Novel Gram-Scale Production of Enantiopure R-Sulforaphane from Tuscan Black Kale Seeds


Abstract
:1. Introduction

2. Results and Discussion
2.1. Purification of Glucoraphanin from Tuscan Black Kale Seeds

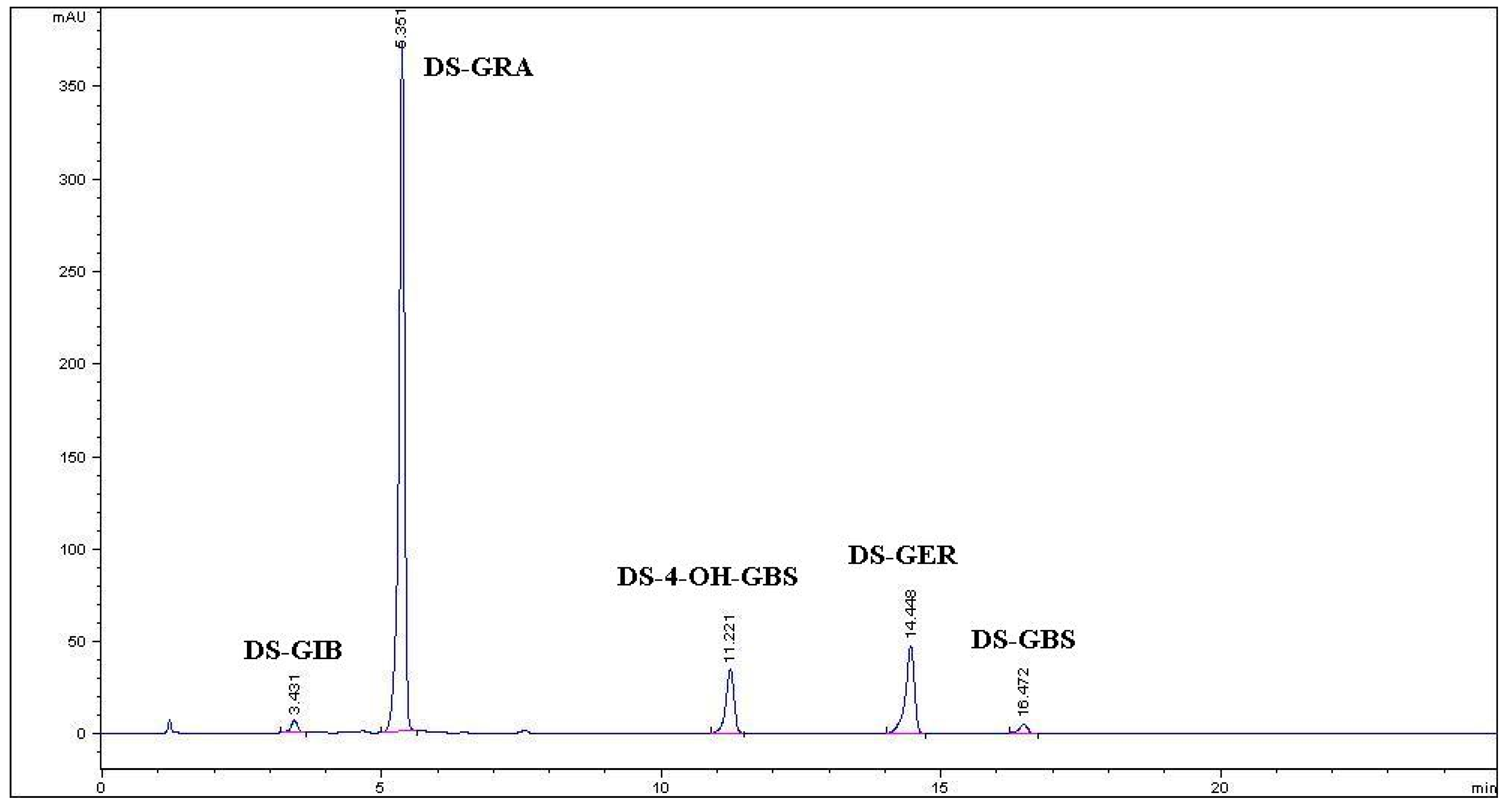{kind=link}
{kind=link}
{kind=link}
| Purification Step | Amount | Aliphatic GLs | Indole-Type GLs | Total GLs | |||
|---|---|---|---|---|---|---|---|
| GIB | GRA | GER | 4-OH-GBS | GBS | |||
| TBK-DSM | 150 g | 0.15 (±0.01) a | 7.66 (±0.62) | 1.25(±0.17) | 0.46 (±0.05) | 0.01 (±0.00) | 9.53 (±0.85) |
