Synthesis and Cytotoxicity of 2,3-Enopyranosyl C-Linked Conjugates of Genistein

Abstract
:1. Introduction

2. Results and Discussion
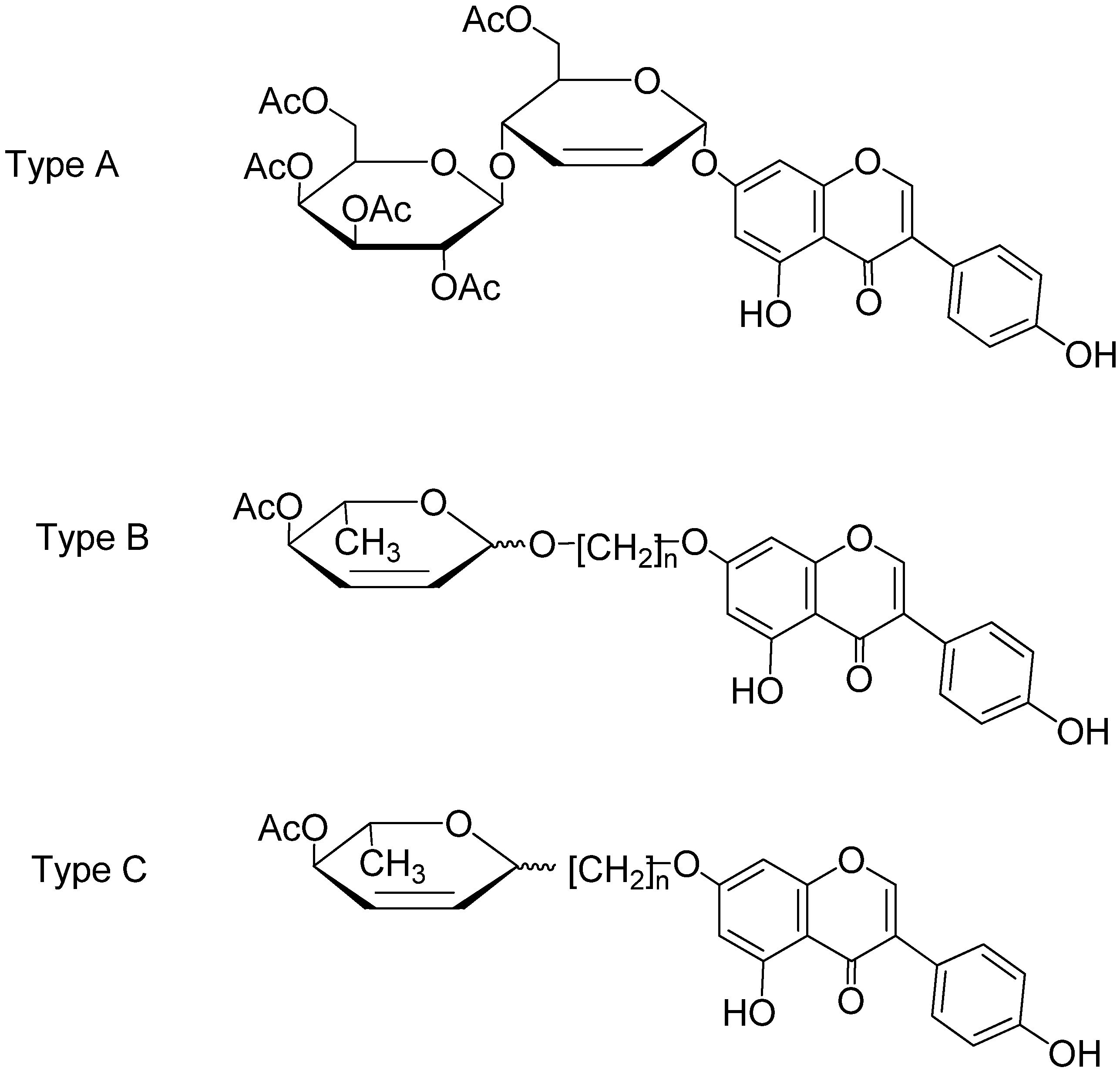
2.1. Synthesis of 2,3-Enopyranosyl C-Linked Conjugates of Genistein

2.2. Anticancer Activity in Vitro


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Line | |
|---|---|---|
| HCT 116 | DU 145 | |
| Genistein | 34.90 ± 9.84 | 47.29 ± 11.78 |
| 15a | 13.04 ± 2.13 | 28.02 ± 9.24 |
| 15b | >25.00 * | >25.00 * |
| 16a | 2.28 ± 0.62 | 4.64 ± 2.52 |
| 16b | 35.51 ±17.30 | 6.85 ± 2.39 |
| 17a | 9.13 ± 6.22 | 10.09 ± 4.61 |
| 17b | 4.69 ± 3.38 | 4.90 ± 2.57 |
| Paclitaxel | (3.5 ± 1.1) × 10−3 | (18.4 ± 3.64) × 10−3 |


3. Experimental
3.1. General Information
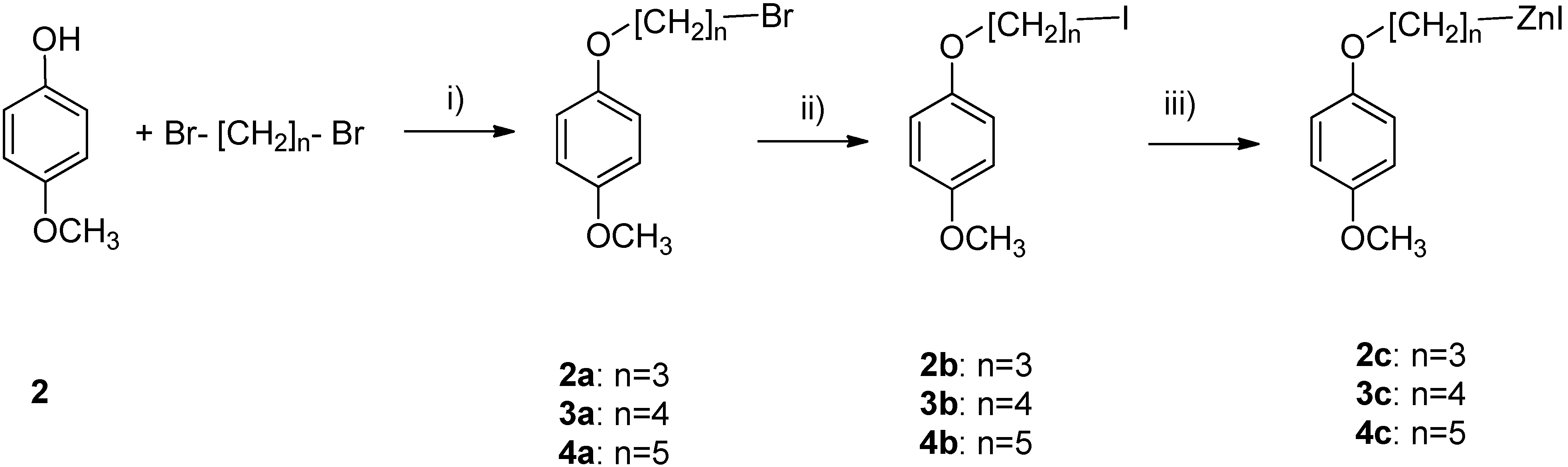
3.2. Synthesis of α,ω-Bromoalkyl Ethers of 4-Methoxyphenol
3.3. Synthesis of α,ω-Iodoalkyl Ethers of 4-Methoxyphenol
3.4. General Procedure for the Preparation of Reformatsky Reagents
3.5. General Procedure for the Synthesis of C-Glycosides
 −57.0° (c 1.37, CHCl3); 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, J = 6.6 Hz, 3H, CH3), 1.70–2.00 (m, 4H, CH2CH2), 2.08 (s, 3H, CH3CO), 3.76 (s, 3H, OCH3), 3.91 (dq, J = 6.6 Hz and 4.9 Hz, 1H, H-5), 3.95 (t, J= 6.3 Hz, 2H, OCH2), 4.21 (m, 1H, H-1), 4.89 (m, 1H, H-4), 5.78 (ddd, J=10.3 Hz, 3.5 Hz and 2.1 Hz, 1H, H-3), 5.92 (ddd, 1H, J = 10.4 Hz, 2.2 Hz and 1.3 Hz, 1H, H-2), 6.83 (s, 4H, H-2ar, H-3ar, H-5ar, H-6ar). 13C-NMR (75 MHz, CDCl3) δ (ppm): 17.2 (CH3), 21.5 (CH3CO), 26.0 (CH2CH2CH2), 30.4 (CH2CH2CH2), 56.0 (OCH3), 68.5 (C-5), 68.7 (CH2O), 69.9 (C-4), 70.1 (C-1), 114.6 (C-3ar, C-5ar), 115.7 (C-2ar, C-6ar), 123.1 (C-3), 134.2 (C-2), 153.4(C-1ar), 154.0 (C-4ar), 171.0 (C=O). HRMS: calcd for [M+Na]+: m/z 359.1471. Found: m/z 359.1473. −65.60° (c 1.74, CHCl3), 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, J = 6.6 Hz, 3H, CH3), 1.50–1.90 (m, 6H, CH2CH2CH2), 2.08 (s, 3H, CH3CO), 3.76 (s, 3H, OCH3), 3.88–3.96 (m, 3H, OCH2; H-5), 4.16 (m, 1H, H-1), 4.89 (m, 1H, H-4), 5.76 (ddd, J = 10.3 Hz, 3.5 Hz and 2.1 Hz, 1H, H-3), 5.92 (ddd, J = 10.4 Hz, 2.1 Hz, and 1.3 Hz, 1H, H-2), 6.83 (s, 4H, H-2ar, H-3ar, H-5ar, H-6ar). 13C-NMR (75 MHz, CDCl3) δ (ppm): 17.2 (CH3), 21.2 (CH3CO), 22.4 (CH2CH2CH2O), 29.2 (CH2CH2CH2O), 33.4 (CH2CH2CH2O), 55.7 (OCH3), 68.3 (C-5), 69.6 (C-4), 69.9 (C-1), 114.5 (C-3ar, C-5ar), 115.4 (C-2ar, C-6ar), 122.6 (C-3), 134.1 (C-2), 153.1 (C-1ar), 153.67 (C-4ar), 170.7 (C=O). HRMS: calcd for [M+Na]+: m/z 359.1471, found: m/z 359.1472. −65.60° (c 1.744, CHCl3) 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.6 Hz, CH3), 1.50–1.90 (m, 6H, CH2CH2CH2), 2.08 (s, 3H, CH3CO), 3.76 (s, 3H, OCH3), 3.88–3.96 (m, 3H, OCH2; H-5), 4.16 (m, 1H, H-1), 4.89 (m, 1H, H-4), 5.76 (ddd, 1H, J = 10.3 Hz, J = 3.5 