3. Experimental
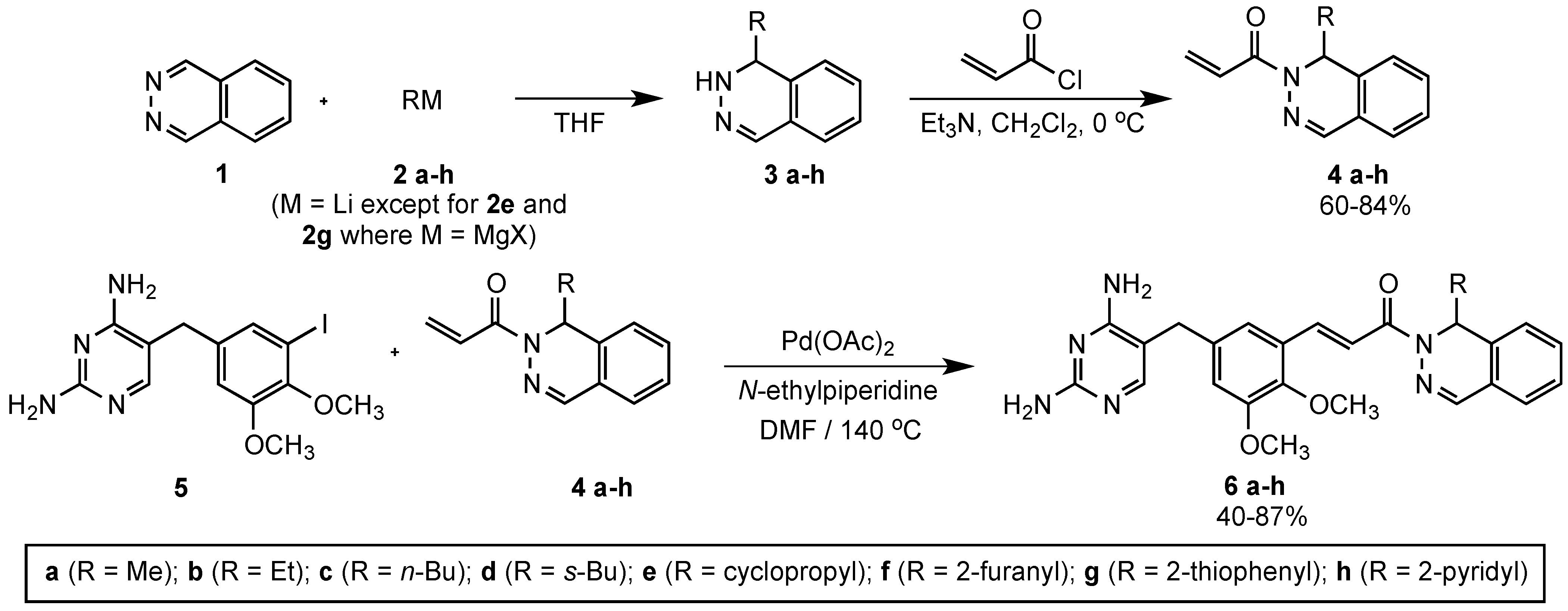
3.2. Synthesis of 1-(Phthalazin-2(1H)-yl)prop-2-en-1-ones 4a‒h
(±)-1-(1-Methylphthalazin-2(1H)-yl)prop-2-en-1-one (4a). A stirred solution of phthalazine (1) (2.00 g, 15.4 mmol) in dry THF (50 mL) was treated dropwise with a solution of methyllithium (2a, 1.5 M in ether, 11.3 mL, 16.9 mmol) over a period of 15 min at –20 °C. The reaction was stirred at this temperature for 45 min and was then poured into saturated NH4Cl (50 mL) and extracted with ethyl acetate (3 × 50 mL). The combined organic extracts were washed with saturated NaCl (100 mL), dried (MgSO4), filtered, and concentrated under vacuum to afford 3a as a dark brown liquid. The crude product 3a was dissolved in dichloromethane (DCM, 50 mL), and triethylamine (1.86 g, 2.56 mL, 18.4 mmol) was added, followed by dropwise addition of acryloyl chloride (1.39 g, 1.25 mL, 15.4 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 2 h. The reaction was then quenched with saturated NaCl (100 mL), the organic layer was separated, and the aqueous layer was extracted with DCM (2 × 50 mL). The combined organic extracts were washed with saturated NaCl (50 mL), dried (MgSO4), filtered, and concentrated to afford the crude product. The crude product was purified on a silica gel column eluted with hexanes:EtOAc (4:1) to afford 4a as a pale yellow liquid (2.60 g, 84%). IR: 1663, 1621 cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.60 (s, 1H), 7.43 (td, J = 7.1, 1.6 Hz, 1H), 7.37–7.23 (complex m, 3H), 7.16 (d, J = 7.7 Hz, 1H), 6.49 (dd, J = 17.5, 2.2 Hz, 1H), 5.90 (q, J = 6.6 Hz, 1H), 5.78 (dd, J = 10.4, 2.2 Hz, 1H), 1.31 (d, J = 6.6 Hz, 3H); 13C-NMR (75 MHz, CDCl3): δ 165.8, 141.4, 135.2, 131.5, 128.0, 127.7, 126.9, 125.42, 125.40, 122.9, 47.1, 20.9.