| Ethanolic extract | 2.29 L | 0.10 (±0.00) | 6.20 (±0.02) | 0.88(±0.00) | 0.19 (±0.07) | 0.10 (±0.00) | 7.47 (±0.09) |
| GLs mix powder
(from DEAE A-25) | 7.19 g | 0.10 (±0.00) | 5.83 (±0.14) | 0.48(±0.06) | − | − | 6.41 (±0.20) |
| Purified GRA | 3.10 g | ||||||
| R-Sulforaphane | 1.09 g | ||||||

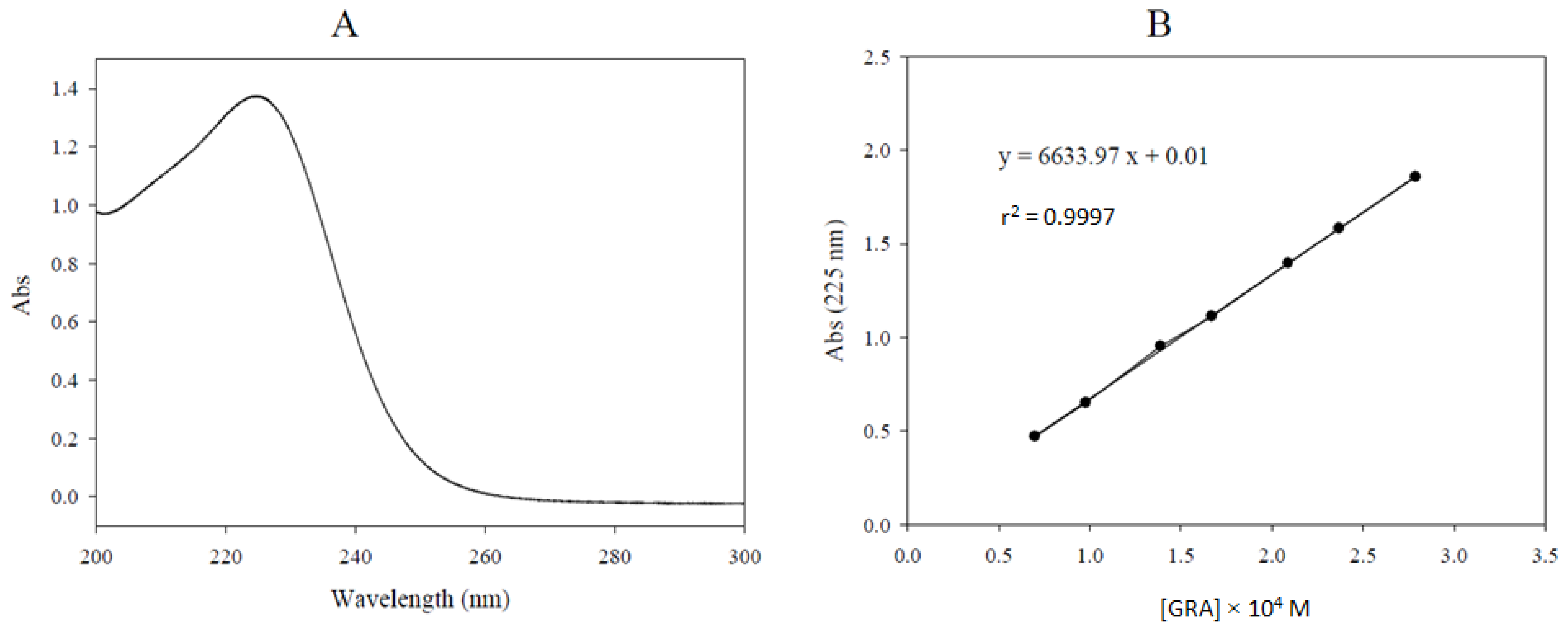
2.2. Glucoraphanin Molar Extinction Coefficient Determination

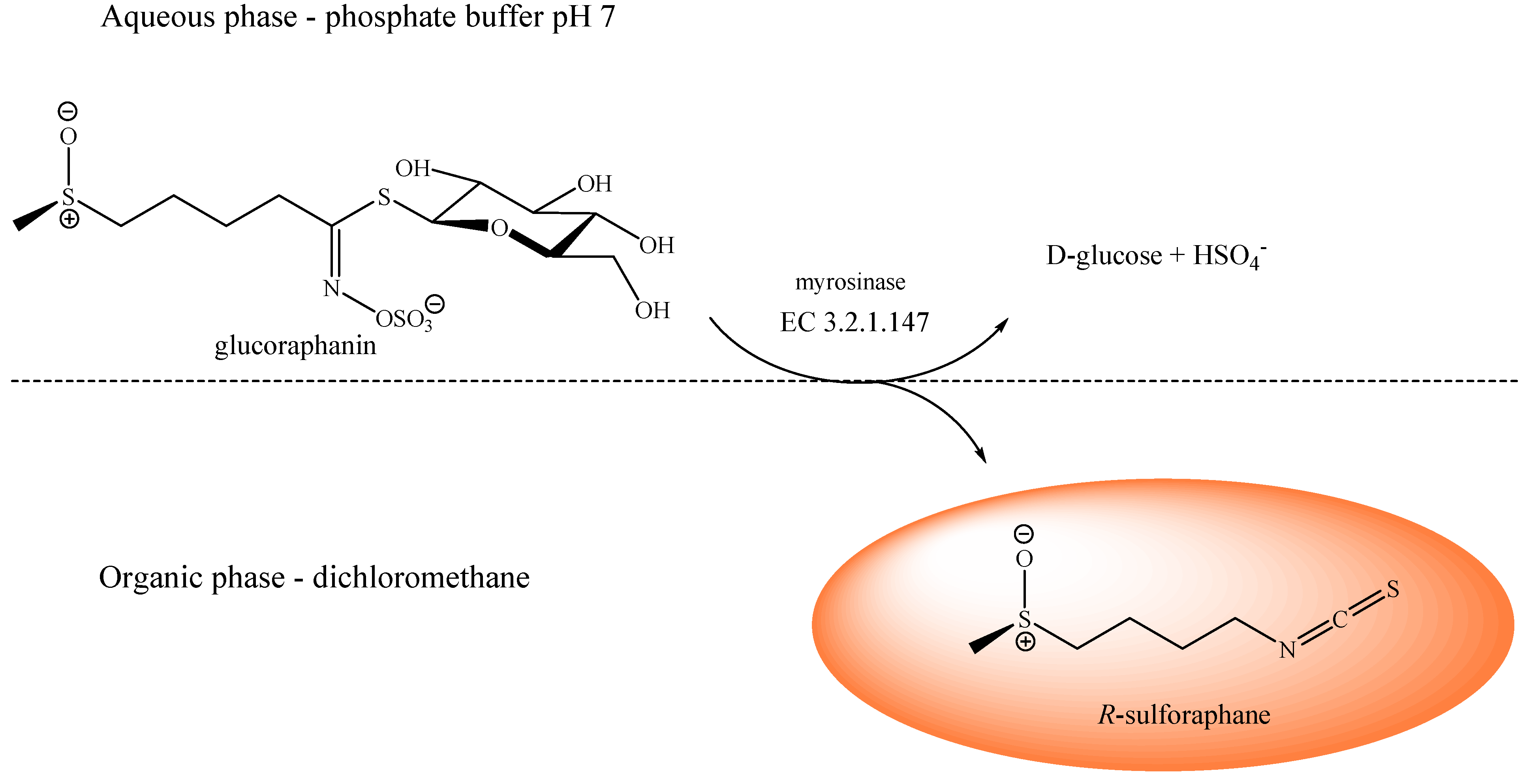
2.3. Production of Enantiopure R-Sulforaphane
2.4. R-Sulforaphane Characterization
 −76 (c 1.3, CHCl3); lit. data: −78.2 (c 0.6, CHCl3) [19]. GC-MS, tR = 18.7 min; EIMS 70 eV m/z (rel. int.): 72 (100), 160 (64), 55 (43), 39 (15), 45 (13), 64 (12), 63 (10), 41(10), 114 (8), 74 (6). The observed data are in agreement with literature values [20]. IR (cm−1): 3426 (O-H from H2O adsorbed), 2923, 2867 (C-H), 2179, 2100 (N=C=S), 1451, 1349 (C-H), 1260 (C-N), 1021 (S=O), 739 (C-H), 688 (C-S), in accordance with literature data [21]. 1H-NMR (400 MHz, CDCl3) δ (ppm): 3.59 (t, Jvic = 6.0 Hz, 2H, CH2N), 2.80–2.66 (m, 2H, CH2S), 2.59 (s, 3H, CH3), 1.97–1.83 (m, 4H, CH2CH2). 13C-NMR (100 MHz, CDCl3) δ (ppm): 53.6 (CH2S), 44.8 (CH2N), 38.7 (CH3), 29.1 (C-2), 20.2 (C-3). NMR data were in accordance with literature data [19].
