Identification and Characterization of Amlexanox as a G Protein-Coupled Receptor Kinase 5 Inhibitor

Abstract
:1. Introduction
2. Results and Discussion
2.1. High-Throughput Screening

2.2. Biochemical Validation
2.3. Cellular Activity


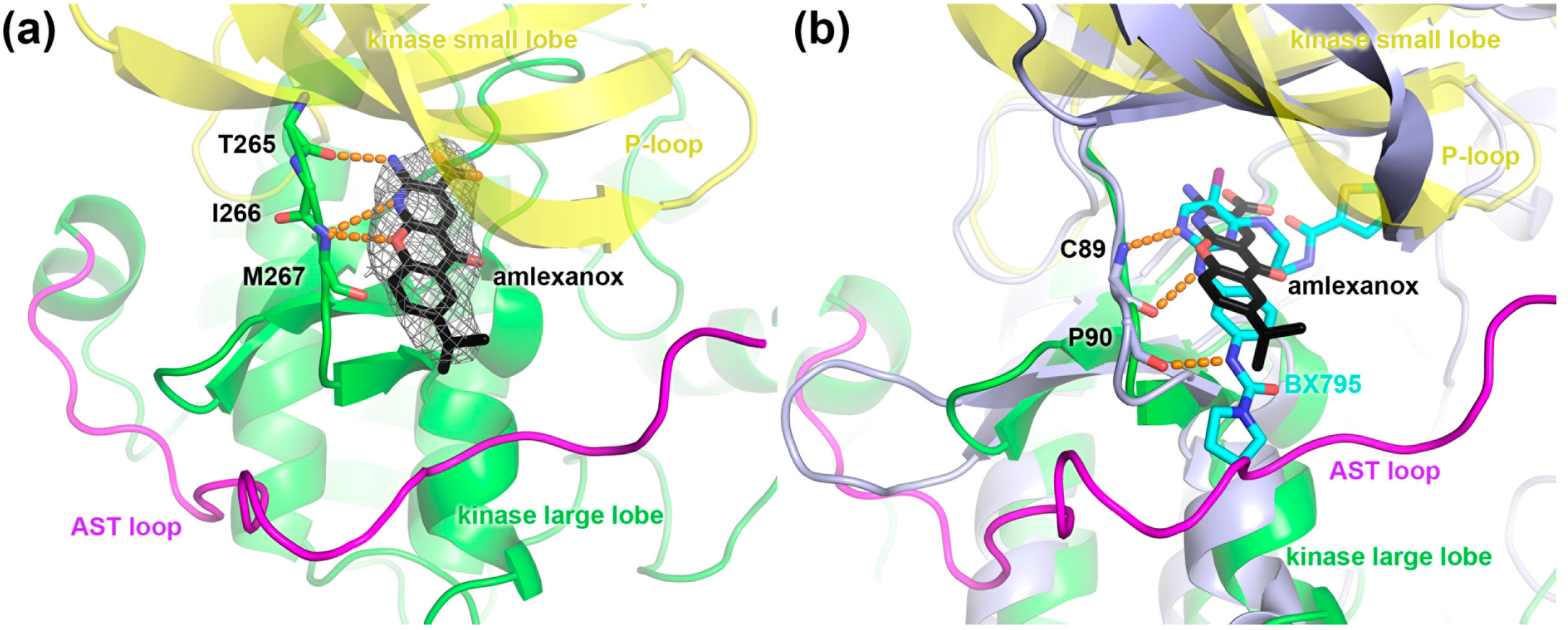
2.4. Crystal Structure of the GRK1 Amlexanox Complex

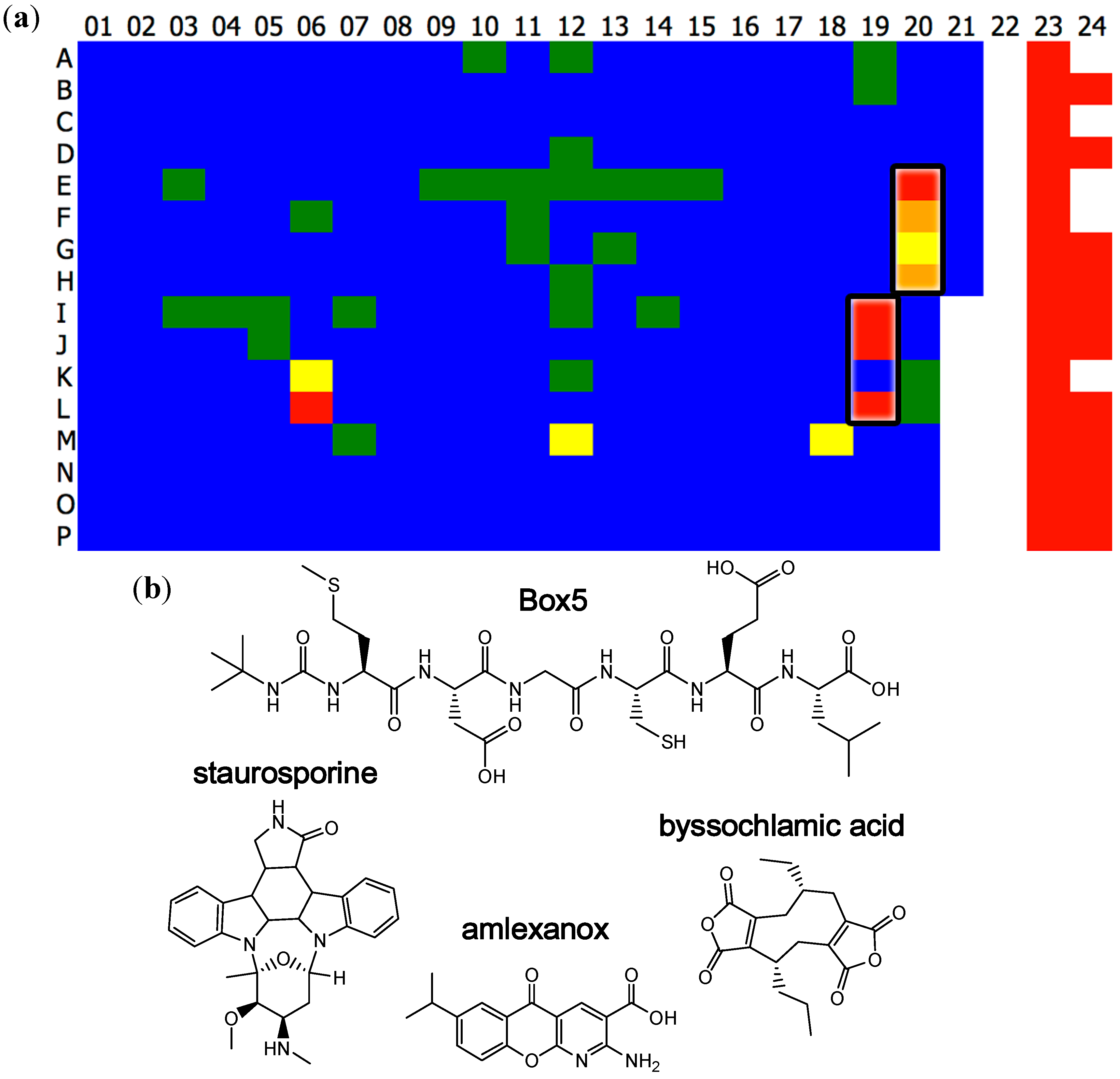{kind=link}
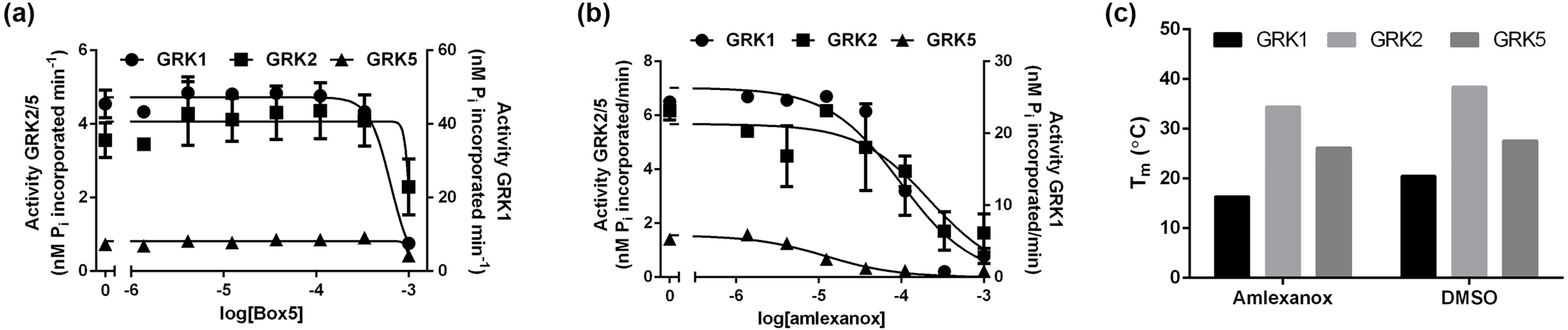{kind=link}
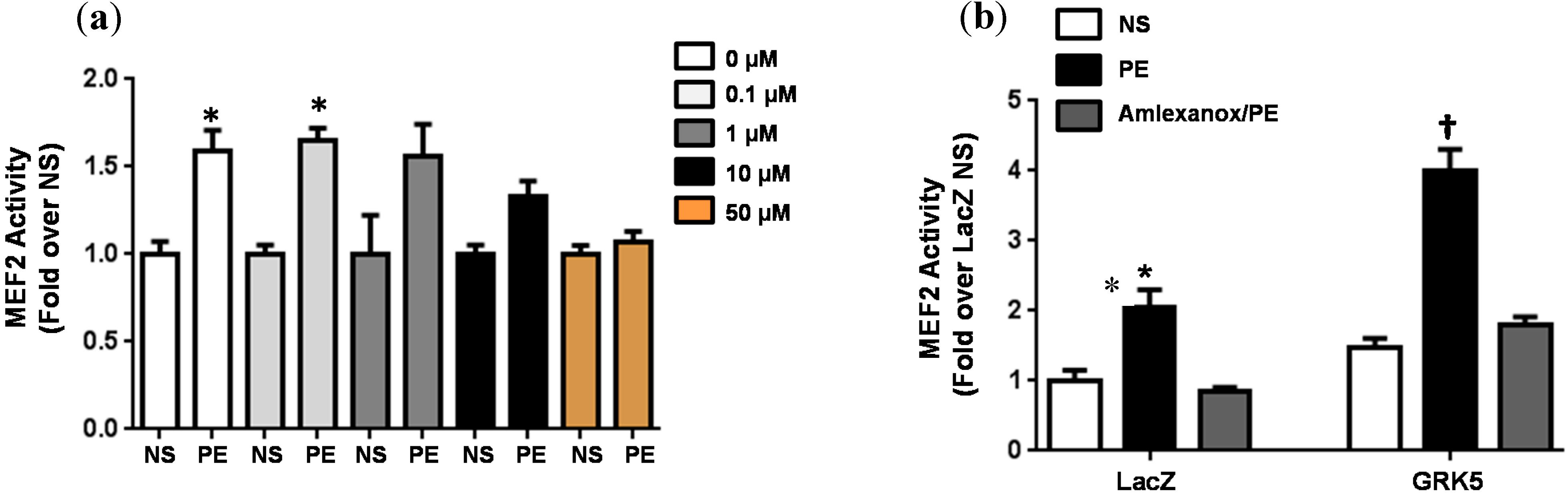{kind=link}
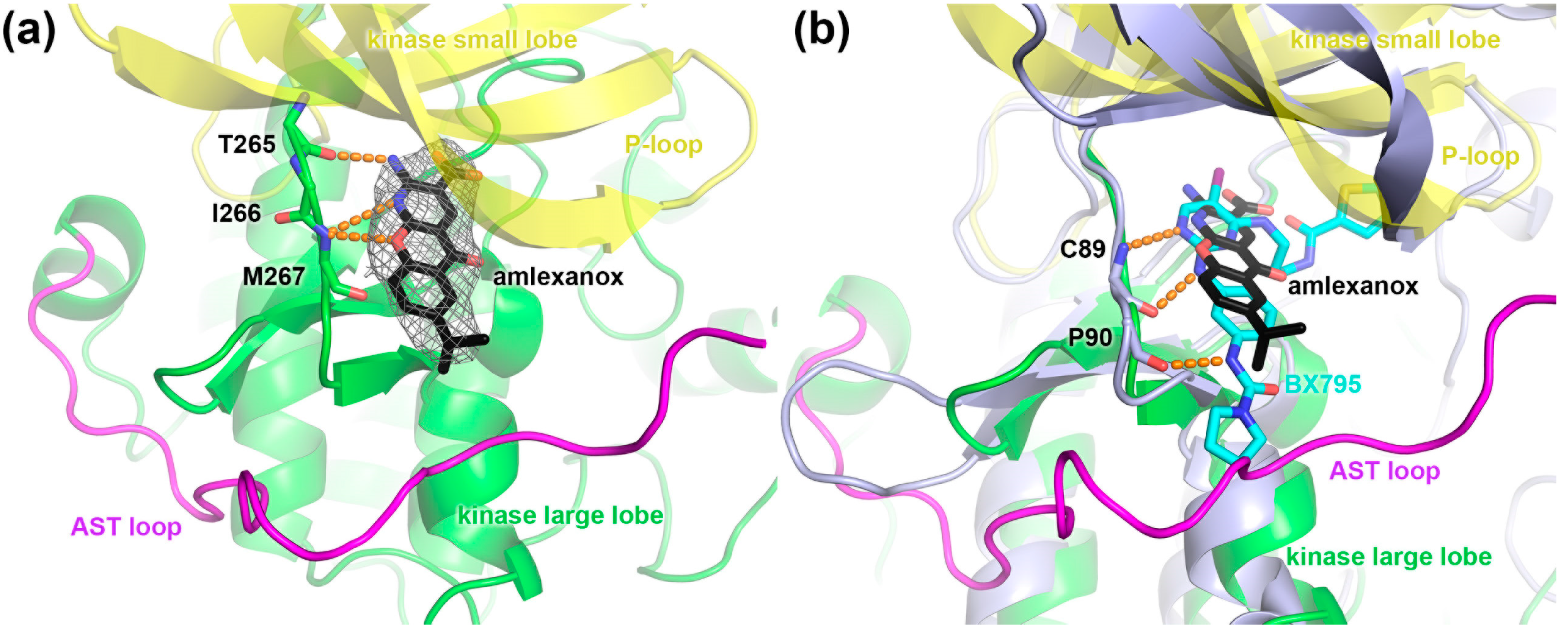{kind=link}
| Protein Complex | GRK1·Amlexanox |
|---|---|
| X-ray source | APS 21-ID-E |
| Wavelength (Å) | 0.9787 |
| Dmin (Å) | 2.82 (2.87–2.82) * |
| Space group | P 21 21 21 |
| Cell constants (Å) | a = 118.1 b = 119.2 c = 174.3 |
| Unique reflections | 60016 (2932) |
| Rmerge (%) | 9.7% (100%) |
| Completeness (%) | 100% (99.9%) |
| <I>/<σI> | 19.5 (1.4) |
| Redundancy | 7.4 (7.1) |
| Refinement resolution (Å) | 25–2.82 (2.88–2.82) |
| Total reflections used | 56928 (3020) |
| RMSD bond lengths (Å) | 0.005 |
| RMSD bond angles (°) | 0.919 |
| Est. coordinate error (Å) | 0.348 |
| Ramachandran plot outliers (%) | 3 (0.15%) |
| Rwork | 24.1 (40.6) |
| Rfree | 26.7 (44.4) |
| Protein atoms | 15786 |
| Water molecules | 54 |
| Inhibitor atoms | 88 |
| Average B-factor (Å2) | 45.1 |
| Protein | 45.5 |
| Inhibitor | 36.2 |
| MolProbity score | 1.27 |
| MolProbity Cβ deviations | 0 |
| MolProbity bad backbone bonds | 0 |
| MolProbity bad backbone angles | 1 |
| PDB Entry | 4WBO |
3. Experimental Section
3.1. Protein Purification
3.2. DSF Screen
3.3. Kinetic Assays
3.5. Luciferase Assay
3.6. GRK1·Paroxetine Crystal Structure Determination
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reiter, E.; Ahn, S.; Shukla, A.K.; Lefkowitz, R.J. Molecular mechanism of beta-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 179–197. [Google Scholar] [CrossRef] [PubMed]
