complexes with fluoride—A B3LYP/6-311+G** study. J. Chem. Inform. Model. 2005, 45, 652–656. [Google Scholar] [CrossRef]
- Szatyłowicz, H.; Krygowski, T.M.; Panek, J.J.; Jezierska, A. H-bonded complexes of aniline with HF/F- and anilide with HF in terms of symmetry-adapted perturbation, atoms in molecules, and natural bond orbitals theories. J. Phys. Chem. 2008, 112, 9895–9905. [Google Scholar] [CrossRef]
- Schleyer, P.R.; Jiao, H.; Hommes, N.J.R.E.; Malkin, V.G.; Malkina, O.L. An evaluation of the aromaticity of inorganic rings: Refined evidence from magnetic properties. J. Am. Chem. Soc. 1997, 119, 12669–12670. [Google Scholar] [CrossRef]
- Albertazzi, E.; Domene, C.; Fowler, P.W.; Heine, T.; Seifert, G.; van Alsenoy, C.; Zerbetto, F. Pentagon adjacency as a determinant of fullerene stability. Phys. Chem. Chem. Phys. 1999, 1, 2913–2918. [Google Scholar]
- Dauben, H.J.; Wilson, J.D.; Laity, J.L. Diamagnetic susceptibility exaltation in hydrocarbons. J. Am. Chem. Soc. 1969, 91, 1991–1998. [Google Scholar] [CrossRef]
- Borosky, G.L.; Laali, K.K. Theoretical study of aza-polycyclic aromatic hydrocarbons (aza-PAHs), modelling carbocations from oxidized metabolites and their covalent adducts with representative nucleophiles. Org. Biomol. Chem. 2005, 3, 1180–1188. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szefler, B.; Diudea, M.V. Quantum-Mechanical Calculations on Molecular Substructures Involved in Nanosystems. Molecules 2014, 19, 15468-15506. https://doi.org/10.3390/molecules191015468
Szefler B, Diudea MV. Quantum-Mechanical Calculations on Molecular Substructures Involved in Nanosystems. Molecules. 2014; 19(10):15468-15506. https://doi.org/10.3390/molecules191015468
Chicago/Turabian StyleSzefler, Beata, and Mircea V. Diudea. 2014. "Quantum-Mechanical Calculations on Molecular Substructures Involved in Nanosystems" Molecules 19, no. 10: 15468-15506. https://doi.org/10.3390/molecules191015468
APA StyleSzefler, B., & Diudea, M. V. (2014). Quantum-Mechanical Calculations on Molecular Substructures Involved in Nanosystems. Molecules, 19(10), 15468-15506. https://doi.org/10.3390/molecules191015468