Hz, J = 2.1Hz, H-3), 5,92 (ddd, 1H, J = 10.4 Hz, J = 2.1 Hz, J = 1.3Hz, H-2), 6.83(s, 4H, H-2ar, H-3ar, H-5ar, H-6ar); 13C-NMR (75 MHz, CDCl3) δ (ppm): 17.23 (CH3), 21.20 (CH3CO) 22.36 (CH2CH2CH2CH2O), 29.23 (CH2CH2CH2CH2O), 33.44 (CH2CH2CH2CH2O), 55.68 (OCH3), 68.33 (C-5), 68.49 (CH2O), 69.65 (C-4), 69.93 (C-1), 114.57 (C-3ar, C-5ar), 115.36 (C-2ar, C-6ar), 122.57 (C-3), 134.11 (C-2), 153.15 (C-1ar), 153.67 (C-4ar) 170.74 (C=O). −54.13°, (c 1.84, CHCl3), 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.0 Hz, CH3), 1.50–1.65 (m, 4H, CH2CH2), 1.71–1.85 (m, 2H, CH2CH2O) 2.07 (s, 3H, CH3CO), 3.57 (dq, 1H, J = 8.7 Hz, J = 6.2 Hz, H-5), 3.76 (s, 3H, OCH3), 3.90 (t, 2H, J = 6.5 Hz, OCH2), 4.15 (m, 1H, H-1), 5.04 (m, 1H, H-4), 5.70 (ddd, 1H, J = 10.3 Hz, J = 2.0 Hz, J = 2.0 Hz, H-2), 5.80 (ddd, J = 10.3, Hz, J = 1.5 Hz, J = 1.5 Hz, H-3) 6.82 (s, 4H, H-2ar, H-3ar, H-5ar, H-6ar). 13C-NMR (150 MHz, CDCl3) δ (ppm): 18.44 (CH3), 21.08 (CH2CH2CH2CH2O), 21.35 (CH3CO), 29.29 (CH2CH2CH2CH2O), 34.96 (CH2CH2CH2CH2O), 55.66 (OCH3), 68.37 (CH2O), 71.27 (C-5), 72.32 (C-4), 74.44 (C-1), 114.56 (C-3ar, C-5ar), 115.38 (C-2ar, C-6ar), 125.71 (C-3), 132.92(C-2), 153.17 (C-1ar), 153.66 (C-4ar) 170.50 (C=O). −58.23° (c 2.23, CHCl3), 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.6 Hz, CH3), 1.38–1.71 (m, 6H, CH2CH2CH2CH2CH2O), 1.78 (q, 2H J = 6.6 Hz, CH2CH2O), 2.08 (s, 3H, CH3CO), 3.76 (s, 3H, OCH3), 3.84–3.95 (m, 3H, OCH2; H-5), 4.14 (m, 1H, H-1), 4.89 (m, 1H, H-4), 5.75 (ddd, 1H, J = 10.3 Hz, J = 3.4 Hz, J = 2.0Hz, H-3), 5.91 (ddd, 1H, J = 10.3 Hz, J = 2.2 Hz, J = 1.2 Hz, H-2), 6.83(s, 4H, H-2ar, H-3ar, H-5ar, H-6ar), 13C-NMR (75 MHz, CDCl3) δ (ppm): 17.23 (CH3), 21.21 (CH3CO), 25.56 (CH2CH2CH2CH2CH2O), 25.98 (CH2CH2CH2CH2CH2O), 29.28 (CH2CH2CH2CH2CH2O), 33.44 (CH2CH2CH2CH2CH2O), 55.68 (OCH3), 68.40 (C-5; CH2O), 69.71 (C-4), 70.06 (C-1), 114.56 (C-3ar, C-5ar), 115.36 (C-2ar, C-6ar), 122.52 (C-3), 134.19 (C-2), 153.18 (C-1ar), 153.64 (C-4ar), 170.75 (C=O). −90.16°, (c 1.96, CHCl3), 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.0 Hz, CH3), 1.35–1.63 (m, 6H, CH2CH2CH2), 1.68–1.84 (m, 2H, CH2CH2O), 2.08 (s, 3H, CH3CO), 3.57 (dq, 1H, J = 8.6 Hz, J = 6.2 Hz, H-5), 3.76 (s, 3H, OCH3), 3.90 (t, 2H, J = 6.6 Hz, OCH2), 4.14 (m, 1H, H-1), 5.04 (m, 1H, H-4), 5,68 (ddd, 1H, J = 10.3 Hz, J = 1.9 Hz, J = 1.9 Hz, H-2), 5.78 (ddd, J = 10.3 Hz, J = 1.6 Hz, J = 1.6 Hz, H-3) 6,82(s, 4H, H-2ar, H-3ar, H-5ar, H-6ar), 13C-NMR (75 MHz, CDCl3) δ (ppm): 18.47(CH3), 21.14 (CH3CO), 24.51 (CH2CH2CH2CH2CH2O), 26.06 (CH2CH2CH2CH2CH2O), 29.27 (CH2CH2CH2CH2CH2O), 35.20 (CH2CH2CH2CH2CH2O) 55.69 (OCH3), 68.44 (CH2O), 71.31 (C-5), 72.32 (C-4), 74.50 (C-1), 114.56 (C-3ar, C-5ar), 115.38 (C-2ar, C-6ar), 125.32 (C-3), 132.06 (C-2), 153.20 (C-1ar), 153.63 (C-4ar), 170.57 (C=O).
