Synthesis and Biological Evaluation of New Pleuromutilin Derivatives as Antibacterial Agents

Abstract
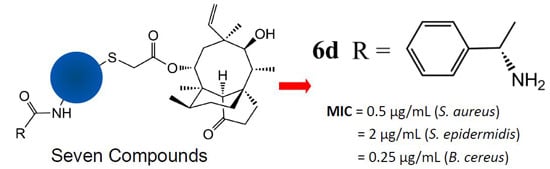
:
1. Introduction

2. Results and Discussion
2.1. Chemistry

2.2. Antibacterial Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | MIC (μg/mL) | |||||
|---|---|---|---|---|---|---|
| MRSA | MRSE | S. aureus | S. epidermidis | E. coli | B. cereus | |
| 5a | 32 | 64 | 32 | 32 | ≥128 | 16 |
| 5b | 64 | ≥128 | 64 | 64 | ≥128 | 32 |
| 5c | 32 | 32 | 16 | 32 | 64 | 4 |
| 6a | 8 | 16 | 4 | 16 | 32 | 2 |
| 6b | 32 | 32 | 32 | 32 | 64 | 16 |
| 6c | 8 | 32 | 8 | 16 | 64 | 8 |
| 6d | 0.5 | 4 | 0.5 | 2 | 32 | 0.25 |
| Tiamulin | 0.5 | 2 | 0.5 | 2 | 16 | 0.25 |
| Comp. | MRSA | MRSE | S. aureus | S. epidermidis | E. col | B. cereus | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 320 | 160 | 320 | 160 | 320 | 160 | 320 | 160 | 320 | 160 | 320 | 160 | |
| 5a | 16.51 | 13.22 | 16.40 | 12.93 | 16.23 | 13.39 | 16.46 | 13.82 | 13.16 | 11.65 | 17.08 | 15.10 |
| 5b | 14.03 | 11.67 | 13.88 | 12.34 | 13.42 | 11.14 | 14.02 | 12.53 | 12.97 | 11.13 | 15.51 | 13.55 |
| 5c | 16.47 | 13.14 | 16.85 | 13.46 | 17.35 | 13.89 | 17.05 | 13.66 | 12.23 | 10.83 | 18.73 | 15.26 |
| 6a | 17.51 | 14.49 | 17.35 | 13.89 | 18.82 | 14.75 | 18.31 | 14.32 | 14.22 | 12.17 | 19.33 | 16.51 |
| 6b | 16.05 | 12.84 | 15.69 | 12.30 | 15.93 | 12.04 | 16.17 | 12.12 | 12.59 | 10.86 | 17.42 | 14.05 |
| 6c | 17.11 | 14.28 | 15.13 | 12.21 | 17.44 | 14.39 | 16.83 | 13.25 | 14.85 | 12.67 | 18.25 | 13.91 |
| 6d | 19.46 | 17.62 | 17.28 | 15.03 | 22.67 | 18.82 | 19.15 | 16.73 | 16.22 | 13.61 | 23.84 | 19.39 |
| Tiamulin | 20.35 | 17.84 | 17.93 | 15.75 | 22.23 | 19.04 | 20.58 | 16.05 | 17.84 | 15.29 | 23.18 | 20.57 |
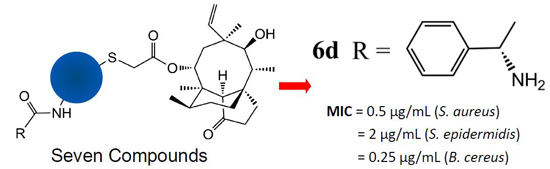
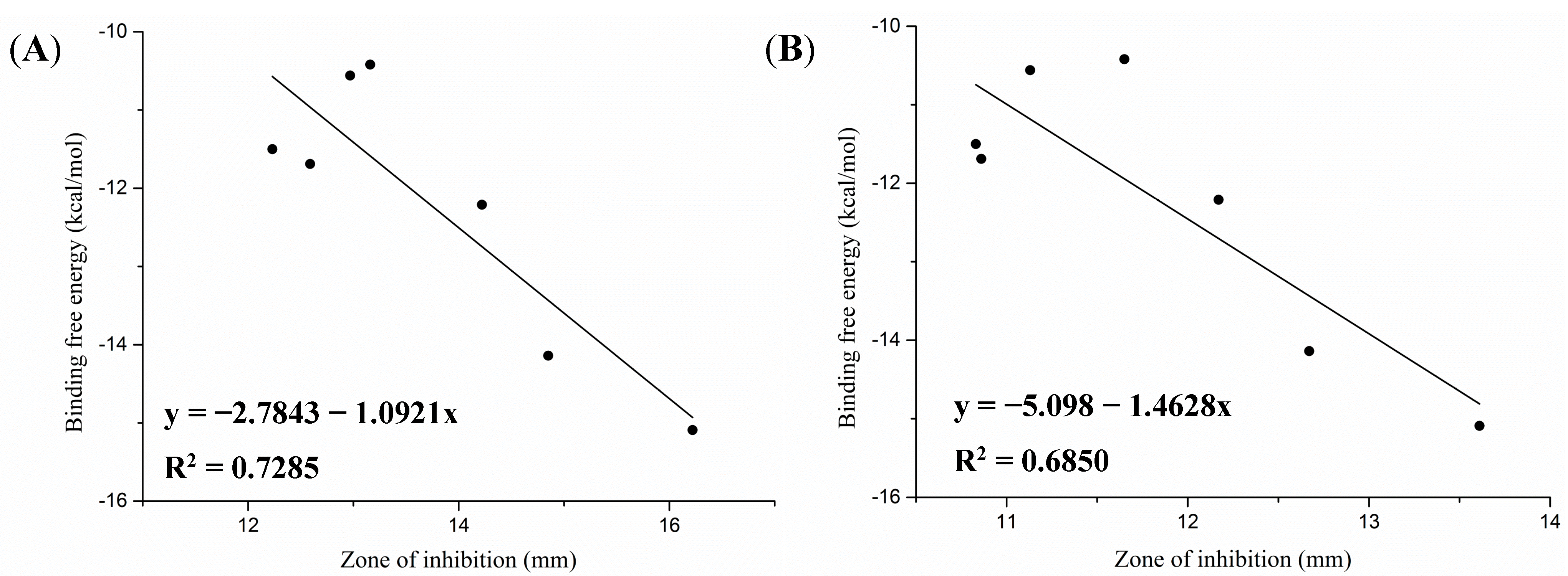
2.3. Molecular Docking Study

| Compound | ΔGb (kcal/mol) | Noncovalent Molecular Interaction | RMSD(Å) | |||
|---|---|---|---|---|---|---|
| Hydro I Interaction | Atom of Compound | Residue | Distance (Å) | |||
| 5a | −10.42 | H-bonding | OH (8-membered ring) | G-2484 | 2.0 | 1.21 |
| H-bonding | N (thiadiazole) | G-2044 | 2.4 | |||
| H-bonding | N (thiadiazole) | G-2044 | 2.7 | |||
| 5b | −10.56 | H-bonding | OH (8-membered ring) | G-2484 | 2.0 | 1.23 |
| H-bonding | N (thiadiazole) | C-2044 | 2.1 | |||