(±)-1-(1-Ethylphthalazin-2(1H)-yl)prop-2-en-1-one (4b). This compound was prepared as above using 1 (2.00 g, 15.4 mmol) and ethyllithium (2b, 1.5 M in dibutyl ether, 11.2 mL, 16.9 mmol), followed by triethylamine (1.86 g, 2.56 mL, 18.4 mmol) and acryloyl chloride (1.39 g, 1.25 mL, 15.4 mmol) to afford 4b (2.63 g, 80%) as a viscous, colorless oil. IR: 1666, 1621 cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.60 (s, 1H), 7.43 (td, J = 7.7, 1.1 Hz, 1H), 7.39–7.28 (complex m, 2H), 7.27 (d, J = 7.1 Hz, 1H), 7.14 (d, J = 7.1 Hz, 1H), 6.48 (dd, J = 17.0, 2.2 Hz, 1H), 5.77 (overlapping dd, J = 10.4, 2.2 Hz, 1H and t, J = 6.6 Hz, 1H), 1.64 (m, 2H), 0.81 (t, J = 7.7 Hz, 3H); 13C-NMR (75 MHz, CDCl3): δ 166.1, 142.1, 133.4, 131.2, 128.1, 127.9, 127.0, 126.4, 125.5, 123.7, 52.3, 28.0, 9.3.
(±)-1-(1-n-Butylphthalazin-2(1H)-yl)prop-2-en-1-one (4c). This compound was prepared as above using 1 (2.00 g, 15.4 mmol) and n-butyllithium (2c, 2.2 M in hexanes, 7.68 mL, 16.9 mmol), followed by triethylamine (1.86 g, 2.56 mL, 18.4 mmol) and acryloyl chloride (1.39 g, 1.25 mL, 15.4 mmol) to afford 4c (3.09 g, 83%) as viscous, colorless oil. IR: 1665, 1621 cm‒1; 1H-NMR (300 MHz, CDCl3): δ 7.62 (s, 1H), 7.44 (td, J = 7.7, 1.6 Hz, 1H), 7.35 (td, J = 7.1, 1.1 Hz, 1H), 7.32 (dd, J = 17.0, 10.4 Hz, 1H), 7.28 (d, J = 7.1 Hz, 1H), 7.16 (d, J = 7.1 Hz, 1H), 6.48 (dd, J = 17.0, 2.2 Hz, 1H), 5.84 (t, J = 6.6 Hz, 1H), 5.78 (dd, J = 10.4, 2,2 Hz, 1H), 1.64 (q, J = 6.6 Hz, 2H), 1.23 (m, 4H), 0.82 (t, J = 6.8 Hz, 3H); 13C-NMR (75 MHz, CDCl3): δ 166.1, 142.4, 134.0, 131.3, 128.2, 127.9, 127.1, 126.4, 125.6, 123.8, 51.2, 34.8, 26.9, 22.4, 13.8.
(±)-1-(1-s-Butylphthalazin-2(1H)-yl)prop-2-en-1-one (4d). This compound was prepared as above using 1 (2.00 g, 15.4 mmol) and s-butyllithium (2d, 1.4 M in cyclohexane, 12.1 mL, 16.9 mmol), followed by triethylamine (1.86 g, 2.56 mL, 18.4 mmol) and acryloyl chloride (1.39 g, 1.25 mL, 15.4 mmol) to afford 4d (3.00 g, 81%) as a viscous, colorless oil. IR: 1663, 1620 cm−1; 1H-NMR (300 MHz, CDCl3, mixture of diastereomers): δ 7.64 and 7.61 (2s, 1H), 7.44 (t, J = 7.7 Hz, 1H), 7.40–7.25 (complex m, 3H), 7.17 (apparent t, J = 7.1 Hz, 1H), 6.46 (d, J = 17.0 Hz, 1H), 5.76 (m, 2H), 1.73 (m, 1H), 1.46 (m, 1H), 1.10 (m, 1H), 0.92 and 0.82 (2t, J = 7.1 Hz, 3H), 0.88 and 0.70 (2d, J = 6.6 Hz, 3H); 13C-NMR (75 MHz, CDCl3, mixture of diastereomers): δ 166.5, 143.4, 143.1, 132.5, 131.6, 131.1, 131.0, 128.2, 127.95, 127.90, 127.5, 127.4, 127.2, 125.4, 124.7, 124.4, 55.74, 55.26, 40.6, 39.9, 25.4, 24.3, 15.0, 14.2, 11.6, 11.4.
(±)-1-(1-Cyclopropylphthalazin-2(1H)-yl)prop-2-en-1-one (4e). To a stirred solution of 1 (2.00 g, 15.4 mmol) in dry THF (50 mL) was added dropwise cyclopropylmagnesium chloride (0.5 M in THF, 33.8 mL, 16.9 mmol) over a period of 10 min at 0 °C. The reaction was stirred at 0 °C for 2 h and was then quenched with saturated NH4Cl (50 mL) and extracted with ethyl acetate (2 × 50 mL). The combined extracts were washed with saturated NaCl, dried (MgSO4), filtered, and concentrated to give 3e as a light brown oil. The crude product 3e was acylated as described for compound 4a using triethylamine (1.86 g, 2.56 mL, 18.4 mmol) and acryloyl chloride (1.39 g, 1.25 mL, 15.4 mmol) in DCM (50 mL) to obtain 4e (2.71 g, 78%) as a pale yellow oil. IR: 1662, 1621 cm−1; 1H-NMR (300 MHz, DMSO-d6): δ 7.66 (s, 1H), 7.45 (td, J = 7.1, 1.1 Hz, 1H), 7.37 (td, J = 7.7, 1.1 Hz, 1H), 7.36 (dd, J = 17.0, 10.4 Hz, 1H), 7.30 (m, 2H), 7.16 (d, J = 7.7 Hz, 1H), 6.48 (dd, J = 17.0, 2.2 Hz, 1H), 5.79 (dd, J = 10.4, 2.2 Hz, 1H), 5.47 (d, J = 7.7 Hz, 1H), 1.17 (m, 1H), 0.66 (quintet, J = 4.9 Hz, 1H), 0.44 (m, 1H), 0.36 (m, 1H); 13C-NMR (100 MHz, CDCl3): δ 166.6, 142.5, 132.9, 131.5, 128.3, 128.1, 127.2, 126.5, 125.5, 124.0, 54.0, 16.7, 3.8, 2.3.