−57.0° (c 1.37, CHCl3); 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, J = 6.6 Hz, 3H, CH3), 1.70–2.00 (m, 4H, CH2CH2), 2.08 (s, 3H, CH3CO), 3.76 (s, 3H, OCH3), 3.91 (dq, J = 6.6 Hz and 4.9 Hz, 1H, H-5), 3.95 (t, J= 6.3 Hz, 2H, OCH2), 4.21 (m, 1H, H-1), 4.89 (m, 1H, H-4), 5.78 (ddd, J=10.3 Hz, 3.5 Hz and 2.1 Hz, 1H, H-3), 5.92 (ddd, 1H, J = 10.4 Hz, 2.2 Hz and 1.3 Hz, 1H, H-2), 6.83 (s, 4H, H-2ar, H-3ar, H-5ar, H-6ar). 13C-NMR (75 MHz, CDCl3) δ (ppm): 17.2 (CH3), 21.5 (CH3CO), 26.0 (CH2CH2CH2), 30.4 (CH2CH2CH2), 56.0 (OCH3), 68.5 (C-5), 68.7 (CH2O), 69.9 (C-4), 70.1 (C-1), 114.6 (C-3ar, C-5ar), 115.7 (C-2ar, C-6ar), 123.1 (C-3), 134.2 (C-2), 153.4(C-1ar), 154.0 (C-4ar), 171.0 (C=O). HRMS: calcd for [M+Na]+: m/z 359.1471. Found: m/z 359.1473. −65.60° (c 1.74, CHCl3), 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, J = 6.6 Hz, 3H, CH3), 1.50–1.90 (m, 6H, CH2CH2CH2), 2.08 (s, 3H, CH3CO), 3.76 (s, 3H, OCH3), 3.88–3.96 (m, 3H, OCH2; H-5), 4.16 (m, 1H, H-1), 4.89 (m, 1H, H-4), 5.76 (ddd, J = 10.3 Hz, 3.5 Hz and 2.1 Hz, 1H, H-3), 5.92 (ddd, J = 10.4 Hz, 2.1 Hz, and 1.3 Hz, 1H, H-2), 6.83 (s, 4H, H-2ar, H-3ar, H-5ar, H-6ar). 13C-NMR (75 MHz, CDCl3) δ (ppm): 17.2 (CH3), 21.2 (CH3CO), 22.4 (CH2CH2CH2O), 29.2 (CH2CH2CH2O), 33.4 (CH2CH2CH2O), 55.7 (OCH3), 68.3 (C-5), 69.6 (C-4), 69.9 (C-1), 114.5 (C-3ar, C-5ar), 115.4 (C-2ar, C-6ar), 122.6 (C-3), 134.1 (C-2), 153.1 (C-1ar), 153.67 (C-4ar), 170.7 (C=O). HRMS: calcd for [M+Na]+: m/z 359.1471, found: m/z 359.1472. −65.60° (c 1.744, CHCl3) 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.6 Hz, CH3), 1.50–1.90 (m, 6H, CH2CH2CH2), 2.08 (s, 3H, CH3CO), 3.76 (s, 3H, OCH3), 3.88–3.96 (m, 3H, OCH2; H-5), 4.16 (m, 1H, H-1), 4.89 (m, 1H, H-4), 5.76 (ddd, 1H, J = 10.3 Hz, J = 3.5 Hz, J = 2.1Hz, H-3), 5,92 (ddd, 1H, J = 10.4 Hz, J = 2.1 Hz, J = 1.3Hz, H-2), 6.83(s, 4H, H-2ar, H-3ar, H-5ar, H-6ar); 13C-NMR (75 MHz, CDCl3) δ (ppm): 17.23 (CH3), 21.20 (CH3CO) 22.36 (CH2CH2CH2CH2O), 29.23 (CH2CH2CH2CH2O), 33.44 (CH2CH2CH2CH2O), 55.68 (OCH3), 68.33 (C-5), 68.49 (CH2O), 69.65 (C-4), 69.93 (C-1), 114.57 (C-3ar, C-5ar), 115.36 (C-2ar, C-6ar), 122.57 (C-3), 134.11 (C-2), 153.15 (C-1ar), 153.67 (C-4ar) 170.74 (C=O). −54.13°, (c 1.84, CHCl3), 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.0 Hz, CH3), 1.50–1.65 (m, 4H, CH2CH2), 1.71–1.85 (m, 2H, CH2CH2O) 2.07 (s, 3H, CH3CO), 3.57 (dq, 1H, J = 8.7 Hz, J = 6.2 Hz, H-5), 3.76 (s, 3H, OCH3), 3.90 (t, 2H, J = 6.5 Hz, OCH2), 4.15 (m, 1H, H-1), 5.04 (m, 1H, H-4), 5.70 (ddd, 1H, J = 10.3 Hz, J = 2.0 Hz, J = 2.0 Hz, H-2), 5.80 (ddd, J = 10.3, Hz, J = 1.5 Hz, J = 1.5 Hz, H-3) 6.82 (s, 4H, H-2ar, H-3ar, H-5ar, H-6ar). 13C-NMR (150 MHz, CDCl3) δ (ppm): 18.44 (CH3), 21.08 (CH2CH2CH2CH2O), 21.35 (CH3CO), 29.29 (CH2CH2CH2CH2O), 34.96 (CH2CH2CH2CH2O), 55.66 (OCH3), 68.37 (CH2O), 71.27 (C-5), 72.32 (C-4), 74.44 (C-1), 114.56 (C-3ar, C-5ar), 115.38 (C-2ar, C-6ar), 125.71 (C-3), 132.92(C-2), 153.17 (C-1ar), 153.66 (C-4ar) 170.50 (C=O). −58.23° (c 2.23, CHCl3), 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.6 Hz, CH3), 1.38–1.71 (m, 6H, CH2CH2CH2CH2CH2O), 1.78 (q, 2H J = 6.6 Hz, CH2CH2O), 2.08 (s, 3H, CH3CO), 3.76 (s, 3H, OCH3), 3.84–3.95 (m, 3H, OCH2; H-5), 4.14 (m, 1H, H-1), 4.89 (m, 1H, H-4), 5.75 (ddd, 1H, J = 10.3 Hz, J = 3.4 Hz, J = 2.0Hz, H-3), 5.91 (ddd, 1H, J = 10.3 Hz, J = 2.2 Hz, J = 1.2 Hz, H-2), 6.83(s, 4H, H-2ar, H-3ar, H-5ar, H-6ar), 13C-NMR (75 MHz, CDCl3) δ (ppm): 17.23 (CH3), 21.21 (CH3CO), 25.56 (CH2CH2CH2CH2CH2O), 25.98 (CH2CH2CH2CH2CH2O), 29.28 (CH2CH2CH2CH2CH2O), 33.44 (CH2CH2CH2CH2CH2O), 55.68 (OCH3), 68.40 (C-5; CH2O), 69.71 (C-4), 70.06 (C-1), 114.56 (C-3ar, C-5ar), 115.36 (C-2ar, C-6ar), 122.52 (C-3), 134.19 (C-2), 153.18 (C-1ar), 153.64 (C-4ar), 170.75 (C=O). −90.16°, (c 1.96, CHCl3), 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.0 Hz, CH3), 1.35–1.63 (m, 6H, CH2CH2CH2), 1.68–1.84 (m, 2H, CH2CH2O), 2.08 (s, 3H, CH3CO), 3.57 (dq, 1H, J = 8.6 Hz, J = 6.2 Hz, H-5), 3.76 (s, 3H, OCH3), 3.90 (t, 2H, J = 6.6 Hz, OCH2), 4.14 (m, 1H, H-1), 5.04 (m, 1H, H-4), 5,68 (ddd, 1H, J = 10.3 Hz, J = 1.9 Hz, J = 1.9 Hz, H-2), 5.78 (ddd, J = 10.3 Hz, J = 1.6 Hz, J = 1.6 Hz, H-3) 6,82(s, 4H, H-2ar, H-3ar, H-5ar, H-6ar), 13C-NMR (75 MHz, CDCl3) δ (ppm): 18.47(CH3), 21.14 (CH3CO), 24.51 (CH2CH2CH2CH2CH2O), 26.06 (CH2CH2CH2CH2CH2O), 29.27 (CH2CH2CH2CH2CH2O), 35.20 (CH2CH2CH2CH2CH2O) 55.69 (OCH3), 68.44 (CH2O), 71.31 (C-5), 72.32 (C-4), 74.50 (C-1), 114.56 (C-3ar, C-5ar), 115.38 (C-2ar, C-6ar), 125.32 (C-3), 132.06 (C-2), 153.20 (C-1ar), 153.63 (C-4ar), 170.57 (C=O).3.6. General Procedure for Deprotection
