Unusual Structure-Energy Correlations in Intramolecular Diels–Alder Reaction Transition States

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
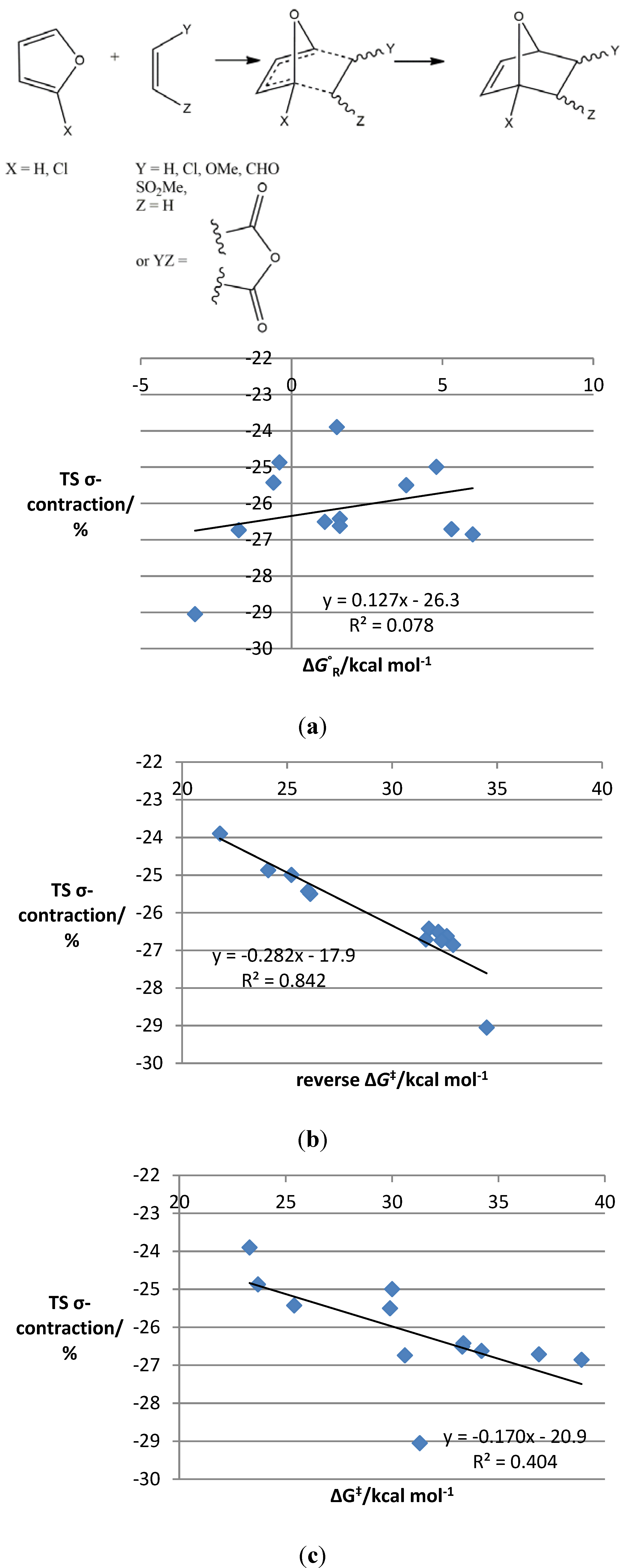
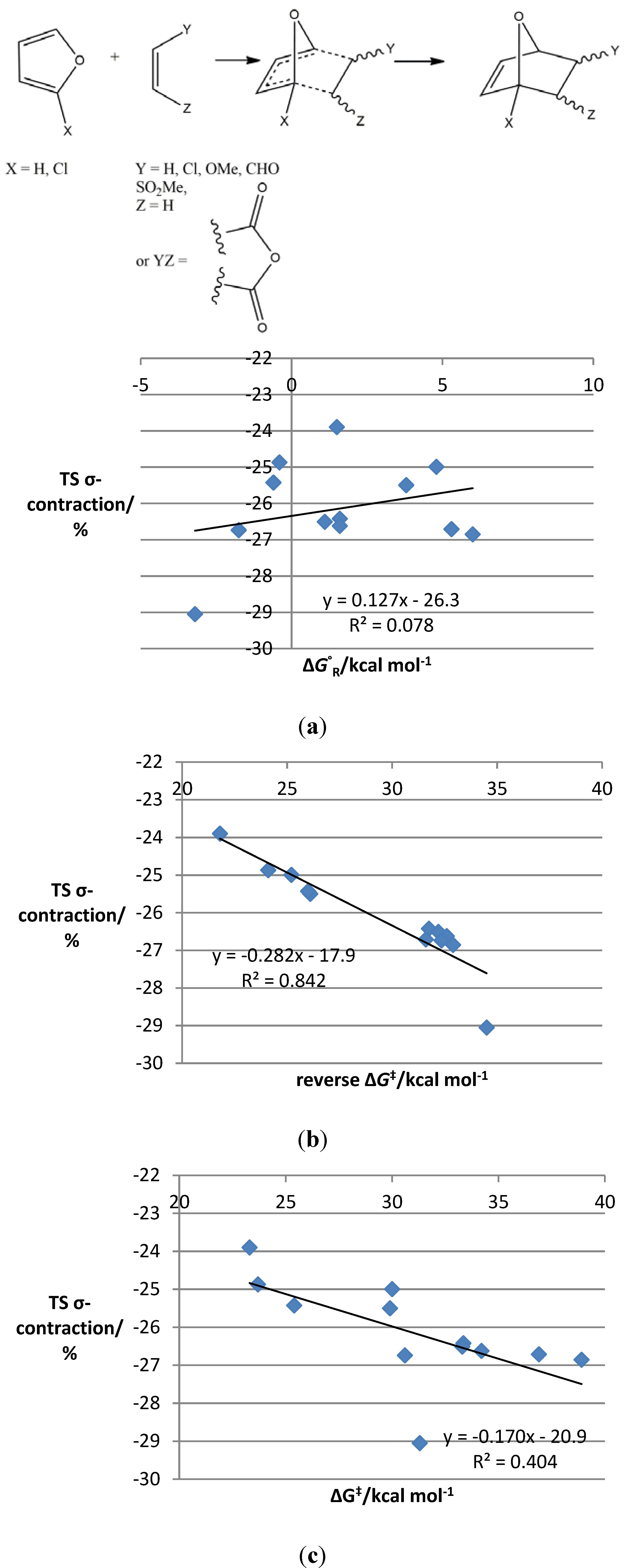{kind=link}
{kind=link}
1. Introduction

2. Results and Discussion



3. Experimental Section
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ciganek, E. The Intramolecular Diels–Alder Reaction. In Organic Reactions; John Wiley & Sons: New York, NY, USA, 2004. [Google Scholar]
- Kappe, C.O.; Murphree, S.S.; Padwa, A. Synthetic Applications of Furan Diels–Alder chemistry. Tetrahedron 1997, 53, 14179–14233. [Google Scholar]
- Vogel, P.; Cossy, J.; Plumet, J.; Arjona, O. Derivatives of 7-oxabicyclo[2.2.1]heptanes in nature and as useful synthetic intermediates. Tetrahedron 1999, 55, 13521–13642. [Google Scholar]
- Padwa, A.; Flick, A.C. Intramolecular Diels–Alder reactions for natural product synthesis. Adv. Heterocycl. Chem. 2013, 110, 1–41. [Google Scholar]
- Cooper, H.D.; Wright, D.L. The tandem ring opening/ring closing metathesis route to oxaspirocycles. Molecules 2013, 18, 2438–2448. [Google Scholar]
- Pitsinos, E.N.; Athinaios, N.; Veroniki, V.P. Enantioselective synthesis of (−)-Laurenditerpenol. Org. Lett. 2012, 14, 4666–4669. [Google Scholar]
- Wang, C.-C.; Li, W.-D.Z. A Convergent and Stereocontrolled Cycloaddition Strategy toward Eudesmane Sesquiterpenoid: Total Synthesis of (±)-6β,14-Epoxyeudesm-4(15)-en-1β-ol. J. Org. Chem. 2012, 77, 4217–4225. [Google Scholar]
- Fischer, P.; Gruner, M.; Jaeger, A.; Kataeva, O.; Metz, P. Total Synthesis of Pamamycin-6498B. Chem. Eur. J. 2011, 17, 13334–13340. [Google Scholar]
- Tanino, K.; Takahashi, M.; Tomata, Y.; Tokura, H.; Uehara, T.; Takashi, N.; Miyashita, M. Total synthesis of solanoeclipon A. Nat. Chem. 2011, 3, 484–488. [Google Scholar]
- Petronijevic, F.R.; Wipf, P. Total Synthesis of (+/−)-Cycloclavine and (+/−)-5-epi-Cycloclavine. J. Am. Chem. Soc. 2011, 133, 7704–7707. [Google Scholar]