(±)-1-(1-(Furan-2-yl)phthalazin-2(1H)-yl)prop-2-en-1-one (4f). To a stirred solution of furan (1.20 g, 17.6 mmol) in dry THF (20 mL) was added dropwise n-butyllithium (2.5 M in hexanes, 7.30 mL, 18.3 mmol) over a period of 30 min at –78 °C. The solution was warmed to –25 °C, and stirring was continued at this temperature for 30 min. The reaction mixture was cooled back to –78 °C, and a solution of 1 (2.00 g, 15.3 mmol) in dry THF (20 mL) was added dropwise over 30 min. The reaction mixture was stirred at this temperature for 2 h. The mixture was poured into saturated NH4Cl (100 mL) and extracted with ethyl acetate (3 × 50 mL). The combined organic extracts were then washed with saturated NaCl (50 mL), dried (MgSO4), filtered, and concentrated under vacuum to afford 3f as a light brown oil. The crude product 3f was dissolved in DCM (30 mL), and triethylamine (2.37 g, 3.26 mL, 23.4 mmol) was added, followed by dropwise addition of acryloyl chloride (1.59 g, 1.43 mL, 17.6 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 2 h. The reaction was then quenched with saturated NaCl (25 mL), and the organic layer was separated. The aqueous layer was extracted with DCM (2 × 30 mL), and the combined organic extracts were washed with saturated NaCl (50 mL), dried (MgSO4), filtered, and concentrated to afford the crude product. The product was purified on a silica gel column eluted with hexanes–EtOAc (7:3) to afford 4f (2.66 g, 60%) as a yellow liquid. IR: 1666, 1616 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.68 (s, 1H), 7.47 (dd, J = 7.4, 1.4 Hz, 1H), 7.41 (td, J = 7.4, 1.2 Hz, 1H), 7.37–7.26 (complex m, 3H), 7.25 (d, J = 1.8 Hz, 1H), 7.04 (s, 1H), 6.52 (dd, J = 17.1, 2.0 Hz, 1H), 6.19 (dd, J = 3.2, 1.8 Hz, 1H), 5.94 (d, J = 3.2 Hz, 1H), 5.80 (dd, J = 10.5, 2.0 Hz, 1H); 13C-NMR (100 MHz, CDCl3): δ 166.2, 149.5, 143.5, 142.0, 131.5, 131.2, 128.7, 128.0, 126.6, 126.1, 126.0, 123.7, 111.7, 111.3, 41.8.
(±)-1-(1-(Thiophen-2-yl)phthalazin-2(1H)-yl)prop-2-en-1-one (4g). This compound was prepared by addition of 2-thiophenylmagnesium bromide, prepared from 2-bromothiophene (1.77 g, 1.69 mL, 21.0 mmol) and magnesium (0.69 g, 28.4 mmol) in dry THF (25 mL), to a solution of 1 (2.50 g, 19.2 mmol) in dry THF (30 mL). Product 3g was acylated using triethylamine (2.80 g, 3.86 mL, 27.7 mmol) and acryloyl chloride (1.90 g, 1.71 mL, 21.0 mmol) in DCM to afford 4g (3.09 g, 60%) as a light yellow liquid. IR: 1662, 1617 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.68 (d, J = 7.6 Hz, 1H), 7.48 (td, J = 7.6, 1.2 Hz, 1H), 7.46-7.32 (complex m, 4H), 7.29 (d, J = 7.4 Hz, 1H), 7.13 (dd, J = 5.1, 3.7 Hz, 1H), 6.51 (dd, J = 17.1, 2.0 Hz, 1H), 5.83 (dd, J = 10.3, 2.0 Hz, 1H), 5.02 (s, 2H); 13C-NMR (100 MHz, CDCl3): δ 166.2, 143.7, 141.8, 132.1, 131.9, 129.1, 128.7, 127.0, 126.8, 126.3, 126.2, 125.84, 125.80, 123.7, 49.1.
(±)-1-(1-(Pyridin-2-yl)phthalazin-2(1H)-yl)prop-2-en-1-one (4h). This compound was prepared by the procedure described for 4f using 2-bromopyridine (1.30 g, 8.22 mmol), n-butyllithium (2.5 M in hexanes, 3.38 mL, 8.45 mmol), and 1 (1.00 g, 7.69 mmol) in dry THF (25 mL). Product 3h was acylated using triethylamine (0.93 g, 1.28 mL, 9.2 mmol) and acryloyl chloride (0.70 g, 0.63 mL, 7.73 mmol) in DCM (30 mL) to afford 4h (1.53 g, 76%) as a light yellow liquid. IR: 1662, 1617 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.48 (dq, J = 4.9, 0.8 Hz, 1H), 7.63 (s, 1H), 7.57 (td, J = 7.6, 1.8 Hz, 1H), 7.47–7.37 (complex m, 3H), 7.34 (td, J = 7.6, 1.8 Hz, 1H), 7.28 (d, J = 7.8 Hz, 2H), 7.08 (ddd, J = 7.6, 4.9, 1.0 Hz, 1H), 6.94 (s, 1H), 6.47 (dd, J = 17.4, 2.0 Hz, 1H), 5.80 (dd, J = 10.3, 2.0 Hz, 1H); 13C-NMR (100 MHz, CDCl3): δ 166.6, 159.6, 149.7, 141.2, 136.8, 132.2, 131.8, 129.0, 128.5, 127.5, 127.1, 126.1, 122.5, 122.2, 120.3, 56.7.