−101.13° (c 1.15, CHCl3); 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, J = 6.6 Hz, 3H, CH3), 1.60–1.80 (m, 4H, CH2CH2), 2.03 (s, 3H, CH3CO), 2.32 (s 1H, OH), 3.66 (m, 2H, CH2OH), 3.92 (dq, J = 6.5 Hz, 4.5 Hz, 1H, H-5), 4.18 (m,1H, H-1), 4.87 (m, 1H, H-4), 5.79 (ddd, J = 10.3 Hz, 3.6 Hz, 2.1 Hz, 1H, H-3), 5.89 (ddd, J = 10.3 Hz, 2.1 Hz, 1.2 Hz, 1H, H-2), 13C-NMR (75 MHz, CDCl3) δ (ppm): 16.76 (CH3), 21.2 (CH3CO), 29.2 (CH2CH2CH2OH), 30.4 (CH2CH2CH2OH), 62.6 (CH2OH), 68.7 (C-5), 69.4 (C-4), 69.9 (C-1), 122.7 (C-3), 134.8 (C-2), 170.7 (C=O). HRMS: calcd for [M+Na]+: m/z 253.1052, found: 253.1050. −126.81° (c 0.82, CHCl3); 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.24 (d, J = 6.0 Hz, 3H, CH3), 1.54–1.63 (m, 1H, CH2(a)CH2), 1.65–1.75 (m, 3H, CH2(b)CH2), 2.08 (s, 3H, CH3CO), 2.42 (s 1H, OH), 3.60 (dq, 1H, J = 8.7 Hz and 6.2 Hz, H-5), 3.64 (m, 2H, CH2OH), 4.19 (m, 1H, H-1), 5.05 (m, 1H, H-4), 5.70 (ddd, J = 10.3 Hz, 2.1 Hz, 2.1 Hz, 1H, H-2), 5.77 (ddd, J = 10.3 Hz, 1.6 Hz, 1.6 Hz, 1H, H-3); 13C-NMR (150 MHz, CDCl3) δ (ppm): 18.4 (CH3), 21.07 (CH3CO), 28.3 (CH2CH2CH2OH), 32.0 (CH2CH2CH2OH), 62.6 (CH2OH), 71.1 (C-5), 72.43 (C-4), 74.5 (C-1), 125.6 (C-3),132.7 (C-2), 170.5 (C=O). HRMS: calcd for [M+Na]+: m/z 253.1052, found: m/z 253.1056. −89.66° (c 1.09, CHCl3), 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.6 Hz, CH3), 1.42–1.72 (m, 6H, CH2CH2CH2), 1.76 (s 1H, OH), 2.08 (s, 3H, CH3CO), 3.65 (t, 2H, J = 6.5 Hz, CH2OH), 3.90 (dq, 1H, J = 6.6 Hz, J = 4.7 Hz, H-5), 4.14 (m, 1H, H-1), 4.88 (m, 1H, H-4), 5.76 (ddd, 1H J = 10.3 Hz, J = 3.6 Hz, J = 2.1 Hz, H-3), 5.91 (ddd, 1H, J = 10.4 Hz, J = 2.3 Hz, J = 1.3 Hz, H-2). 13C-NMR (150 MHz, CDCl3) δ (ppm): 16.88 (CH3), 21.18 (CH3CO), 21.90 (CH2CH2CH2CH2OH), 32.48 (CH2CH2CH2CH2OH), 33.35 (CH2CH2CH2CH2OH), 62.62 (CH2OH), 68.52 (C-5), 69.62 (C-4), 69.92 (C-1), 122.50 (C-3), 134.10 (C-2), 170.77 (C=O). −88.73° (c 1.63, CHCl3), 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.6 Hz, CH3), 1.33–1.70 (m, 8H, CH2CH2CH2CH2), 1.73 (s 1H, OH), 2.08 (s, 3H, CH3CO), 3.64 (t, 2H, J = 6.8 Hz, CH2OH), 3.89 (dq, 1H, J = 6.5 Hz, J = 4.7 Hz, H-5), 4.13 (m, 1H, H-1), 4.88 (m, 1H, H-4), 5.75 (ddd, 1H, J = 10.3 Hz, J = 3.5 Hz, J = 2.1 Hz, H-3), 5.91 (ddd, 1H, J = 9.4 Hz, J = 2.3 Hz, J = 1.2 Hz, H-2). 13C-NMR (75 MHz, CDCl3) δ (ppm): 16.94 (CH3), 21.15 (CH3CO), 25.48 (CH2CH2CH2CH2CH2OH), 25.59 (CH2CH2CH2CH2CH2OH), 33.56 (CH2CH2CH2CH2CH2OH), 33.75 (CH2CH2CH2CH2CH2OH), 62.67 (CH2OH), 68.35 (C-5), 69.71 (C-4), 70.05 (C-1), 122.48 (C-3), 134.14 (C-2), 170.74 (C=O). −119.32° (c 1.294, CHCl3), 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.4 Hz, CH3), 1.30–1.65 (m, 8H, CH2CH2CH2CH2), 1.79 (s 1H, OH), 2.08 (s, 3H, CH3CO), 3,57 (dq, 1H, J = 8.7, J = 6.2Hz, H-5), 3.64 (t, 2H, J = 6.5 Hz, CH2OH), 4.14 (m, 1H, H-1), 5.04 (m, 1H, H-4), 5.68 (ddd, 1H, J = 10.3 Hz, J = 2.0 Hz, J = 2.0 Hz, H-2), 5.79 (ddd, 1H, J = 10.3 Hz, J = 1.5 Hz, J = 1.5 Hz, H-3); 13C-NMR (75 MHz, CDCl3) δ (ppm): 18.43 (CH3), 21.12 (CH3CO), 25.48 (CH2CH2CH2CH2CH2OH), 25.70 (CH2CH2CH2CH2CH2OH), 33.58 (CH2CH2CH2CH2CH2OH), 35.18 (CH2CH2CH2CH2CH2OH), 62.78 (CH2OH), 71.31 (C-5), 72.30 (C-4), 74.50 (C-1), 125.28 (C-3), 133.03 (C-2), 170.61 (C=O).3.7. General Procedure for the Transformation of Hydroxyl Group to Bromide
−62.20° (c 0.947, CHCl3); 1H-NMR (600 MHz, CDCl3) δ (ppm): 1,24 (d, J = 6.6 Hz, 3H, CH3), 1.62–2.15 (m, 4H, CH2CH2), 2.09 (s, 3H, CH3CO), 3.48 (m, 2H, CH2Br), 3.90 (dq, J = 6.5 Hz and J = 4.7 Hz, 1H, H-5), 4.18 (m, 1H, H-1), 4.89 (m, 1H, H-4), 5.79 (ddd, J = 10.3 Hz, 3.5 Hz, and 2.0 Hz, 1H, H-3), 5.89 (ddd, J = 10.3 Hz, 2.1 Hz and 1.1 Hz, 1H, H-2); 13C-NMR (150 MHz, CDCl3) δ (ppm): 16.9 (CH3), 21.2 (CH3CO), 29.0 (CH2CH2CH2Br), 31.9 (CH2CH2CH2Br), 33.8 (CH2Br), 68.5 (C-5), 69.4 (C-4), 69.5 (C-1), 123.1 (C-3), 134.7 (C-2), 170.7 (C=O). HRMS: calcd for [M+Na]+: m/z 315.0208, found: m/z 315.0205. −112.58° (c 1.27, CHCl3); 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.23 (d, J = 6.3 Hz, 3H, CH3), 1.54–1.81 (m, 2H, CH2CH2CH2Br), 1.91–2.05 (m, 2H, CH2CH2CH2Br), 2.08 (s, 3H, CH3CO), 3.44 (t, 2H, J = 6.7 Hz, CH2Br), 3.56 (dq, 1H J = 8.7 Hz and 6.2 Hz, H-5), 4.18 (m, 1H, H-1), 5.03 (m, 1H, H-4), 5.71 (ddd, J = 10.4 Hz, 1.7 Hz, and 1.7 Hz, 1H, H-2), 5.77 (ddd, J = 10.3 Hz, 1.2 Hz, and 1.2 Hz, 1H, H-3); 13C-NMR (150 MHz, CDCl3) δ (ppm): 18.4 (CH3), 21.1 (CH3CO), 28.1 (CH2CH2CH2Br), 33.6 (CH2CH2CH2Br), 33.9 (CH2Br), 71.1 (C-5), 72.34(C-4), 74.0 (C-1), 126.0 (C-3), 132.5 (C-2), 170.5 (C=O). HRMS: calcd for [M+Na]+: m/z 315.0208, found: m/z 315.0204. −52.20° (c 1.63, CHCl3), 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.3Hz, CH3), 1.47–1.73 (m, 4H, CH2CH2), 1.83–1.98 (tt, 2H, J = 7.0 Hz, CH2CH2Br) 2.08 (s, 3H, CH3CO), 3.42 (t, 2H, J = 6,8 Hz, CH2Br), 3.89 (m, 1H, H-5), 4.14 (m, 1H, H-1), 4.88 (m, 1H, H-4), 5.77 (ddd, 1H, J = 10.3 Hz, J = 3.7 Hz, J = 2.2Hz, H-3), 5.90 (m, 1H, H-2); 13C-NMR (150 MHz, CDCl3) δ (ppm): 16.91 (CH3), 21.19 (CH3CO), 24.40 (CH2CH2CH2CH2Br), 32.57 (CH2CH2CH2CH2Br), 32.76 (CH2Br), 33.52 (CH2CH2CH2CH2Br), 68.52 (C-5), 69.58 (C-4), 69.77 (C-1), 122.77 (C-3), 133.90 (C-2), 170.69 (C=O). −84.67° (c 1.807, CHCl3); 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.23 (d, 3H, J = 6.3 Hz, CH3), 1.49–1.62 (m, 4H, CH2CH2), 1.82–1.96 (m, 2H, CH2CH2Br), 2.08 (s, 3H, CH3CO), 3.41 (t, 2H, J = 6.8 Hz, CH2Br), 3.56 (dq, 1H, J = 8.8 J = 6.1 Hz, H-5), 4.14(m, 1H, H-1), 5.03 (m, 1H, H-4), 5.69 (ddd, 1H, J = 10.3 Hz, J = 2.0 Hz, J = 2.0Hz, H-2), 5.77 (ddd, 1H, J = 10.6 Hz, J = 1.8 Hz, J = 1.8Hz, H-3). 