−76 (c 1.3, CHCl3); lit. data: −78.2 (c 0.6, CHCl3) [19]. GC-MS, tR = 18.7 min; EIMS 70 eV m/z (rel. int.): 72 (100), 160 (64), 55 (43), 39 (15), 45 (13), 64 (12), 63 (10), 41(10), 114 (8), 74 (6). The observed data are in agreement with literature values [20]. IR (cm−1): 3426 (O-H from H2O adsorbed), 2923, 2867 (C-H), 2179, 2100 (N=C=S), 1451, 1349 (C-H), 1260 (C-N), 1021 (S=O), 739 (C-H), 688 (C-S), in accordance with literature data [21]. 1H-NMR (400 MHz, CDCl3) δ (ppm): 3.59 (t, Jvic = 6.0 Hz, 2H, CH2N), 2.80–2.66 (m, 2H, CH2S), 2.59 (s, 3H, CH3), 1.97–1.83 (m, 4H, CH2CH2). 13C-NMR (100 MHz, CDCl3) δ (ppm): 53.6 (CH2S), 44.8 (CH2N), 38.7 (CH3), 29.1 (C-2), 20.2 (C-3). NMR data were in accordance with literature data [19].2.5. Discussion
3. Experimental
3.1. Chemicals
3.2. Plant Source
3.3. Glucoraphanin Extraction and Purification
3.4. R-Sulforaphane Production
3.5. Glucosinolate Profiling and Quantification by HPLC-PDA Analysis
3.6. Glucoraphanin Molar Extinction Coefficient Determination
3.7. NMR Analysis of Glucoraphanin and Sulforaphane
3.8. HPLC-PDA Analysis of Sulforaphane
3.9. IR of Sulforaphane
3.10. GC/MS Analysis of Sulforaphane
3.11. Optical Rotation of Sulforaphane
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.T.; Iavicoli, I.; di Paola, R.; Koverech, A.; Cuzzocrea, S.; Rizzarelli, E.; Calabrese, E.J. Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 753–783. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: http://clinicaltrials.gov/ct2/results?term=sulforaphane&Search=Search (accessed on 16 April 2014).
- Vergara, F.; Wenzler, M.; Hansen, B.G.; Kliebenstein, D.J.; Halkier, B.A.; Gershenzon, J.; Schneider, B. Determination of the absolute configuration of the glucosinolate methyl sulfoxide group reveals a stereospecific biosynthesis of the side chain. Phytochemistry 2008, 69, 2737–2742. [Google Scholar] [CrossRef]
- Li, S.Z.; Royce, S.G.; Ververis, K.; Karagiannis, T.C. Cell cycle effects of L-sulforaphane, a major antioxidant from cruciferous vegetables: The role of the anaphase promoting complex. Hell. J. Nucl. Med. 2013, 17, 11–16. [Google Scholar]
- Schmid, H.; Karrer, P. Synthese der racemischen und der optisch aktiven Formen des Sulforaphans. Helv. Chim. Acta 1948, 31, 1497–1505. [Google Scholar] [CrossRef]
- Iori, R.; Bernardi, R.; Gueyrard, D.; Rollin, P.; Palmieri, S. Formation of glucoraphanin by chemoselective oxidation of natural glucoerucin: A chemoenzymatic route to sulforaphane. Bioorg. Med. Chem. Lett. 1999, 9, 1047–1048. [Google Scholar]
- Chen, X.; Li, Z.; Sun, X.; Ma, H.; Chen, X.; Ren, J.; Hu, K. New method for the synthesis of sulforaphane and related isothiocyanates. Synthesis 2011, 24, 3991–3996. [Google Scholar]
- Abdull Razis, A.F.; Iori, R.; Ioannides, C. The natural chemopreventive phytochemical R-sulforaphane is a far more potent inducer of the carcinogen-detoxifying enzyme systems in rat liver and lung than the S-isomer. Int. J. Cancer 2011, 128, 2775–2782. [Google Scholar] [CrossRef]
- Abdull Razis, A.F.; Noor, N.M. Cruciferous vegetables: Dietary phytochemicals for cancer prevention. Asian Pac. J. Cancer Prev. 2013, 14, 1565–1570. [Google Scholar] [CrossRef]