- Homan, K.T.; Tesmer, J.J. Structural insights into G protein-coupled receptor kinase function. Curr. Opin. Cell Biol. 2014, 27, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Eckhart, A.D.; Ozaki, T.; Tevaearai, H.; Rockman, H.A.; Koch, W.J. Vascular-targeted overexpression of G protein-coupled receptor kinase-2 in transgenic mice attenuates β-adrenergic receptor signaling and increases resting blood pressure. Mol. Pharmacol. 2002, 61, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Liggett, S.B.; Cresci, S.; Kelly, R.J.; Syed, F.M.; Matkovich, S.J.; Hahn, H.S.; Diwan, A.; Martini, J.S.; Sparks, L.; Parekh, R.R.; et al. A GRK5 polymorphism that inhibits β-adrenergic receptor signaling is protective in heart failure. Nat. Med. 2008, 14, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Anis, Y.; Leshem, O.; Reuveni, H.; Wexler, I.; Ben Sasson, R.; Yahalom, B.; Laster, M.; Raz, I.; Ben Sasson, S.; Shafrir, E.; et al. Antidiabetic effect of novel modulating peptides of G-protein-coupled kinase in experimental models of diabetes. Diabetologia 2004, 47, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Krilov, L.; Nguyen, A.; Miyazaki, T.; Unson, C.G.; Williams, R.; Lee, N.H.; Ceryak, S.; Bouscarel, B. Dual mode of glucagon receptor internalization: Role of PKCalpha, GRKs and beta-arrestins. Exp. Cell Res. 2011, 317, 2981–2994. [Google Scholar] [CrossRef] [PubMed]
- Iino, M.; Furugori, T.; Mori, T.; Moriyama, S.; Fukuzawa, A.; Shibano, T. Rational design and evaluation of new lead compound structures for selective betaARK1 inhibitors. J. Med. Chem. 2002, 45, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Kassack, M.U.; Hogger, P.; Gschwend, D.A.; Kameyama, K.; Haga, T.; Graul, R.C.; Sadee, W. Molecular modeling of G-protein coupled receptor kinase 2: Docking and biochemical evaluation of inhibitors. AAPS Pharmsci. 2000, 2, 9–21. [Google Scholar] [CrossRef]