3.3. Synthesis of 2,4-Diaminopyrimidine 5
2,4-Diamino-5(5-iodo-3,4-dimethoxybenzyl)pyrimidine (
5). This compound was prepared in 60% yield from morpholine and acrylonitrile on a 0.47-mol scale according to the literature procedure [
15], mp 217–218 °C (lit. [
15] mp 217–218 °C). IR: 3467, 3315, 3140, 1638 cm-1;
1H-NMR (300 MHz, DMSO-
d6): δ 7.57 (s, 1H), 7.14 (d,
J = 1.8 Hz, 1H), 6.98 (d,
J = 1.8 Hz, 1H), 6.16 (br s, 2H), 5.77 (br s, 2H), 3.77 (s, 3H), 3.66 (s, 3H), 3.54 (s, 2H);
13C-NMR (75 MHz, DMSO-
d6): δ 162.4, 162.1, 156.0, 152.0, 146.3, 138.9, 129.1, 113.8, 105.2, 92.4, 59.8, 55.8, 31.7.
3.4. Synthesis of Drug Candidates 6
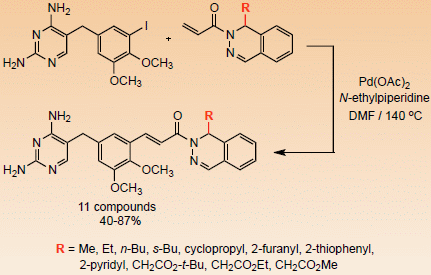
(±)-(E)-3-(5-((2,4-Diaminopyrimidin-5-yl)methyl)-2,3-dimethoxyphenyl)-1-(1-methylphthalazin-2(1H)-yl)prop-2-en-1-one (6a). To a stirred solution of 5 (1.00 g, 2.59 mmol) in dry DMF (6 mL) was added a solution of 4a (0.57 g, 2.85 mmol) in DMF (2 mL), followed by N-ethylpiperidine (0.32 g, 0.40 mL, 2.84 mmol) and Pd(OAc)2 (20 mg, 0.089 mmol). The reaction was heated at 140 °C for 20 h and then cooled using an ice bath. The product was purified by directly pouring the crude reaction mixture onto a 50 cm × 2.5 cm silica gel chromatography column slurry packed with CH2Cl2. Impurities were eluted using CH2Cl2, and the final product was collected using CH2Cl2/MeOH/Et3N (97:3:1). Evaporation of the solvent gave an oily, yellow-brown foam, which was dried under high vacuum for a period of 2 h. Methanol (3 mL) was added, followed by ether (10 mL), to crystallize the product, and the mixture was allowed to cool for 4 h. The product was filtered and dried under vacuum to afford 6a (0.97 g, 82%) as an off-white solid, mp 153–155 °C. IR: 3612, 3308, 3192, 1634, 1600 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 7.93 (s, 1H), 7.88 (d, J = 16.5 Hz, 1H), 7.64 (d, J = 16.5 Hz, 1H), 7.62–7.35 (complex m, 5H), 7.29 (s, 1H), 7.15 (br s, 2H), 7.04 (s, 1H), 6.64 (br s, 2H), 5.89 (m, 1H), 3.81 (s, 3H), 3.76 (s, 3H), 3.65 (s, 2H), 1.23 (d, J = 6.0 Hz, 3H); 13C-NMR (100 MHz, DMSO-d6): δ 165.5, 163.0, 158.6, 152.6, 148.6, 146.2, 142.1, 136.6, 135.3, 135.1, 132.0, 128.2, 127.9, 126.1, 126.0, 123.0, 118.7, 118.0, 114.9, 107.2, 60.8, 55.8, 46.8, 32.0, 21.1. Anal. Calcd for C25H26N6O3·3.9 H2O: C, 56.79; H, 6.44; N, 15.89. Found: C, 56.75; H, 6.32; N, 15.63.
(±)-(E)-3-(5-((2,4-Diaminopyrimidin-5-yl)methyl)-2,3-dimethoxyphenyl)-1-(1-ethylphthalazin-2(1H)-yl)prop-2-en-1-one (6b). This compound was prepared as above using 5 (1.00 g, 2.59 mmol), 4b (0.61 g, 2.85 mmol), N-ethylpiperidine (0.32 g, 0.40 mL, 2.85 mmol), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) to give 6b (0.98 g, 80%) as an off-white solid, mp 232–234 °C. IR: 3472, 3325, 3179, 1635, 1598 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 7.92 (s, 1H), 7.88 (d, J = 15.9 Hz, 1H), 7.65 (d, J = 15.9 Hz, 1H), 7.62 (s, 1H), 7.58-7.43 (complex m, 3H), 7.40 (d, J = 7.1 Hz, 1H), 7.26 (s, 1H), 7.00 (s, 1H), 6.21 (br s, 2H), 5.80 (t, J = 6.6 Hz, 1H), 5.77 (br s, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 3.61 (s, 2H), 1.61 (m, 2H), 0.74 (t, J = 7.7 Hz, 3H); 13C-NMR (100 MHz, DMSO-d6): δ 165.7, 162.4, 162.2, 155.9, 152.5, 146.0, 142.6, 136.7, 133.2, 131.7, 128.3, 127.8, 126.7, 126.1, 123.7, 118.4, 117.9, 114.7, 105.8, 60.8, 55.8, 51.7, 32.5, 27.8, 9.3 (1 aromatic C unresolved). Anal. Calcd for C26H28N6O3: C, 65.83; H, 5.99; N, 17.72. Found: C, 65.84; H, 5.96; N, 17.63.