- Xue, Y.-P.; Li, W.-D.Z. Formal Total Synthesis of (+/−)-Estrone via the Furano Diene Approach. J. Org. Chem. 2011, 76, 57–64. [Google Scholar]
- O’Keefe, M.B.; Mans, D.M.; Kaelin, D.E.; Martin, S.F. Total Synthesis of Isokidamycin. J. Am. Chem. Soc. 2010, 132, 15528–15530. [Google Scholar]
- Mitsuru, S.; Hayashi, Y. Chemistry of Epoxyquinols A, B, and C and Epoxytwinol A. Eur. J. Org. Chem. 2007, 23, 3783–3800. [Google Scholar]
- Sparks, S.M.; Chen, C.-L.; Martin, S.F. Tandem Intramolecular Benzyne-furan cycloadditions. Total synthesis of vineomycinone B2 methyl ester. Tetrahedron 2007, 63, 8619–8635. [Google Scholar]
- Morton, G.E.; Barrett, A.G.M. Interative Benzyne-Furan Cycloaddition Reactions: Studies toward the Total Synthesis of ent-Sch 47554 and ent-Sch 47555. Org. Lett. 2006, 8, 2859–2861. [Google Scholar]
- Padwa, A.; Ginn, J.D. Studies on the Synthesis of (+/−)-Stenine: A Combined Intramolecular [4 + 2]- Cycloaddition/Rearrangement Cascade. J. Org. Chem. 2005, 70, 5197–5206. [Google Scholar]
- Takadoi, M.; Katoh, T.; Tadashi, I.; Terashima, S. Synthetic studies of himbacine, a potent agonist of the muscarinic M2 subtype receptor 1. Stereoselective total synthesis and antagonistic activity of enantiomeric pairs of himbacine and (2'S,6'R)-diepihimbacine, 4-epihimbacine, and novel himbacine congeners. Tetrahedron 2002, 58, 9903–9923. [Google Scholar]
- Mitsuru, S.; Yamaguchi, J.; Kakeya, H.; Osada, H.; Hayashi, Y. Total Synthesis of (+)-Epoxyquinols A and B. Angew. Chem. Int. Ed. 2002, 41, 3192–3194. [Google Scholar]
- Rae, R.L.; Żurek, J.M.; Paterson, M.J.; Bebbington, M.W.P. Halogenation effects in intramolecular furan Diels–Alder reactions: Broad scope synthetic and computational studies. Org. Biomol. Chem. 2013, 11, 7946–7952. [Google Scholar]
- Fleming, I. Molecular Orbitals and Organic Chemical Reactions; Wiley: Chichester, UK, 2010. [Google Scholar]
- Houk, K.N. The frontier molecular orbital theory of cycloaddition reaction. Acc. Chem. Res. 1975, 8, 361–369. [Google Scholar]