- Kelsey, N.A.; Wilkins, H.M.; Linseman, D.A. Nutraceutical antioxidants as novel neuroprotective agents. Molecules 2010, 15, 7792–7814. [Google Scholar] [CrossRef]
- Liang, H.; Yuan, Q.; Xiao, Q. Purification of sulforaphane from Brassica oleracea seed meal using low-pressure column chromatography. J. Chromatogr. B 2005, 828, 91–96. [Google Scholar] [CrossRef]
- Liang, H.; Li, C.; Yuan, Q.; Vriesekoop, F. Separation and purification of sulforaphane from broccoli seeds by solid phase extraction and preparative high-performance liquid chromatography. J. Agric. Food Chem. 2007, 55, 8047–53. [Google Scholar] [CrossRef]
- Li, C.; Liang, H.; Yuana, Q.; Houa, X. Optimization of sulforaphane separation from broccoli seeds by macroporous resins. Separ. Sci. Technol. 2008, 43, 609–623. [Google Scholar] [CrossRef]
- Sasaki, K.; Neyazaki, M.; Shindo, K.; Ogawa, T.; Momose, M. Quantitative profiling of glucosinolates by LC-MS analysis reveals several cultivars of cabbage and kale as promising candidates for sulforaphane production. J. Chromatogr. B 2012, 903, 171–176. [Google Scholar]
- Abdull Razis, A.F.; Bagatta, M.; de Nicola, G.R.; Iori, R.; Ioannides, C. Intact glucosinolates modulate hepatic cytochrome P450 and phase II conjugation activities and may contribute directly to the chemopreventive activity of cruciferous vegetables. Toxicology 2010, 277, 74–85. [Google Scholar] [CrossRef]
- Clarke, D.B. Glucosinolates, structures and analysis in food. Anal. Method. Instrum. 2010, 2, 301–416. [Google Scholar] [CrossRef]
- Fahey, J.F.; Wade, K.L.; Stephenson, K.K.; Chou, F.E. Separation and purification of glucosinolates from crude plant homogenates by high-speed counter-current chromatography. J. Chromatogr. A 2003, 996, 85–93. [Google Scholar]
- Khiar, N.; Werner, S.; Mallouk, S.; Lieder, F.; Alcudia, A.; Fernández, I. Enantiopure sulforaphane analogues with various substituents at the sulfinyl sulfur: Asymmetric synthesis and biological activities. J. Org. Chem. 2009, 74, 6002–6009. [Google Scholar] [CrossRef]
- Chiang, W.C.K.; Pusateri, D.J.; Leitz, R.E.A. Gas chromatography/mass spectrometry method for the determination of sulforaphane and sulforaphane nitrile in broccoli. J. Agric. Food Chem. 1998, 46, 1018–1021. [Google Scholar] [CrossRef]
- Wu, H.; Liang, H.; Yuan, Q.; Wang, T.; Yan, X. Preparation and stability investigation of the inclusion complex of sulforaphane with hydroxypropyl-β-cyclodextrin. Carbohydr. Polym. 2010, 82, 613–617. [Google Scholar]
- Lai, R.H.; Keck, A.S.; Wallig, M.A.; West, L.G.; Jeffery, E.H. Evaluation of the safety and bioactivity of purified and semi-purified glucoraphanin. Food Chem. Toxicol. 2008, 46, 195–202. [Google Scholar] [CrossRef]
- Agerbirk, N.; Olsen, C.E.; Sørensen, H. Initial and final products, nitriles, and ascorbigens produced in myrosinase-catalyzed hydrolysis of indole glucosinolates. J. Agric. Food Chem. 1998, 46, 1563–1571. [Google Scholar] [CrossRef]