(±)-(E)-1-(1-n-Butylphthalazin-2(1H)-yl)-3-(5-((2,4-diaminopyrimidin-5-yl)methyl)-2,3-dimethoxyphenyl)prop-2-en-1-one (6c). This compound was prepared as above using 5 (1.00 g, 2.59 mmol), 4c (0.69 g, 2.85 mmol), N-ethylpiperidine (0.32 g, 0.40 mL, 2.85 mmol), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) to give 6c (0.97 g, 75%) as an off-white solid, mp 112–114 °C. IR: 3474, 3329, 3177, 1637, 1603 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 7.94 (s, 1H), 7.87 (d, J = 15.9 Hz, 1H), 7.63 (d, J = 15.9 Hz, 1H), 7.61 (s, 1H), 7.52 (m, 2H), 7.45 (d, J = 7.1 Hz, 1H), 7.39 (d, J = 7.1 Hz, 1H), 7.25 (s, 1H), 7.00 (s, 1H), 6.22 (br s, 2H), 5.84 (t, J = 6.6 Hz, 1H), 5.78 (br s, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 3.60 (s, 2H), 1.58 (m, 2H), 1.17 (m, 4H), 0.79 (t, J = 6.6 Hz, 3H); 13C-NMR (100 MHz, DMSO-d6): δ 165.6, 162.22, 162.18, 155.6, 152.5, 146.0, 142.8, 136.6 (2C), 133.6, 131.7, 128.2, 127.8, 126.5, 126.0, 123.6, 118.3, 117.8, 114.7, 105.8, 60.8, 55.7, 50.5, 34.3, 32.4, 26.6, 21.9, 13.8. Anal. Calcd for C28H32N6O3·0.8 H2O: C, 65.30; H, 6.58; N, 16.32. Found: C, 65.24; H, 6.39; N, 16.22.
(±)-(E)-1-(1-s-Butylphthalazin-2(1H)-yl)-3-(5-((2,4-diaminopyrimidin-5-yl)methyl)-2,3-dimethoxyphenyl)prop-2-en-1-one (6d). This compound was prepared as above using 5 (1.00 g, 2.59 mmol), 4d (0.69 g, 2.85 mmol), N-ethylpiperidine (0.32 g, 0.40 mL, 2.85 mmol), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) to give 6d (0.93 g, 72%) as an off-white solid, mp 122–124 °C. IR: 3469, 3371, 3214, 1634, 1603 cm−1; 1H-NMR (400 MHz, DMSO-d6, mixture of diastereomers): δ 7.97 and 7.95 (2s, 1H), 7.86 (d, J = 15.9 Hz, 1H), 7.66 (2d, J = 15.9 Hz, 1H), 7.60 (s, 1H), 7.59-7.42 (complex m, 3H), 7.37 (apparent t, J = 7.7 Hz, 1H), 7.27 (s, 1H), 7.00 (s, 1H), 6.32 (br s, 2H), 5.86 (br s, 2H), 5.74 (d, J = 6.6 Hz, 1H), 3.79 (s, 3H), 3.74 (s, 3H), 3.60 (s, 2H), 1.64 (m, 1H), 1.39 (m, 1H), 0.94 (m, 1H), 0.87 and 0.78 (2t, J = 7.1 Hz, 3H), 0.81 and 0.65 (2d, J = 7. 1 Hz, 3H); 13C-NMR (100 MHz, DMSO-d6, mixture of diastereomers) δ 165.9, 165.6, 162.3, 161.8, 154.8, 152.5, 146.0, 143.8, 143.5, 136.6, 136.4, 132.0, 131.4, 131.1, 128.3, 127.8, 127.5, 127.4, 125.9, 124.4, 124.2, 118.4, 117.9, 114.7, 105.9, 60.8, 55.7, 54.5, 32.4, 24.9, 24.0, 15.0, 14.2, 11.4, 11.3. Anal. Calcd for C28H32N6O3·1.0 H2O: C, 64.85; H, 6.61; N, 16.20. Found: C, 64.83; H, 6.43; N, 16.28.
(±)-(E)-1-(1-Cyclopropylphthalazin-2(1H)-yl)-3-(5-((2,4-diaminopyrimidin-5-yl)methyl)-2,3-dimethoxyphenyl)prop-2-en-1-one (6e). This compound was prepared as above using 5 (1.00 g, 2.59 mmol), 4e (0.64 g, 2.85 mmol), N-ethylpiperidine (0.32 g, 0.40 mL, 2.85 mmol), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) to give 6e (0.90 g, 72%) as an off-white solid, mp 155–157 °C. IR: 3464, 3359, 3202, 1636, 1602 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 7.99 (s, 1H), 7.87 (d, J = 15.9 Hz, 1H), 7.66 (d, J = 15.9 Hz, 1H), 7.59 (s, 1H), 7.53 (d, J = 7.1 Hz, 2H), 7.46 (t, J = 7.1 Hz, 2H), 7.28 (s, 1H), 7.02 (s, 1H), 6.64 (br s, 2H), 6.16 (br s, 2H), 5.42 (d, J = 8.8 Hz, 1H), 3.80 (s, 3H), 3.75 (s, 3H), 3.62 (s, 2H), 1.13 (m, 1H), 0.55 (m, 1H), 0.44 (m, 2H), 0.33 (m, 1H); 13C-NMR (100 MHz, DMSO-d6): δ 165.9, 162.6, 160.4, 152.5, 152.1, 146.1, 142.9, 136.7, 135.9, 132.8, 131.8, 128.3, 127.8, 126.6, 125.9, 123.7, 118.6, 118.1, 114.8, 106.5, 60.8, 55.8, 53.4, 32.2, 16.7, 4.0, 2.0. Anal. Calcd for C27H28N6O3·2.2 H2O: C, 61.87; H, 6.23; N, 16.03. Found: C, 61.88; H, 6.32; N, 16.00.