| H-bonding | N (thiadiazole) | G-2044 | 2.2 | |||
| 5c | −11.50 | H-bonding | OH (8-membered ring) | G-2484 | 2.1 | 1.08 |
| H-bonding | C=O (ester) | G-2044 | 2.7 | |||
| H-bonding | C=O (ester) | G-2044 | 2.8 | |||
| 6a | −12.21 | H-bonding | OH (8-membered ring) | G-2484 | 1.9 | 1.00 |
| H-bonding | C=O (ester) | G-2044 | 2.5 | |||
| H-bonding | C=O (ester) | G-2044 | 2.5 | |||
| H-bonding | NH2 (terminal) | C-2565 | 2.1 | |||
| H-bonding | NH2 (terminal) | C-2565 | 2.7 | |||
| 6b | −11.69 | H-bonding | OH (8-membered ring) | G-2484 | 1.9 | 1.00 |
| H-bonding | C=O (ester) | G-2044 | 2.5 | |||
| H-bonding | C=O (ester) | G-2044 | 2.5 | |||
| H-bonding | N (thiadiazole) | G-2044 | 2.1 | |||
| H-bonding | N (thiadiazole) | G-2044 | 2.3 | |||
| H-bonding | NH2 (terminal) | C-2565 | 2.6 | |||
| 6c | −14.14 | H-bonding | OH (8-membered ring) | G-2484 | 2.1 | 1.02 |
| H-bonding | C=O (ester) | G-2044 | 2.3 | |||
| H-bonding | C=O (ester) | G-2044 | 2.3 | |||
| H-bonding | N (thiadiazole) | G-2044 | 2.3 | |||
| H-bonding | NH2 (phenylglycinamide) | C-2565 | 1.6 | |||
| 6d | −15.09 | H-bonding | OH (8-membered ring) | G-2484 | 2.1 | 1.02 |
| H-bonding | C=O (ester) | G-2044 | 2.3 | |||
| H-bonding | C=O (ester) | G-2044 | 2.3 | |||
| H-bonding | N (thiadiazole) | G-2044 | 2.2 | |||
| H-bonding | NH2 (phenylglycinamide) | C-2565 | 2.3 | |||
| Cation–π interaction | N (pyrrolidine) | G-2045 | 3.7 | |||


2.4. Prediction of ADMET Properties
| Comp. | ADMET Parameter | pKa | c Log p | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Peff (cm/s) a | Log BB b | Log PS c | PPB (%) d | Vd (L/kg) e | LD50 (mg/kg) f | Acid | Base | ||
| 5a | 6.39 × 10−4 | 0.29 | −1.7 | 99.27 | 4.15 | 1400 | 10.40 | - | 6.29 |
| 5b | 6.39 × 10−4 | 0.29 | −1.7 | 99.27 | 4.15 | 1400 | 10.40 | - | 6.29 |
| 5c | 6.39 × 10−4 | 0.29 | −1.7 | 99.27 | 4.15 | 1400 | 10.40 | - | 6.29 |
| 6a | 5.12 × 10−4 | 0.02 | −2.3 | 97.14 | 3.15 | 730 | 10.30 | 7.70 | 3.88 |
| 6b | 5.12 × 10−4 | 0.02 | −2.3 | 97.14 | 3.15 | 730 | 10.30 | 7.70 | 3.88 |
| 6c | 5.41 × 10−4 | 0.2 | −2.4 | 98.44 | 1.60 | 970 | 7.80 | 11.80 | 4.64 |
| 6d | 5.41 × 10−4 | 0.2 | −2.4 | 98.44 | 1.60 | 970 | 7.80 | 11.80 | 4.64 |
3. Experimental Section
3.1. General
3.2. Synthesis
3.2.1. General Procedure for the Synthesis of Compounds 5a–c
3.2.2. General Procedure for the Synthesis of Compounds 6a–d