- Sauer, J.; Sustmann, R. Mechanistic aspects of Diels–Alder reactions: A critical survey. Angew. Chem. Int. Ed. Engl. 1980, 19, 779–807. [Google Scholar]
- Brown, F.K.; Houk, K.N.; Burnell, D.J.; Valenta, Z. Modeling Control of Facial Stereoselectivity. Diels–Alder Cycloadditions of Unsymmetrically Substituted Cyclopentadienes. J. Org. Chem. 1987, 52, 3050–3059. [Google Scholar]
- Robiette, R.; Marchand-Brynaert, J.; Peeters, D. Influence of Diene Substitution on Transition State Stabilization in Diels–Alder Reaction. J. Org. Chem. 2002, 67, 6823–6826. [Google Scholar]
- James, N.C.; Um, J.M.; Padias, A.B.; Hall, H.K., Jr.; Houk, K.N. Computational investigation of the competition between the concerted Diels–Alder reaction and formation of diradicals in reactions of acrylonitrile with non-polar dienes. J. Org. Chem. 2013, 78, 6582–6592. [Google Scholar]
- Crawford, K.R.; Bur, S.K.; Straub, C.S.; Padwa, A. Intramolecular Cyclization Reactions of 5-Halo and 5-Nitro-Substituted Furans. Org. Lett. 2003, 5, 3337–3340. [Google Scholar]
- Padwa, A.; Crawford, K.R.; Straub, C.S.; Pieniazek, S.N.; Houk, K.N. Halo Substituent Effects on Intramolecular Cycloadditions involving Furanyl Amides. J. Org. Chem. 2006, 71, 5432–5439. [Google Scholar]
- Pienazek, S.N.; Houk, K.N. The Origin of the Halogen Effect on Reactivity and Reversibility of Diels–Alder Cycloadditions involving Furan. Angew. Chem. Int. Ed. 2006, 45, 1442–1445. [Google Scholar]
- Nakamura, N.; Takahasi, I.; Yamada, S.; Dobashi, Y.; Kitagawa, O. Intramolecular Diels–Alder reaction of N-allyl 2-furoyl amides: Effect of strain and amide rotational isomerisation. Tetrahedron Lett. 2011, 52, 53–55. [Google Scholar]
- Tantillo, D.J.; Houk, K.N.; Jung, M.E. Origins of Stereoselectivity in Intramolecular cycloadditions of dienes and dienophiles linked by ester and amide tethers. J. Org. Chem. 2001, 66, 1938–1940. [Google Scholar]
- Williams, A. Free Energy Relationships in Organic and Bioorganic Chemistry; RSC Publishing: Cambridge, UK, 2003. [Google Scholar]