13C-NMR (150 MHz, CDCl3) δ (ppm): 18.45(CH3), 21.11(CH3CO), 23.45 (CH2CH2CH2CH2Br), 32.72 (CH2Br), 33.56 (CH2CH2CH2CH2Br), 34.30 (CH2CH2CH2CH2Br), 71.22 (C-5), 72.33 (C-4), 74.26 (C-1) 125.65 (C-3), 132.73 (C-2), 170.51 (C=O). −100.93° (c 0.86, CHCl3); 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.6 Hz, CH3), 1.36–1.70 (m, 6H, CH2CH2CH2), 1.88 (q, 2H, J = 7.0 Hz, CH2CH2Br) 2.08 (s, 3H, CH3CO), 4.02 (t, 2H, J = 6.7 Hz, CH2Br), 3.91 (dq, 1H, J = 6.5 Hz, J = 4.7 Hz, H-5), 4.13 (m, 1H, H-1), 4.88 (m, 1H, H-4), 5.77 (ddd, 1H, J = 10.6 Hz, J = 3.5 Hz, J = 1.7Hz, H-3), 5.94 (ddd, 1H, J = 10.6 Hz, J = 2.3 Hz, J = 1.2 Hz, H-2). 13C-NMR (150 MHz, CDCl3) δ (ppm): 16.94 (CH3), 21.19 (CH3CO),24.91 (CH2CH2CH2CH2CH2Br), 28.88 (CH2CH2CH2CH2CH2Br), 32.63 (CH2CH2CH2CH2CH2Br), 33.45 (CH2CH2CH2CH2CH2Br), 33.74 (CH2Br), 68.42 (C-5), 69.63 (C-4), 69.90 (C-1),122.58 (C-3), 134.07 (C-2), 170.69 (C=O). −126.07° (c 1.417, CHCl3), 1H-NMR (300 MHz, CDCl3) δ (ppm): 1.24 (d, 3H, J = 6.4 Hz, CH3), 1.35–1.62 (m, 6H, CH2CH2CH2), 1.80–1.93 (m, 2H, CH2CH2Br) 2.08 (s, 3H, CH3CO), 3.41 (t, 2H, J = 6.8 Hz, CH2Br), 3,57 (dq, 1H, J = 8.6 Hz, J = 6.2 Hz, H-5), 4.14 (m, 1H, H-1), 5.04 (m, 1H, H-4), 5.69 (ddd, 1H, J = 10.3 Hz, J = 2.0 Hz, J = 2.0 Hz, H-2), 5.78 (ddd, 1H, J = 10.3 Hz, J = 1.5 Hz, J = 1.5Hz, H-3); 13C-NMR (75 MHz, CDCl3) δ (ppm): 18.46 (CH3), 21.14 (CH3CO), 23.87 (CH2CH2CH2CH2CH2Br), 28.10 (CH2CH2CH2CH2CH2Br), 32.65 (CH2CH2CH2CH2CH2Br), 33.83 (CH2Br), 35.03 (CH2CH2CH2CH2CH2Br), 71.27 (C-5), 72.32 (C-4), 74.40 (C-1), 125.46 (C-3), 132.94 (C-2), 170.56 (C=O).3.8. General Procedure for Synthesis of 2,3-Enopyranosyl C-Linked Conjugates of Genistein
−35.82° (c 0.67, CHCl3), HRMS: calcd for [M+Na]+: 489.5, found: m/z = 489.0. 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.27 (d, 3H, J = 6.6 Hz, CH3), 1.68–2.05 (m, 4H, CH2CH2), 2.10 (s, 3H, CH3CO), 3.91 (dq, 1H, J = 6.6, J = 4.6 Hz, H-5), 4.06 (t, 2H, J = 6.1 Hz, OCH2), 4.24 (m, 1H, H-1), 4.91 (m, 1H, H-4), 5.81 (ddd, 1H, J = 10.3 Hz, J = 3.6 Hz, J = 2.0 Hz, H-3), 5.94 (ddd, 1H, J = 10.3 Hz, J = 2.1 Hz, J = 1.1 Hz, H-2), 6.29 (s, 1H, 4'-OH), 6,36 (d, 1H, J = 2.4 Hz,, H-8), 6,38 (d, 1H, J = 2.4 Hz, H-6), 6.84 (AA'XX', 2H, J = 8.7 Hz, 2H, H-3'g, H-5'g), 7.34 (AA'XX', 2H, J = 8.7 Hz,, 2H, H-2'g, H-6'g), 7.84 (s, 1H, H-2g), 12.80 (s, 1H, 5g-OH); 13C-NMR (150 MHz, CDCl3) δ (ppm): 16.82 (CH3), 21.19 (CH3CO), 25.23 (CH2CH2CH2O), 29,91 (CH2CH2CH2O), 68.24 (C-5), 68.72 (CH2O), 69.53 (C-4), 69.55 (C-1), 92.82 (C-8g), 98.64 (C-6g), 106.12 (C-4a), 115.67 (C-3'g, C-5'g), 122.52 (C-1'g), 122.85 (C-3), 123.71 (C-3g), 130.22 (C-2'g, C-6'g), 133.82 (C-2), 152.80 (C-2g), 156.26 (C-4'g), 157.94 (C-8a), 162.50 (C-5g), 164.99 (C-7g) 171.00 (C=O), 180.94 (C-4g). −74.69° (c 0.64, CHCl3) HRMS: calcd for [M+Na]+: 489.5, found: m/z = 489.0, 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.26 (d, 3H, J = 6.2 Hz, CH3), 1.59-2.00 (m, 4H, CH2CH2) 2.10 (s, 3H, CH3CO), 3.61 (dq, 1H, J = 8.6, J = 6.2 Hz, H-5), 4.07 (t, 2H, J = 6.3 Hz, CH2O), 4.25 (m, 1H, H-1), 5.07 (m, 1H, H-4), 5.54 (s, 1H, 4'-OH), 5.73 (ddd, 1H J = 10.3 Hz J = 1.8 Hz, H-2), 5.81 (ddd, 1H, J = 10.3 Hz, J = 1.4 Hz, J = 1.4 Hz, H-3), 6.36 (d, 1H, J = 2.2 Hz, H-8), 6.39 (d, 1H, J = 2.2 Hz, H-6), 6.87 (AA'XX', 2H, J = 8.6 Hz, 2H, H-3'g, H-5'g), 7.37 (AA'XX', 2H, J=8.6 Hz, 2H, H-2'g, H-6'g), 7.85(s, 1H, H-2g), 12.80(s, 1H, 5g-OH); 13C-NMR (150 MHz, CDCl3) δ (ppm): 18.50 (CH3), 21.17 (CH3CO), 24.32 (CH2CH2CH2O), 31.49 (CH2CH2CH2O), 68.48 (CH2O), 71.26 (C-5), 72.43 (C-4), 74.08 (C-1), 92.87 (C-8g), 98.68 (C-6g), 106.19 (C-4a), 115.65 (C-3'g, C-5'g), 122.94 (C-1'g), 123.69 (C-3g) 125.99 (C-3), 130.33 (C-2'g, C-6'g), 132.65 (C-2), 152.77 (C-2g), 156.04 (C-4'g), 157.99 (C-8a), 162.33 (C-5g), 165.06 (C-7g), 170.74 (C=O), 180.90 (C-4g). −37.51°(c 1.27, CHCl3), HRMS: calcd for [M+Na]+: 503.5, found: m/z = 503.1; 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.27 (d, 3H, J = 6.6 Hz, CH3), 1.5–2.00 (m, 6H, CH2CH2CH2), 2.09 (s, 3H, CO-CH3), 3.95 (dq, 1H, J = 6.6 Hz, J = 4.4 Hz, H-5), 4.01 (t, 2H, J = 6.1 Hz, OCH2), 4.19 (m, 1H, H-1), 4.90 (m, 1H, H-4), 5.79 (ddd, 1H, J = 10.3 Hz, J = 3.6 Hz, J = 2.0 Hz, H-3), 5.94 (ddd, 1H, J = 10.3 Hz, J = 2.0 Hz, J = 1.1Hz, H-2), 6.35 (d, 1H, J = 2.2 Hz, H-8), 6.37 (d, 1H, J = 2.2Hz, H-6), 6.71 (s, 1H, 4'-OH), 6.87 (AA'XX', 2H, J = 8.6 Hz, 2H, H-3'g, H-5'g), 7.35 (AA'XX', 2H, J = 8.4 Hz, 2H, H-2'g, H-6'g), 7.83 (s, 1H, H-2g), 12.82 (s, 1H, 5g-OH), 13C-NMR (150 MHz, CDCl3) δ (ppm): 16.84 (CH3), 21.21 (CH3CO), 22.24 (CH2CH2CH2CH2O), 28.77 (CH2CH2CH2CH2O), 33.32 (CH2CH2CH2CH2O), 68.40 (C-5), 68.70 (CH2O), 69.62 (C-4), 69.80 (C-1), 92.79 (C-8g), 98.58 (C-6g), 106.09 (C-4a), 115.65 (C-3'g, C-5'g), 122.41(C-1'g) 122.58 (C-3), 123.68 (C-3g), 130.18 (C-2'g, C-6'g) 134,02 (C-2), 152.71 (C-2g), 156.47 (C-4’g), 157.91 (C-8a), 162.52 (C-5g), 164.99 (C-7g) 170.99 (C=O), 180.90 (C-4g). −47.20° (c 1.017, CHCl3), HRMS: [M+Na]+: m/z = 503.1; 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.25 (d, 3H, J = 6.2 Hz, CH3), 1.52–1.66 (m, 4H, CH2CH2), 1.77–1.88 (m, 2H, CH2CH2O), 2.09 (s, 3H, CH3CO), 3.59 (dq, 1H, J = 8.8 Hz, J = 6.2 Hz, J = 6.2 Hz, H-5), 4.01(t, 2H, J = 6.5 Hz, J = 6.2 Hz, OCH2), 4.18 (m, 1H, H-1), 5.06 (m, 1H, H-4), 5.71 (ddd, 1H, J = 