3.3. Antibacterial Activity
3.3.1. MIC Determination
3.3.2. Oxford Cup Assay
3.4. Molecular Modeling Studies
3.5. ADMET Prediction
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kavanagh, F.; Hervey, A.; Robbins, W.J. Antibiotic Substances From Basidiomycetes: VIII. Pleurotus Multilus (Fr.) Sacc. and Pleurotus Passeckerianus Pilat. Proc. Natl. Acad. Sci. USA 1951, 37, 570–574. [Google Scholar]
- Arigoni, D. Structure of a new type of terpene. Gazz. Chem. Ital. 1962, 92, 884–901. [Google Scholar]
- Birch, A.J.; Holzapfel, C.W.; Richards, R.W. Diterpenoid nature of pleuromutilin. Chem. Ind. 1963, 14, 374–375. [Google Scholar]
- Shang, R.F.; Liu, Y.; Xin, Z.J.; Guo, W.Z.; Guo, Z.T.; Hao, B.C.; Liang, J.P. Synthesis and antibacterial evaluation of novel pleuromutilin derivatives. Eur. J. Med. Chem. 2013, 63, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Z.; Liu, Y.H.; Chen, J.X. Pleuromutilin and its derivatives—The lead compounds for novel antibiotics. Mini Rev. Med. Chem. 2012, 12, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Moody, M.N.; Morrison, L.K.; Tyring, S.K. Retapamulin: What is the role of this topical antimicrobial in the treatment of bacterial infections in atopic dermatitis? Skin Ther. Lett. 2010, 15, 1–4. [Google Scholar]
- Sader, H.S.; Paukner, S.; Ivezic-Schoenfeld, Z.; Biedenbach, D.J.; Schmitz, F.J.; Jones, R.N. Antimicrobial activity of the novel pleuromutilin antibiotic BC-3781 against organisms responsible for community-acquired respiratory tract infections (CARTIs). J. Antimicrob. Chemother. 2012, 67, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.F.; Wang, J.T.; Guo, W.Z.; Liang, J.P. Efficient antibacterial agents: A review of the synthesis, biological evaluation and mechanism of pleuromutilin derivatives. Curr. Top. Med. Chem. 2013, 13, 3013–3025. [Google Scholar] [CrossRef] [PubMed]
- Novak, R.; Shlaes, D.M. The pleuromutilin antibiotics: A new class for human use. Curr. Opin. Invest. Drugs 2010, 11, 182–191. [Google Scholar]
- Schlunzen, F.; Pyetan, E.; Fucini, P.; Yonath, A.; Harms, J.M. Inhibition of peptide bond formation by pleuromutilins: The structure of the 50S ribosomal subunit from Deinococcus radiodurans in complex with tiamulin. Mol. Microbiol. 2004, 54, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Davidovich, C.; Bashan, A.; Auerbach-Nevo, T.; Yaggie, R.D.; Gontarek, R.R.; Yonath, A. Induced-fit tightens pleuromutilins, binding to ribosomes and remote interactions enable their selectivity. Proc. Natl. Acad. Sci. USA 2007, 104, 4291–4296. [Google Scholar] [CrossRef] [PubMed]