- Abdull Razis, A.F.; Bagatta, M.; de Nicola, G.R.; Iori, R.; Ioannides, C. Induction of epoxide hydrolase and glucuronosyl transferase by isothiocyanates and intact glucosinolates in precision-cut rat liver slices: Importance of side-chain substituent and chirality. Arch. Toxicol. 2011, 85, 919–927. [Google Scholar] [CrossRef]
- Giacoppo, S.; Galuppo, M.; Iori, R.; de Nicola, G.R.; Cassata, G.; Bramanti, P.; Mazzon, E. Protective role of (RS)-glucoraphanin bioactivated with myrosinase in an experimental model of multiple sclerosis. CNS Neurosci. Ther. 2013, 19, 577–584. [Google Scholar] [CrossRef]
- Galuppo, M.; Iori, R.; de Nicola, G.R.; Ferrantelli, V.; Bramanti, P.; Mazzon, E. Anti-inflammatory and anti-apoptotic effects of (RS)-glucoraphanin bioactivated with myrosinase in murine sub-acute and acute MPTP-induced Parkinson’s Disease. Bioorg. Med. Chem. 2013, 21, 5532–5547. [Google Scholar] [CrossRef]
- Roselli, C.; Perly, B.; Rollin, P. Inclusion Compounds of Natural Precursors of Isothiocyanates in Cyclodextrins, Their Preparation and Use. International application PCT/FR99/00720, 29 March 1999. [Google Scholar]
- Franklin, S.J.; Dickinson, S.E.; Karlage, K.L.; Bowden, G.T.; Myrdal, P.B. Stability of sulforaphane for topical formulation. Drug. Dev. Ind. Pharm. 2013, 40, 494–502. [Google Scholar]
- Pessina, A.; Thomas, R.M.; Palmieri, S.; Luisi, P.L. An improved method for the purification of myrosinase and its physicochemical characterization. Arch. Biochem. Biophys. 1990, 280, 383–389. [Google Scholar] [CrossRef]
- European Economic Community, Commisssion Regulation, EEC No. 1864/90. Oilseeds-determination of glucosinolates high performance liquid chromatography. Off. J. Eur. Comm. 1990, L170, 27–34.
- De Nicola, G.R.; Bagatta, M.; Pagnotta, E.; Angelino, D.; Gennari, L.; Ninfali, P.; Rollin, P.; Iori, R. Comparison of bioactive phytochemical content and release of isothiocyanates in selected brassica sprouts. Food Chem. 2013, 141, 297–303. [Google Scholar] [CrossRef]
- De Nicola, G.R.; Nyegue, M.; Montaut, S.; Iori, R.; Menut, C.; Tatibouet, A.; Rollin, P.; Ndoyé, C.; Amvam Zollo, P.H. Profile and quantification of glucosinolates in Pentadiplandra brazzeana Baillon. Phytochemistry 2012, 73, 51–56. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Nicola, G.R.; Rollin, P.; Mazzon, E.; Iori, R. Novel Gram-Scale Production of Enantiopure R-Sulforaphane from Tuscan Black Kale Seeds. Molecules 2014, 19, 6975-6986. https://doi.org/10.3390/molecules19066975
De Nicola GR, Rollin P, Mazzon E, Iori R. Novel Gram-Scale Production of Enantiopure R-Sulforaphane from Tuscan Black Kale Seeds. Molecules. 2014; 19(6):6975-6986. https://doi.org/10.3390/molecules19066975
Chicago/Turabian StyleDe Nicola, Gina Rosalinda, Patrick Rollin, Emanuela Mazzon, and Renato Iori. 2014. "Novel Gram-Scale Production of Enantiopure R-Sulforaphane from Tuscan Black Kale Seeds" Molecules 19, no. 6: 6975-6986. https://doi.org/10.3390/molecules19066975
APA StyleDe Nicola, G. R., Rollin, P., Mazzon, E., & Iori, R. (2014). Novel Gram-Scale Production of Enantiopure R-Sulforaphane from Tuscan Black Kale Seeds. Molecules, 19(6), 6975-6986. https://doi.org/10.3390/molecules19066975