10.3 Hz, J = 2.1 Hz, J = 2.1 Hz, J = 6.2 Hz, H-2), 5.79 (ddd, J = 10.3 Hz, J = 1.6 Hz, J = 1.6 Hz, J = 6.2 Hz, H-3), 6,35 (d, 1H, J = 2.1 Hz, J = 6.2 Hz, H-8), 6.37 (d, 1H, J = 2.1 Hz, J = 6.2 Hz, H-6), 6.02 (s, 1H, 4'-OH), 6.85 (AA'XX', 2H, J = 9.0 Hz, 2H, H-3'g, H-5'g), 7.35 (AA'XX', 2H, J = 8.4 Hz, 2H, H-2'g, H-6'g), 7.83 (s, 1H, H-2g), 12.79 (s, 1H, 5g-OH); 13C-NMR (150 MHz, CDCl3) δ (ppm): 18.48 (CH3), 21.18 (CH2CH2CH2CH2O), 21.29 (CH3CO), 28.92 (CH2CH2CH2CH2O), 34.87 (CH2CH2CH2CH2O), 68.50 (CH2O), 71.38 (C-5), 72.44 (C-4), 74.48 (C-1), 92.88 (C-8g), 98.70 (C-6g), 106.15 (C-4a), 115.71 (C-3'g, C-5'g), 122.70 (C-1'g), 122.73 (C-3g), 125.62 (C-3), 130.29 (C-2'g, C-6'g), 132,86 (C-2), 152.82 (C-2g), 156.82 (C-4'g), 157.99 (C-8a), 162.56 (C-5g), 165.11 (C-7g), 170.85 (C=O), 180.9 (C-4g). −39.09° (c 0.66, CHCl3), HRMS: calcd for [M+Na]+: 517.2 found: m/z = 517.5; 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.25 (d, 3H, J = 6.6 Hz, CH3), 1.44–1.72 (m, 6H, CH2CH2CH2), 1.83 (q, 2H, J = 6.8 Hz, CH2CH2O), 2.09 (s, 3H, CH3CO), 3.91 (dq, 1H, J = 6.6 J = 4.8 Hz, H-5), 4.02 (t, 2H, J = 6.1 Hz, OCH2), 4.15 (m, 1H, H-1), 4.90 (m, 1H, H-4), 5.77 (ddd, 1H J = 10.4 Hz, J = 3.5 Hz, J = 2.1 Hz, H-3), 5.91 (ddd, 1H, J = 10.3 Hz, J = 2.3 Hz, J = 1.3 Hz, H-2), 6.36 (d, 1H, J = 2.2 Hz, H-8), 6.38 (d, 1H, J = 2.2 Hz, H-6), 6.90 (AA'XX', 2H, J = 8.4 Hz, H-3'g, H-5'g), 7.41 (AA'XX', 2H, J = 8.4 Hz, H-2'g, H-6'g), 7.85 (s, 1H, H-2g), 12.81 (s, 1H, 5g-OH); 13C-NMR (150 MHz, CDCl3) δ (ppm): 16.92 (CH3), 21.28 (CH3CO), 25.43 (CH2CH2CH2CH2CH2O), 25.86 (CH2CH2CH2CH2CH2O), 28.84 (CH2CH2CH2CH2CH2O), 33.59 (CH2CH2CH2CH2CH2O), 68.50 (C-5), 68.66 (CH2O), 69.74 (C-4), 69.99 (C-1), 92.85 (C-8g), 98.64 (C-6g), 106.07 (C-4a), 115.71 (C-3'g, C-5'g), 122.46 (C-1'g, C-3), 123.70 (C-3g), 130.24 (C-2'g, C-6'g) 134.21 (C-2), 152.83 (C-2g), 156.40 (C-4'g), 157.95 (C-8a), 162.46 (C-5g), 165.09 (C-7g), 171.12 (C=O), 180.96 (C-4g). −65.45° (c 0.64, CHCl3), HRMS: calcd for [M+Na]+: 517.5 found: 517.2; 1H-NMR (600 MHz, CDCl3) δ (ppm): 1.25 (d, 3H, J = 6.2 Hz, CH3), 1.42–1.62 (m, 6H, CH2CH2CH2), 1.78–1.85(m, 2H, CH2CH2O), 2.09 (s, 3H, CH3CO), 3.57 (dq, 1H, J = 8.7 Hz, J = 6.2 Hz, H-5), 4.01 (t, 2H, J = 6.5 OCH2), 4.16 (m, 1H, H-1), 5.05 (m, 1H, H-4), 5.67 (s, 1H, 4'-OH), 5.70 (ddd, 1H, J = 10.4 Hz, J = 2.1 Hz, H-2), 5.79 (ddd, J = 10.3 Hz, J = 1.6 Hz, H-3), 6.36 (d, 1H, J = 2.2 Hz, H-8), 6,38 (d, 1H, J = 2.2 Hz, H-6), 6.85 (AA'XX', 2H, H-3'g, H-5'g J = 8.8 Hz), 7.36 (d, 2H, H-2'g, H-6'g J = 8.7 Hz), 7.84 (s, 1H, H-2g), 12.79 (s, 1H, 5g-OH); 13C-NMR (150 MHz, CDCl3) δ (ppm): 18.49 (CH3ram), 21.18 (CH3CO), 24.43 (CH2CH2CH2CH2CH2O), 25.96 (CH2CH2CH2CH2CH2O), 28.85 (CH2CH2CH2CH2CH2O), 35.15 (CH2CH2CH2CH2CH2O), 68.44 (CH2O), 71.41 (C-5), 72.41 (C-4), 74.55 (C-1), 92.90 (C-8g), 98.68 (C-6g), 106.13 (C-4a), 115.68 (C-3’g, C-5’g), 122.83 (C-1’g), 123.71 (C-3g), 125.45 (C-3), 130.31 (C-2'g, C-6'g), 133,02 (C-2), 152.79 (C-2g), 156.11 (C-4'g), 158.00 (C-8a), 162.59 (C-5g), 165.16 (C-7g), 170.80 (C=O), 180.94 (C-4g).3.9. Anticancer Activity in Vitro
3.9.1. Cell Viability
3.9.2. Cell Cycle Analysis
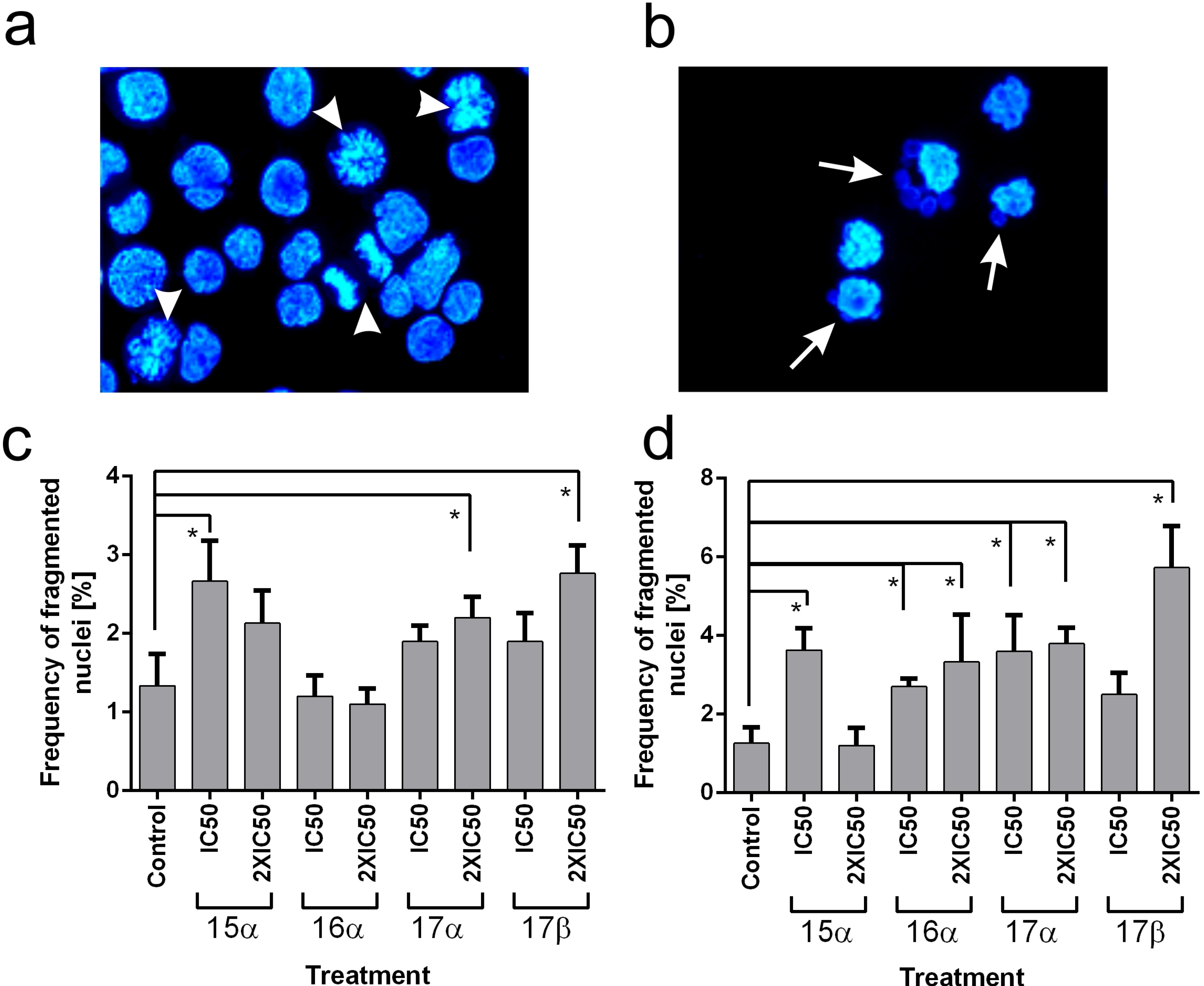
3.9.3. Microscope Analysis
3.9.4. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferrier, R.J.; Hoberg, J.O. Synthesis and reactions of unsaturated sugars. Adv. Carbohydr. Chem. Biochem. 2003, 58, 55–119. [Google Scholar] [CrossRef]
- Fraser-Reid, B. Some progeny of 2,3-unsaturated sugars-they little resemble grandfather glucose-10 years later. Acc. Chem. Res. 1985, 18, 347–354. [Google Scholar] [CrossRef]
- Ferrier, R.J. Unsaturated sugars. Adv. Carbohydr. Chem. Biochem. 1969, 24, 199–266. [Google Scholar]
- Grynkiewicz, G.; Szeja, W.; Boryski, J. Synthetic analogs of natural glycosides in drug discovery and development. Acta Pol. Pharm. 2008, 65, 655–76. [Google Scholar]
- Sears, P.; Wong, C.H. Carbohydrate Mimetics: A New Strategy for Tackling the Problem of Carbohydrate-Mediated Biological Recognition. Angew. Chem. Int. Ed. Engl. 1999, 38, 2300–2324. [Google Scholar] [CrossRef]
- Witczak, Z.J.; Nieforth, K.A. Carbohydrates in drug design. Eur. J. Med. Chem. 1997, 32, 842. [Google Scholar]
- Dorgan, B.J.; Jackson, R.F.W. Synthesis of C-linked glycosyl amino acid derivatives using organozinc reagents. Synlett 1996, 859–861. [Google Scholar]
- Liu, Z.J.; Zhou, M.; Min, J.M.; Zhang, L.H. Syntheses of 5'-O-glycosylnucleosides. Tetrahedron Asymmetr. 1999, 10, 2119–2127. [Google Scholar] [CrossRef]
- Seeberger, P.H.; Bilodeau, M.T.; Danishefsky, S.J. Synthesis of biologically important oligosaccharides and other glycoconjugates by the glycal assembly method. Aldrichim. Acta 1997, 30, 75–92. [Google Scholar]
- Danishefsky, S.J.; Bilodeau, M.T. Glycals in organic synthesis: The evolution of comprehensive strategies for the assembly of oligosaccharides and glycoconjugates of biological consequence. Angew. Chem. Int. Ed. 1996, 35, 1380–1419. [Google Scholar] [CrossRef]
- Schmidt, R.R.; Angerbauer, R. Simple de-novo synthesis of reactive pseudoglycals (Hex-2-enopyranosides)-stereospecific α-glycoside coupling. Angew. Chem. Int. Ed. 1997, 16, 783–784. [Google Scholar]