(±)-(E)-3-(5-((2,4-Diaminopyrimidin-5-yl)methyl)-2,3-dimethoxyphenyl)-1-(1-(furan-2-yl)phthalazin-2(1H)-yl)prop-2-en-1-one (6f). This compound was prepared as above using 5 (1.50 g, 3.88 mmol), 4f (1.05 g, 4.15 mmol), N-ethylpiperidine (0.48 g, 0.58 mL, 4.27 mmol), and Pd(OAc)2 (30 mg, 0.134 mmol) in dry DMF (10 mL) to give 6f (0.82 g, 42%), as a brown solid, mp 242–244 °C. IR: 3439, 3336, 3181, 1639, 1601 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 8.01 (s, 1H), 7.91 (d, J = 16.0 Hz, 1H), 7.66 (d, J = 16.0 Hz, 1H), 7.61-7.49 (complex m, 6H), 7.32 (s, 1H), 7.17 (br s, 2H), 7.09 (s, 1H), 7.05 (s, 1H), 6.67 (br s, 2H), 6.33 (m, 1H), 5.99 (dd, J = 3.1, 0.6 Hz, 1H), 3.81 (s, 3H), 3.76 (s, 3H), 3.65 (s, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 165.6, 163.1, 158.1, 152.59, 152.56, 147.8, 146.3, 143.0, 142.4, 137.2, 135.1, 132.1, 130.2, 129.0, 127.8, 127.2, 126.3, 123.7, 118.8, 117.7, 115.1, 110.4, 107.6, 107.4, 60.8, 55.8, 47.4, 32.0. Anal. Calcd for C28H26N6O4·4.6 H2O·0.1 Et2O: C, 56.77; H, 6.07; N, 13.99. Found: C, 56.44; H, 5.85; N, 13.72.
(±)-(E)-3-(5-((2,4-Diaminopyrimidin-5-yl)methyl)-2,3-dimethoxyphenyl)-1-(1-(thiophen-2-yl)-phthalazin-2(1H)-yl)prop-2-en-1-one (6g). This compound was prepared as above using 5 (1.00 g, 2.59 mmol), 4g (0.77 g, 2.85 mmol), N-ethylpiperidine (0.32 g, 0.40 mL, 2.85 mmol), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) to give 6g (0.93 g, 68%) as a brown solid, mp 235–237 °C. IR: 3452, 3345, 3179, 1637, 1602 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 8.06 (s, 1H), 7.94 (d, J = 16.0 Hz, 1H), 7.70–7.50 (complex m, 6H), 7.40 (d, J = 4.1 Hz, 1H), 7.31 (s, 1H), 7.27 (s, 1H), 7.04 (s, 1H), 6.96 (br s, 2H), 6.89 (s, 1H), 6.66 (s, 1H), 6.47 (br s, 2H), 3.81 (s, 3H), 3.77 (s, 3H), 3.64 (s, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 165.7, 162.9, 159.0, 152.6, 149.4, 146.3, 143.8, 142.4, 137.4, 135.4, 132.2, 132.1, 129.0, 127.7, 127.1, 126.5, 126.32, 126.27, 126.0, 123.4, 118.8, 117.7, 115.1, 107.9, 60.8, 55.8, 48.8, 32.1. Anal. Calcd for C28H26N6O3S·4.3 H2O: C, 55.67; H, 5.77; N, 13.91. Found: C, 55.99; H, 5.75; N, 13.82.
(±)-(E)-3-(5-((2,4-Diaminopyrimidin-5-yl)methyl)-2,3-dimethoxyphenyl)-1-(1-(pyridin-2-yl)phthalazin-2(1H)-yl)prop-2-en-1-one (6h). This compound was prepared as above using 5 (1.50 g, 3.88 mmol), 4h (1.10 g, 4.18 mmol), N-ethylpiperidine (0.48 g, 0.58 mL, 4.27 mmol), and Pd(OAc)2 (30 mg, 0.134 mmol) in dry DMF (10 mL) to give 6h (0.80 g, 40%) as a brown solid, mp 177–179 °C. IR: 3459, 3347, 3216, 1648, 1611 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 8.43 (d, J = 4.1 Hz, 1H), 7.89 (s, 1H), 7.85 (d, J = 16.0 Hz, 1H), 7.76 (d, J = 16.0 Hz, 1H), 7.74 (td, J = 7.4, 1.6 Hz, 1H), 7.61 (m, 2H), 7.50 (m, 2H), 7.42 (m, 2H), 7.30 (s, 1H), 7.23 (dd, J = 6.7, 5.0 Hz, 1H), 7.01 (d, J = 1.4 Hz, 1H), 6.95 (s, 1H), 6.48 (br s, 2H), 6.01 (br s, 2H), 3.79 (s, 3H), 3.73 (s, 3H), 3.63 (s, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 166.0, 162.4, 161.1, 159.6, 153.5, 152.5, 149.3, 146.1, 140.9, 137.1, 136.9, 136.2, 132.0, 131.8, 128.6, 127.7, 127.5, 126.3, 122.9, 122.8, 120.2, 118.4, 117.8, 114.9, 106.2, 60.8, 56.0, 55.8, 32.3. Anal. Calcd for C29H27N7O3·2.4 H2O: C, 61.67; H, 5.68; N, 17.36. Found: C, 61.58; H, 5.47; N, 17.43.
3.5. Synthesis of Esters 9a–c
(±)-t-Butyl 2-(2-Acryloylphthalazin-2(1H)-yl)acetate (9a). To a stirred solution of tert-butyl acetate (2.67 g, 3.08 mL, 23.0 mmol) in dry THF (40 mL) at –78 °C was added dropwise n-butyllithium (2.5 M in hexanes, 7.68 mL, 19.2 mmol) over a period of 30 min. The solution was warmed to –25 °C and stirred at this temperature for a period of 30 min. To this reaction mixture was added a solution of 1 (2.00 g, 15.4 mmol) in dry THF (25 mL), and stirring was continued for an additional 30 min at 0 °C. The reaction mixture was poured into saturated NH4Cl (100 mL) and extracted with EtOAc (3 × 50 mL). The organic extracts were washed with saturated NaCl (50 mL), dried (MgSO4) and concentrated to afford 8 as a light brown oil. The crude product 8 was then dissolved in DCM (50 mL), and triethylamine (1.86 g, 2.56 mL, 18.4 mmol) was added, followed by dropwise addition of acryloyl chloride (1.39 g, 1.25 mL, 15.4 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for a period of 2 h. The reaction was quenched with saturated NaCl (50 mL), and the organic layer was separated. The aqueous layer was extracted with DCM (2 × 50 mL) and the combined organic layers were washed with saturated NaCl (50 mL), dried (MgSO4), filtered, and concentrated to afford the crude product. The crude product was purified on a silica gel column eluted with hexanes–EtOAc (4:1) to afford 9a (4.00 g, 87%) as a colorless liquid. IR: 1727, 1668, 1621 cm−1; 1H-NMR (300 MHz, CDCl3): δ 8.64 (s, 1H), 7.44 (td, J = 7.7 Hz, 1H), 7.38 (td, J = 7.7, 1.6 Hz, 1H), 7.36-7.26 (complex m, 3H), 6.49 (dd, J = 17.0, 2.2 Hz, 1H), 6.25 (t, J = 7.1 Hz, 1H), 5.79 (dd, J = 10.4, 2.2 Hz, 1H), 2.58 (d, J = 7.1 Hz, 2H), 1.37 (s, 9H); 13C-NMR (75 MHz, CDCl3): δ 168.7, 166.0, 142.0, 132.5, 131.6, 128.7, 128.4, 126.9, 126.7, 125.7, 123.6, 80.9, 48.0, 40.7, 27.8.