- Homan, K.T.; Larimore, K.; Elkins, J.M.; Szklarz, M.; Knapp, S.; Tesmer, J.J.G. Identification and structure-function analysis of sub-family selective G protein-coupled receptor kinase inhibitors. ACS Chem. Biol. 2014, in press. [Google Scholar]
- Homan, K.T.; Wu, E.; Wilson, M.W.; Singh, P.; Larsen, S.D.; Tesmer, J.J. Structural and functional analysis of g protein-coupled receptor kinase inhibition by paroxetine and a rationally designed analog. Mol. Pharmacol. 2014, 85, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.M.; Homan, K.T.; Chen, J.; Wu, E.K.; Hinkle, P.M.; Huang, Z.M.; Chuprun, J.K.; Song, J.; Gao, E.; Cheung, J.Y.; et al. Paroxetine is a direct inhibitor of G protein-coupled receptor kinase 2 and increases myocardial contractility. ACS Chem. Biol. 2012, 7, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Gold, J.I.; Gao, E.; Shang, X.; Premont, R.T.; Koch, W.J. Determining the absolute requirement of G protein-coupled receptor kinase 5 for pathological cardiac hypertrophy: Short communication. Circ. Res. 2012, 111, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Gold, J.I.; Martini, J.S.; Hullmann, J.; Gao, E.; Chuprun, J.K.; Lee, L.; Tilley, D.G.; Rabinowitz, J.E.; Bossuyt, J.; Bers, D.M.; et al. Nuclear translocation of cardiac G protein-Coupled Receptor kinase 5 downstream of select Gq-activating hypertrophic ligands is a calmodulin-dependent process. PLoS One 2013, 8, e57324. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Lee, B.H.; Jung, H.; Yun, C.S.; Ha, J.D.; Kim, H.R.; Chae, C.H.; Lee, J.H.; Seo, H.W.; Oh, K.S. Design and synthesis of novel 3-(benzo[d]oxazol-2-yl)-5-(1-(piperidin-4-yl)-1H-pyrazol-4-yl)pyridin-2-amine derivatives as selective G-protein-coupled receptor kinase-2 and -5 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 6711–6716. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Chiang, S.H.; Decker, S.J.; Chang, L.; Uhm, M.; Larsen, M.J.; Rubin, J.R.; Mowers, J.; White, N.M.; Hochberg, I.; et al. An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat. Med. 2013, 19, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Tamaoki, T.; Nomoto, H.; Takahashi, I.; Kato, Y.; Morimoto, M.; Tomita, F. Staurosporine, a potent inhibitor of phospholipid/Ca++dependent protein kinase. Biochem. Biophys. Res. Commun. 1986, 135, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Jenei, V.; Sherwood, V.; Howlin, J.; Linnskog, R.; Safholm, A.; Axelsson, L.; Andersson, T. A t-butyloxycarbonyl-modified Wnt5a-derived hexapeptide functions as a potent antagonist of Wnt5a-dependent melanoma cell invasion. Proc. Natl. Acad. Sci. USA 2009, 106, 19473–19478. [Google Scholar] [CrossRef] [PubMed]
- McGovern, S.L.; Caselli, E.; Grigorieff, N.; Shoichet, B.K. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002, 45, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006, 49, 7274–7277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Matkovich, S.J.; Duan, X.; Gold, J.I.; Koch, W.J.; Dorn, G.W. Nuclear effects of G-protein receptor kinase 5 on histone deacetylase 5-regulated gene transcription in heart failure. Circ.: Heart Fail. 2011, 4, 659–668. [Google Scholar] [CrossRef]