- Williams, N.R.; Wander, J.D. The Carbohydrates in Chemistry and Biochemistry; Academic Press: New York, NY, USA, 1980; p. 761. [Google Scholar]
- Borrachero-Moya, P.; Cabrera-Escribano, F.; Gomez-Guillen, M.; Paredes-Leon, M.D.R. Synthesis of 4-(4,6-di-O-benzyl-2,3-dideoxy-beta-d-erythro-hex-2-enopyranosyl)pyrazoles from 3,4,6-tri-O-acetyl-d-glucal. Carbohydr. Res. 1998, 308, 181–190. [Google Scholar] [CrossRef]
- Schmidt, M.A.; Talley, J.J.; Scaros, M.G.; Yonan, P.K. Palladium-catalyzed coupling of aryl halides and aryl triflates to itaconate diesters-a convenient preparation of E-benzylidenesuccinate diesters. Chem. Indust. 1995, 62, 105–114. [Google Scholar]
- Polkowski, K.; Popiolkiewicz, J.; Krzeczynski, P.; Ramza, J.; Pucko, W.; Zegrocka-Stendel, O.; Boryski, J.; Skierski, J.S.; Mazurek, A.P.; Grynkiewicz, G. Cytostatic and cytotoxic activity of synthetic genistein glycosides against human cancer cell lines. Cancer Lett. 2004, 203, 59–69. [Google Scholar] [CrossRef]
- Popiolkiewicz, J.; Polkowski, K.; Skierski, J.S.; Mazurek, A.P. In vitro toxicity evaluation in the development of new anticancer drugs-genistein glycosides. Cancer Lett. 2005, 229, 67–75. [Google Scholar] [CrossRef]
- Rusin, A.; Zawisza-Puchalka, J.; Kujawa, K.; Gogler-Piglowska, A.; Wietrzyk, J.; Switalska, M.; Glowala-Kosinska, M.; Gruca, A.; Szeja, W.; Krawczyk, Z.; et al. Synthetic conjugates of genistein affecting proliferation and mitosis of cancer cells. Bioorg. Med. Chem. 2011, 19, 295–305. [Google Scholar] [CrossRef]
- Rusin, A.; Gogler, A.; Glowala-Kosinska, M.; Bochenek, D.; Gruca, A.; Grynkiewicz, G.; Zawisza, J.; Szeja, W.; Krawczyk, Z. Unsaturated genistein disaccharide glycoside as a novel agent affecting microtubules. Bioorg. Med. Chem. Lett. 2009, 19, 4939–4943. [Google Scholar] [CrossRef]
- Gogler-Piglowska, A.; Rusin, A.; Bochenek, D.; Krawczyk, Z. Aneugenic effects of the genistein glycosidic derivative substituted at C7 with the unsaturated disaccharide. Cell Biol. Toxicol. 2012, 28, 331–342. [Google Scholar]
- Priebe, W.; Fokt, I.; Grynkiewicz, G. Glycal Derivatives. In Glycoscience, Chemistry and Chemical Biology, 2nd ed.; Springer-Verlag: Berlin, Heidelberg, Germany, 2008; pp. 699–735. [Google Scholar]
- Levy, D.E.; Tang, C. The Chemistry of C-Glycosides; Elsevier Science Ltd: Tarrytown, NY, USA, 1995; p. 291. [Google Scholar]
- Lown, J.W. Discovery and development of anthracycline antitumor antibiotics. Chem. Soc. Rev. 1993, 22, 165–176. [Google Scholar]
- Ravishankar, R.; Surolia, A.; Vijayan, M.; Lim, S.; Kishi, Y. Preferred conformation of C-lactose at the free and peanut lectin bound states. J. Am. Chem. Soc. 1998, 120, 11297–11303. [Google Scholar] [CrossRef]
- Rubinstenn, G.; Sinay, P.; Berthault, P. Evidence of conformational heterogeneity for carbohydrate mimetics. NMR study of methyl beta-C-lactoside in aqueous solution. J. Phys. Chem. A 1997, 101, 2536–2540. [Google Scholar] [CrossRef]
- Espinosa, J.F.; Montero, E.; Vian, A.; Garcia, J.L.; Dietrich, H.; Schmidt, R.R.; Martin-Lomas, M.; Imberty, A.; Canada, F.J.; Jimenez-Barbero, J. Escherichia coli beta-galactosidase recognizes a high-energy conformation of C-lactose, a nonhydrolizable substrate analogue. NMR and modeling studies of the molecular complex. J. Am. Chem. Soc. 1998, 120, 1309–1318. [Google Scholar] [CrossRef]
- Sparks, M.A.; Williams, K.W.; Whitesides, G.M. Neuraminidase-resistant hemagglutination inhibitors-acrylamide copolymers containing a C-glycoside of N-acetylneuraminic acid. J. Med. Chem. 1993, 36, 778–783. [Google Scholar] [CrossRef]
- Nagy, J.O.; Wang, P.; Gilbert, J.H.; Schaefer, M.E.; Hill, T.G.; Callstrom, M.R.; Bednarski, M.D. Carbohydrate materials bearing neuraminidase-resistant C-glycosides of sialic-acid strongly inhibit the in vitro infectivity of influenza-virus. J. Med. Chem. 1992, 35, 4501–4502. [Google Scholar] [CrossRef]
- Bertozzi, C.R.; Cook, D.G.; Gonzalezscarano, F.; Bednarski, M.D. Carbon-linked galactosphingolipid analogs bind specifically to Hiv-1 Gp120-applications for immunotargeting. J. Cell. Biochem. 1993, 382–382. [Google Scholar]
- Suhadolnik, R.J. Nucleoside Antibiotics; Wiley-Interscience: New York, NY, USA, 1970; p. 480. [Google Scholar]
- Bertozzi, C.; Bednarski, M. Synthesis of C-Glycosides: Stable Mimics of O-Glycosidic Linkages. In Modern Methods in Carbohydrate Synthesis; Harwood Academic Publishers: Amsterdam, Poland, 1996; pp. 316–351. [Google Scholar]
- Stick, R.V. The Synthesis of Novel Enzyme Inhibitors and Their Use in Defining the Active Sites of Glycan Hydrolases. In Topics in Current Chemistry-Glycoscience; Springer Verlag: Berlin, Heilderberg, Germany, 1997; pp. 187–213. [Google Scholar]
- Weatherman, R.V.; Mortell, K.I.; Chervenak, M.; Kiessling, L.L.; Toone, E.J. Specificity of C-glycoside complexation by mannose glucose specific lectins. Biochemistry 1996, 35, 3619–3624. [Google Scholar]
- Sutherlin, D.P.; Stark, T.M.; Hughes, R.; Armstrong, R.W. Generation of C-glycoside peptide ligands for cell surface carbohydrate receptors using a four-component condensation on solid support. J. Org. Chem. 1996, 61, 8350–8354. [Google Scholar]