(±)-2-(2-Acryloylphthalazin-2(1H)-yl)acetic Acid (10). To a stirred solution of 9a (1.50 g, 5.00 mmol) in benzene (25 mL) was added Bi(OTf)3 (0.164 g, 0.25 mmol, 5 mol%), and the solution was refluxed for a period of 2 h. To this solution was added EtOAc (50 mL), followed by H2O (50 mL). The organic layer was washed with saturated NaCl (50 mL), dried (MgSO4), filtered, and concentrated to afford 10 (1.15 g, 94%) as a pale yellow solid, mp 142–145 °C. IR: 3400, 1734, 1670 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 12.4 (s, 1H), 7.93 (s, 1H), 7.56–7.43 (complex m, 3H), 7.39 (d, J = 7.4 Hz, 1H), 7.24 (dd, J = 17.2, 10.5 Hz, 1H), 6.34 (dd, J = 17.2, 2.1 Hz, 1H), 6.13 (t, J = 6.8 Hz, 1H), 5.86 (dd, J = 10.5, 2.1 Hz, 1H), 2.51 (m, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 170.7, 165.1, 142.8, 132.3, 131.9, 128.9, 128.7, 127.0, 126.4, 126.3, 123.4, 47.5, 39.5. Attempts to further purify this compound failed to yield material with sufficient purity for biological testing.
(±)-Ethyl 2-(2-Acryloylphthalazin-2(1H)-yl)acetate (9b). To a stirred solution of 10 (1.00 g, 4.10 mmol) in ethanol (25 mL) was added Bi(OTf)3 (0.134 g, 0.20 mmol, 5 mol%), and the mixture was refluxed for a period of 2 h. The solution was concentrated and purified using a silica gel column eluted with hexanes–EtOAc (4:1) to afford 9b (1.06 g, 95%) as a colorless, viscous liquid. IR: 1733, 1667, 1621 cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.66 (s, 1H), 7.45 (td, J = 7.1, 1.1 Hz, 1H), 7.38 (td, J = 7.7, 1.1 Hz, 1H), 7.35–7.23 (complex m, 3H), 6.49 (dd, J = 17.0, 2.2 Hz, 1H), 6.29 (t, J = 6.6 Hz, 1H), 5.81 (dd, J = 10.4, 1.6 Hz, 1H), 4.06 (q, J = 7.1 Hz, 2H), 2.65 (m, 2H), 1.19 (t, J = 7.1 Hz, 3H); 13C-NMR (75 MHz, CDCl3): δ 169.5, 166.1, 142.1, 132.3, 131.7, 128.9, 128.6, 126.8, 126.6, 125.8, 123.6, 60.7, 47.9, 39.5, 14.0.
(±)-Methyl 2-(2-Acryloylphthalazin-2(1H)-yl)acetate (9c). To a stirred solution of 10 (1.00 g, 4.10 mmol) in methanol (25 mL) was added Bi(OTf)3 (0.134 g, 0.20 mmol, 5 mol%), and the reaction was refluxed for 2 h. The solution was concentrated and purified using a silica gel column eluted with hexanes:EtOAc (4:1) to afford 9c (1.00 g, 95%) as colorless, viscous liquid. IR: 1732, 1663, 1618 cm−1; 1H-NMR (300 MHz, CDCl3): δ 7.67 (s, 1H), 7.45 (td, J = 7.7, 1.6 Hz, 1H), 7.38 (td, J = 7.7, 1.6 Hz, 1H), 7.34–7.27 (complex m, 3H), 6.49 (dd, J = 17.0, 2.2 Hz, 1H), 6.28 (t, J = 7.7 Hz, 1H), 5.81 (dd, J = 10.4, 1.6 Hz, 1H), 3.61 (s, 3H), 2.66 (m, 2H); 13C-NMR (75 MHz, CDCl3): δ 169.9, 166.1, 142.1, 132.2, 131.8, 128.9, 128.6, 126.7, 126.4, 125.8, 123.4, 51.7, 47.9, 39.2.