- McKinsey, T.A.; Zhang, C.L.; Lu, J.; Olson, E.N. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000, 408, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.; Berendsen, H.J. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins 1998, 30, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.; Kitao, A.; Berendsen, H.J. Model-free methods of analyzing domain motions in proteins from simulation: A comparison of normal mode analysis and molecular dynamics simulation of lysozyme. Proteins 1997, 27, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Tesmer, J.J.; Tesmer, V.M.; Lodowski, D.T.; Steinhagen, H.; Huber, J. Structure of human G protein-coupled receptor kinase 2 in complex with the kinase inhibitor balanol. J. Med. Chem. 2010, 53, 1867–1870. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.M.; Yeow, R.Y.; Schoenau, C.; Huber, J.; Tesmer, J.J. Molecular mechanism of selectivity among G protein-coupled receptor kinase 2 inhibitors. Mol. Pharmacol. 2011, 80, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Boguth, C.A.; Singh, P.; Huang, C.C.; Tesmer, J.J. Molecular basis for activation of G protein-coupled receptor kinases. EMBO J. 2010, 29, 3249–3259. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Helgason, E.; Phung, Q.T.; Quan, C.L.; Iyer, R.S.; Lee, M.W.; Bowman, K.K.; Starovasnik, M.A.; Dueber, E.C. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl. Acad. Sci. USA 2012, 109, 9378–9383. [Google Scholar] [CrossRef] [PubMed]
- Papermaster, D.S. Preparation of retinal rod outer segments. Methods Enzymol. 1982, 81, 48–52. [Google Scholar] [PubMed]
- Lodowski, D.T.; Tesmer, V.M.; Benovic, J.L.; Tesmer, J.J. The structure of G protein-coupled receptor kinase (GRK)-6 defines a second lineage of GRKs. J. Biol. Chem. 2006, 281, 16785–16793. [Google Scholar] [CrossRef] [PubMed]