- Garcia, I.G.; Stevenson, C.E.M.; Uson, I.; Meyers, C.L.F.; Walsh, C.T.; Lawson, D.M. The crystal structure of the novobiocin biosynthetic enzyme novp: The first representative structure for the TylF O-methyltransferase superfamily. J. Mol. Biol. 2010, 395, 390–407. [Google Scholar] [CrossRef]
- Osman, H.; Larsen, D.S.; Simpson, J. Synthesis of orthogonally protected d-olivoside, 1,3-di-O-acetyl-4-O-benzyl-2,6-dideoxy-d-arabinopyranose, as a C-glycosyl donor. Tetrahedron 2009, 65, 4092–4098. [Google Scholar]
- Wang, J.H.; Li, J.; Czyryca, P.G.; Chang, H.W.; Kao, J.; Chang, C.W.T. Synthesis of an unusual branched-chain sugar, 5-C-methyl-l-idopyranose for SAR studies of pyranmycins: Implication for the future design of aminoglycoside antibiotics. Bioorg. Med. Chem. Lett. 2004, 14, 4389–4393. [Google Scholar] [CrossRef]
- Ilas, J.; Anderluh, P.S.; Dolenc, M.S.; Kikelj, D. Recent advances in the synthesis of 2H-1,4-benzoxazin-3-(4H)-ones and 3,4-dihydro-2H-1,4-benzoxazines. Tetrahedron 2005, 61, 7325–7348. [Google Scholar] [CrossRef]
- Allen, C.F.H.; Gates, J.W. Ortho-normal-butoxynitrobenzene-ether, butyl ortho-nitrophenyl. Org. Synth. 1945, 25, 9–11. [Google Scholar] [CrossRef]
- Batcho, A.D.; Leimgruber, W. Indoles from 2-methylnitrobenzenes by condensation with formamide acetals followed by reduction: 4-Benzyloxyindole [1H-indole, 4-(phenylmethoxy)-]. Org. Synth. 1985. [Google Scholar] [CrossRef]
- McKillop, A.; Fiaud, J.C.; Hug, R.P. Use of phase-transfer catalysis for synthesis of phenol ethers. Tetrahedron 1974, 30, 1379–1382. [Google Scholar] [CrossRef]
- Vogel, A.I.; Tatchell, A.R.; Furnis, B.S.; Hannaford, A.J.; Smith, P.W.G. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Longman Scientific & Technical: Essex, UK, 2006. [Google Scholar]
- Orsini, F.; Pelizzoni, F. Synthesis of C-glycosyl compounds-the reaction of acetylated glycals with tert-butoxycarbonylmethylzinc bromide. Carbohydr. Res. 1993, 243, 183–189. [Google Scholar] [CrossRef]
- Pearce, A.J.; Ramaya, S.; Thorn, S.N.; Bloomberg, G.B.; Walter, D.S.; Gallagher, T. C-glycosyl tyrosines. Synthesis and incorporation into C-glycopeptides. J. Org. Chem. 1999, 64, 5453–5462. [Google Scholar] [CrossRef]
- Cook, M.J.; Fletcher, M.J.E.; Gray, D.; Lovell, P.J.; Gallagher, T. Highly stereoselective addition of organozinc reagents to pentopyranose derived glycals: Effect of protecting group and assignment of C-glycoside stereochemistry. Tetrahedron 2004, 60, 5085–5092. [Google Scholar] [CrossRef]
- Szeja, W.; Puchałka, J.; Swierk, P.; Hendrich, A.B.; Grynkiewicz, G. Selective alkylation of genistein and daidzein. Chem. Biol. Interface 2013, 3, 95–106. [Google Scholar]
- Cobas, C.; Domínguez, S.; Larin, N.; Iglesias, I.; Geada, C.; Seoane, F.; uxa Sordo, M.; Monje, P.; Fraga, S.; Cobas, R.; et al. Program MestReNova, version 8.1.4–12489 ed; Mestrelab Research S.L: Santiago de Compostela, Spain, 2013. [Google Scholar]
- Hashimoto, K.; Nakajima, Y.; Matsumura, S.; Chatani, F. An in vitro micronucleus assay with size-classified micronucleus counting to discriminate aneugens from clastogens. Toxicol. In Vitro 2010, 24, 208–216. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Zegrocka-Stendel, O.; Pucko, W.; Ramza, J.; Koscielecka, A.; Kolodziejski, W.; Wozniak, K. X-ray and 13C CP MAS investigations of structure of two genistein derivatives. J. Mol. Struct. 2004, 694, 121–129. [Google Scholar] [CrossRef]
- Yadav, J.S.; Reddy, B.V.S.; Chand, P.K. Sc(OTf)(3)-catalyzed C-glycosidation of glycals: A facile synthesis of allyl glycosides, glycosyl cyanides and glycosyl azides. Tetrahedron Lett. 2001, 42, 4057–4059. [Google Scholar] [CrossRef]
- Ansari, A.A.; Lahiri, R.; Vankar, Y.D. The carbon-Ferrier rearrangement: An approach towards the synthesis of C-glycosides. ARKIVOC 2013, ii, 316–362. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 15a–17a, 15b–17b are available from the authors.
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Szeja, W.; Grynkiewicz, G.; Bieg, T.; Swierk, P.; Byczek, A.; Papaj, K.; Kitel, R.; Rusin, A. Synthesis and Cytotoxicity of 2,3-Enopyranosyl C-Linked Conjugates of Genistein. Molecules 2014, 19, 7072-7093. https://doi.org/10.3390/molecules19067072
Szeja W, Grynkiewicz G, Bieg T, Swierk P, Byczek A, Papaj K, Kitel R, Rusin A. Synthesis and Cytotoxicity of 2,3-Enopyranosyl C-Linked Conjugates of Genistein. Molecules. 2014; 19(6):7072-7093. https://doi.org/10.3390/molecules19067072
Chicago/Turabian StyleSzeja, Wieslaw, Grzegorz Grynkiewicz, Tadeusz Bieg, Piotr Swierk, Anna Byczek, Katarzyna Papaj, Radosław Kitel, and Aleksandra Rusin. 2014. "Synthesis and Cytotoxicity of 2,3-Enopyranosyl C-Linked Conjugates of Genistein" Molecules 19, no. 6: 7072-7093. https://doi.org/10.3390/molecules19067072
APA StyleSzeja, W., Grynkiewicz, G., Bieg, T., Swierk, P., Byczek, A., Papaj, K., Kitel, R., & Rusin, A. (2014). Synthesis and Cytotoxicity of 2,3-Enopyranosyl C-Linked Conjugates of Genistein. Molecules, 19(6), 7072-7093. https://doi.org/10.3390/molecules19067072