3.6. Synthesis of Drug Candidates 11a–c
t-Butyl (±)-(E)-2-(2-(3-(5-((2,4-Diaminopyrimidin-5-yl)methyl)-2,3-dimethoxyphenyl)acryloyl)-phthal-azin-2(1H)-yl)acetate (11a). This compound was prepared as described for 6a using 5 (1.00 g, 2.59 mmol), 9a (0.86 g, 2.85 mmol), N-ethylpiperidine (0.32 g, 0.40 mL, 2.85 mmol), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) to give 11a (1.12 g, 78%) as an off-white solid, mp 185–187 °C. IR: 3361, 3187, 3068, 1698, 1672, 1638 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 7.96 (s, 1H), 7.89 (d, J = 15.9 Hz, 1H), 7.64 (d, J = 15.9 Hz, 1H), 7.62–7.46 (complex m, 6H), 7.41 (d, J = 7.1 Hz, 1H), 7.32 (s, 1H), 7.06 (s, 1H), 7.02 (br s, 2H), 6.18 (t, J = 6.6 Hz, 1H), 3.82 (s, 3H), 3.75 (s, 3H), 3.66 (s, 2H), 2.50 (m, 2H), 1.30 (s, 9H); 13C-NMR (100 MHz, DMSO-d6): δ 168.4, 163.54, 163.46, 156.4, 152.6, 146.3, 144.5, 142.5, 136.8, 134.6, 132.1, 131.9, 128.7, 127.9, 126.5, 126.2, 123.5, 118.8, 117.9, 115.1, 108.0, 80.4, 60.8, 55.9, 47.9, 40.7, 31.8, 27.5. Anal. Calcd for C30H34N6O5·6.2 H2O: C, 53.75; H, 5.90; N, 12.54. Found: C, 53.71; H, 5.53; N, 12.56.
Ethyl (±)-(E)-2-(2-(3-(5-((2,4-Diaminopyrimidin-5-yl)methyl)-(2,3-dimethoxyphenyl)acryloyl)-phthal-azin-2(1H)-yl)acetate (11b). This compound was prepared as above using 5 (1.00 g, 2.59 mmol), 9b (0.78 g, 2.85 mmol), N-ethylpiperidine (0.32 g, 0.40 mL, 2.85 mmol), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) to give 11b (1.01 g, 74%) as a pale yellow solid, mp 113–115 °C. IR: 3473, 3352, 3185, 1728, 1651, 1614 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 7.98 (s, 1H), 7.88 (d, J = 15.9 Hz, 1H), 7.61 (d, J = 15.9 Hz, 1H), 7.60 (s, 1H), 7.52 (complex m, 3H), 7.40 (d, J = 7.1 Hz, 1H), 7.27 (s, 1H), 7.02 (s, 1H), 6.47 (br s, 2H), 6.20 (t, J = 6.6 Hz, 1H), 6.00 (br s, 2H), 3.96 (q, J = 7.1 Hz, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 3.61 (s, 2H), 2.59 (m, 2H), 1.09 (t, J = 7.1 Hz, 3H); 13C-NMR (100 MHz, DMSO-d6): δ 169.2, 165.7, 162.4, 161.1, 153.6, 152.5, 146.1, 142.5, 136.9, 136.2, 132.0 (2C), 128.8, 127.7, 126.4, 126.2, 123.5, 118.4, 117.7, 114.9, 106.2, 60.8, 60.3, 55.8, 47.8, 39.4, 32.3, 13.8. Anal. Calcd for C28H30N6O5·2.1 H2O: C, 59.17; H, 6.06; N, 14.79. Found: C, 59.16; H, 5.74; N, 14.60.
Methyl (±)-(E)-2-(2-(3-(5-((2,4-Diaminopyrimidin-5-yl)methyl)-2,3-dimethoxyphenyl)acryloyl)-phthal-azin-2(1H)-yl)acetate (11c). This compound was prepared as above using 5 (1.00 g, 2.59 mmol), 9c (0.74 g, 2.85 mmol), N-ethylpiperidine (0.32 g, 0.40 mL, 2.85 mmol), and Pd(OAc)2 (20 mg, 0.089 mmol) in dry DMF (8 mL) to give 11c (1.04 g, 78%) as a pale yellow solid, mp 158–160 °C. IR: 3477, 3370, 3192, 1720, 1653, 1614 cm−1; 1H-NMR (400 MHz, DMSO-d6): δ 7.98 (s, 1H), 7.87 (d, J = 15.9 Hz, 1H), 7.60 (d, J = 15.9 Hz, 1H), 7.59-7.46 (complex m, 4H), 7.39 (d, J = 7.1 Hz, 1H), 7.28 (s, 1H), 7.03 (s, 1H), 6.86 (br s, 2H), 6.37 (br s, 2H), 6.20 (t, J = 6.6 Hz, 1H), 3.80 (s, 3H), 3.75 (s, 3H), 3.62 (s, 2H), 3.51 (s, 3H), 2.60 (m, 2H); 13C-NMR (100 MHz, DMSO-d6): δ 169.6, 165.7, 162.8, 159.4, 152.5, 150.1, 146.2, 142.5, 136.9, 135.6, 132.01, 131.97, 128.8, 127.8, 126.3 (2C), 123.4, 118.6, 117.7, 115.0, 106.9, 60.8, 55.8, 51.6, 47.8, 32.1. Anal. Calcd for C27H28N6O5∙3.7 H2O: C, 55.61; H, 6.12; N, 14.41. Found: C, 55.63; H, 6.32; N, 14.43.
3.7. Biological Potency Measurements
Measurements of the MIC and the K
i utilized a racemic mixture of each compound and have been described previously [
9,
10,
12,
15,
16]. In brief, MIC values were based on standardized cultures of
B. anthracis Sterne strain as prescribed by the CLSI [
19]. Evaluation of growth utilized spectrophotometric values of turbidity at 600 nm and on visual inspection for assessment of bacterial growth. The lowest concentration that yielded no growth was assigned as the MIC. Evaluation of the enzymatic activity and inhibition utilized purified DHFR protein cloned from
B. anthracis Sterne strain and expressed recombinantly in
E. coli BL21 (DE3) cells. The protein preparation utilized an
N-terminal His-tag, which was determined to not interfere with the enzymatic activity assay and was left intact for the current studies. The reaction was reconstituted, including the NADPH co-factor, and was initiated by the addition of the dihydrofolate substrate. The reaction was carried out at 30 °C, and the linear rate was monitored for 2.8 min. These rates were plotted as a function of inhibitor concentration, and the 50% activity point was calculated using a 4-parameter curve fit. These IC
50 values were converted to K
i values using the Cheng-Prusoff equation [
20].






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