- Huisgen, R.; Schug, R. New Mechanistic Criterion for Early and Late Transition States. J. Am. Chem. Soc. 1976, 98, 7819–7821. [Google Scholar]
- Gajewski, J.J.; Peterson, K.B.; Kagel, J.R. Transition State Structure Variation in the Diels–Alder reaction from Secondary Deuterium Kinetic Isotope effects: The reaction of a nearly symmetrical diene and dienophile is nearly synchronous. J. Am. Chem. Soc. 1987, 109, 5545–5546. [Google Scholar]
- Frisch, M.J.T.G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Nyden, M.R.; Petersson, G.A. Complete Basis Set Correlation Energies 1. The Asymptotic Convergence of Pair Natural Orbital Expansions. J. Chem. Phys. 1981, 75, 1843–1862. [Google Scholar]
- Ochterski, J.W.; Petersson, G.A.; Montgomery, J.A. A complete basis set model chemistry 5. Extensions to six or more heavy atoms. J. Chem. Phys. 1996, 104, 2598–2619. [Google Scholar]
- Montgomery, J.A.; Frisch, M.J.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J. Chem. Phys. 1999, 110, 2822–2827. [Google Scholar]
- Montgomery, J.A.; Frisch, M.J.; Ochterski, J.W.; Petersson, G.A. A complete basis set model chemistry. VII. Use of the minimum population localization method. J. Chem. Phys. 2000, 112, 6532–6542. [Google Scholar]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żurek, J.M.; Rae, R.L.; Paterson, M.J.; Bebbington, M.W.P. Unusual Structure-Energy Correlations in Intramolecular Diels–Alder Reaction Transition States. Molecules 2014, 19, 15535-15545. https://doi.org/10.3390/molecules191015535
Żurek JM, Rae RL, Paterson MJ, Bebbington MWP. Unusual Structure-Energy Correlations in Intramolecular Diels–Alder Reaction Transition States. Molecules. 2014; 19(10):15535-15545. https://doi.org/10.3390/molecules191015535
Chicago/Turabian StyleŻurek, Justyna M., Robert L. Rae, Martin J. Paterson, and Magnus W. P. Bebbington. 2014. "Unusual Structure-Energy Correlations in Intramolecular Diels–Alder Reaction Transition States" Molecules 19, no. 10: 15535-15545. https://doi.org/10.3390/molecules191015535