- Long, K.S.; Hansen, L.H.; Jakobsen, L.; Vester, B. Interaction of pleuromutilin derivatives with the ribosomal peptidyl transferase center. Antimicrob. Agent. Chemother. 2006, 50, 1458–1462. [Google Scholar] [CrossRef]
- Popiolek, L.; Kosikowska, U.; Mazur, L.; Dobosz, M.; Malm, A. Synthesis and antimicrobial evaluation of some novel 1,2,4-triazole and 1,3,4-thiadiazole derivatives. Med. Chem. Res. 2013, 22, 3134–3147. [Google Scholar] [CrossRef] [PubMed]
- Aliabadi, A.; Eghbalian, E.; Kiani, A. Synthesis and evaluation of the cytotoxicity of a series of 1,3,4-thiadiazole based compounds as anticancer agents. Iran J. Basic Med. Sci. 2013, 16, 1133–1138. [Google Scholar] [PubMed]
- Chandrakantha, B.; Isloor, A.M.; Shetty, P.; Fun, H.K.; Hegde, G. Synthesis and biological evaluation of novel substituted 1,3,4-thiadiazole and 2,6-di aryl substituted imidazo [2,1-b] [1,3,4] thiadiazole derivatives. Eur. J. Med. Chem. 2014, 71, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.F.; Pu, X.Y.; Xu, X.M.; Xin, Z.J.; Zhang, C.; Guo, W.Z.; Liu, Y.; Liang, J.P. Synthesis and Biological Activities of Novel Pleuromutilin Derivatives with a Substituted Thiadiazole Moiety as Potent Drug-Resistant Bacteria Inhibitors. J. Med. Chem. 2014, 57, 5664–5678. [Google Scholar] [CrossRef] [PubMed]
- Marialke, J.; Tietze, S.; Apostolakis, J. Similarity based docking. J. Chem. Inf. Model. 2008, 48, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Vyas, V.K.; Gupta, N.; Ghate, M.; Patel, S. Design, synthesis, pharmacological evaluation and in silico ADMET prediction of novel substituted benzimidazole derivatives as angiotensin II–AT1 receptor antagonists based on predictive 3D QSAR models. SAR QSAR Environ. Res. 2014, 25, 117–146. [Google Scholar] [CrossRef] [PubMed]
- Lanevskij, K.; Japertasa, P.; Didziapetrisa, R.; Petrauskasa, A. Ionization-specific QSAR models of blood-brain penetration of drugs. Chem. Biodivers. 2009, 6, 2050–2054. [Google Scholar] [CrossRef] [PubMed]
- Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Marcus, D.H.; Donald, E.C.; David, C.L.; Tim, V.; Eva, Z.; Geoffrey, R.H. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Stierand, K.; Maaß, P.; Rarey, M. Molecular complexes at a glance: Automated generation of two-dimensional complex diagrams. Bioinformatics 2006, 22, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Delano, W.L.; Ultsch, M.H.; De Vos, A.M.; Wells, J.A. Convergent solutions to binding at a protein-protein interface. Science 2000, 287, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.J.; Xu, X.J. ADME evaluation in drug discovery. 3. Modeling blood-brain barrier partitioning using simple molecular descriptors. J. Chem. Inf. Comput. Sci. 2003, 43, 2137–2152. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, N.; Mladenovic, M.; Mihailovic, M.; Arambasic, J.; Uskokovic, A.; Stankovic, V.; Mihailovic, V.; Katanic, J.; Matic, S.; Solujic, S.; et al. Synthesis and toxicological studies of in vivo anticoagulant activity of novel 3-(1-aminoethylidene)chroman-2,4-diones and 4-hydroxy-3-(1-iminoethyl)-2H-chromen-2-ones combined with a structure-based 3-D pharmacophore model. Eur. J. Pharm. Sci. 2014, 55, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 2–4, 5a–c and 6a–d are available from the corresponding author.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shang, R.-F.; Wang, G.-H.; Xu, X.-M.; Liu, S.-J.; Zhang, C.; Yi, Y.-P.; Liang, J.-P.; Liu, Y. Synthesis and Biological Evaluation of New Pleuromutilin Derivatives as Antibacterial Agents. Molecules 2014, 19, 19050-19065. https://doi.org/10.3390/molecules191119050
Shang R-F, Wang G-H, Xu X-M, Liu S-J, Zhang C, Yi Y-P, Liang J-P, Liu Y. Synthesis and Biological Evaluation of New Pleuromutilin Derivatives as Antibacterial Agents. Molecules. 2014; 19(11):19050-19065. https://doi.org/10.3390/molecules191119050
Chicago/Turabian StyleShang, Ruo-Feng, Guan-Hua Wang, Xi-Ming Xu, Si-Jie Liu, Chao Zhang, Yun-Peng Yi, Jian-Ping Liang, and Yu Liu. 2014. "Synthesis and Biological Evaluation of New Pleuromutilin Derivatives as Antibacterial Agents" Molecules 19, no. 11: 19050-19065. https://doi.org/10.3390/molecules191119050
APA StyleShang, R.-F., Wang, G.-H., Xu, X.-M., Liu, S.-J., Zhang, C., Yi, Y.-P., Liang, J.-P., & Liu, Y. (2014). Synthesis and Biological Evaluation of New Pleuromutilin Derivatives as Antibacterial Agents. Molecules, 19(11), 19050-19065. https://doi.org/10.3390/molecules191119050