- Brinks, H.; Boucher, M.; Gao, E.; Chuprun, J.K.; Pesant, S.; Raake, P.W.; Huang, Z.M.; Wang, X.; Qiu, G.; Gumpert, A.; et al. Level of G protein-coupled receptor kinase-2 determines myocardial ischemia/reperfusion injury via pro- and anti-apoptotic mechanisms. Circ. Res. 2010, 107, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Martini, J.S.; Raake, P.; Vinge, L.E.; DeGeorge, B.R., Jr.; Chuprun, J.K.; Harris, D.M.; Gao, E.; Eckhart, A.D.; Pitcher, J.A.; Koch, W.J. Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 12457–12462. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997, 276, 307–326. [Google Scholar]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Sample Availability: Amlexanox, Box5, staurosporine are commercially available from Toronto Research Chemicals, CalBiochem, and Sigma, respectively. (R)-(−)-phenylephrine hydrochloride (PE) was purchased from Sigma Aldrich.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Homan, K.T.; Wu, E.; Cannavo, A.; Koch, W.J.; Tesmer, J.J.G. Identification and Characterization of Amlexanox as a G Protein-Coupled Receptor Kinase 5 Inhibitor. Molecules 2014, 19, 16937-16949. https://doi.org/10.3390/molecules191016937
Homan KT, Wu E, Cannavo A, Koch WJ, Tesmer JJG. Identification and Characterization of Amlexanox as a G Protein-Coupled Receptor Kinase 5 Inhibitor. Molecules. 2014; 19(10):16937-16949. https://doi.org/10.3390/molecules191016937
Chicago/Turabian StyleHoman, Kristoff T., Emily Wu, Alessandro Cannavo, Walter J. Koch, and John J. G. Tesmer. 2014. "Identification and Characterization of Amlexanox as a G Protein-Coupled Receptor Kinase 5 Inhibitor" Molecules 19, no. 10: 16937-16949. https://doi.org/10.3390/molecules191016937
APA StyleHoman, K. T., Wu, E., Cannavo, A., Koch, W. J., & Tesmer, J. J. G. (2014). Identification and Characterization of Amlexanox as a G Protein-Coupled Receptor Kinase 5 Inhibitor. Molecules, 19(10), 16937-16949. https://doi.org/10.3390/molecules191016937