Drug-Induced Conformational Population Shifts in Topoisomerase-DNA Ternary Complexes

Abstract
:
1. Introduction
2. Results and Discussion
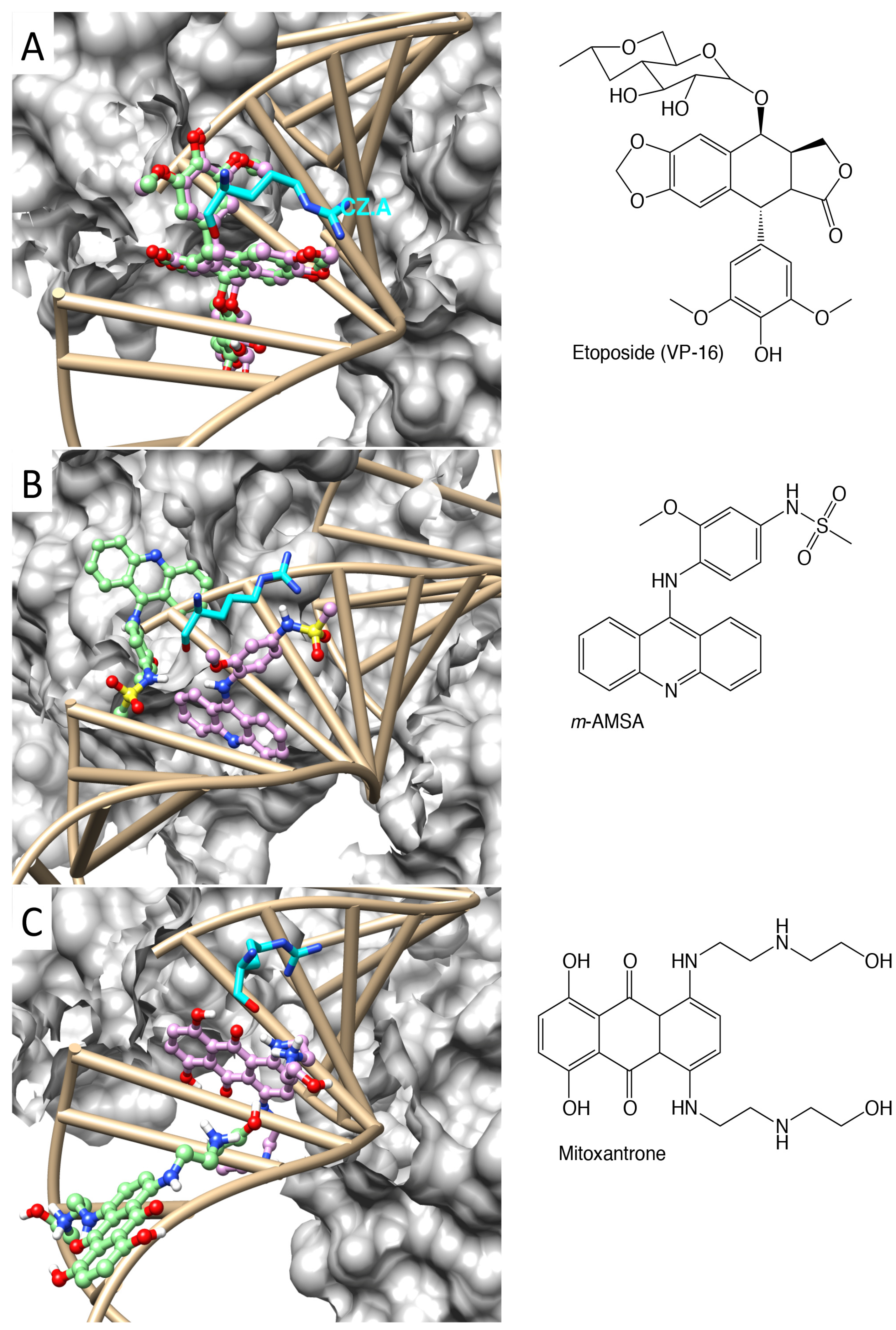
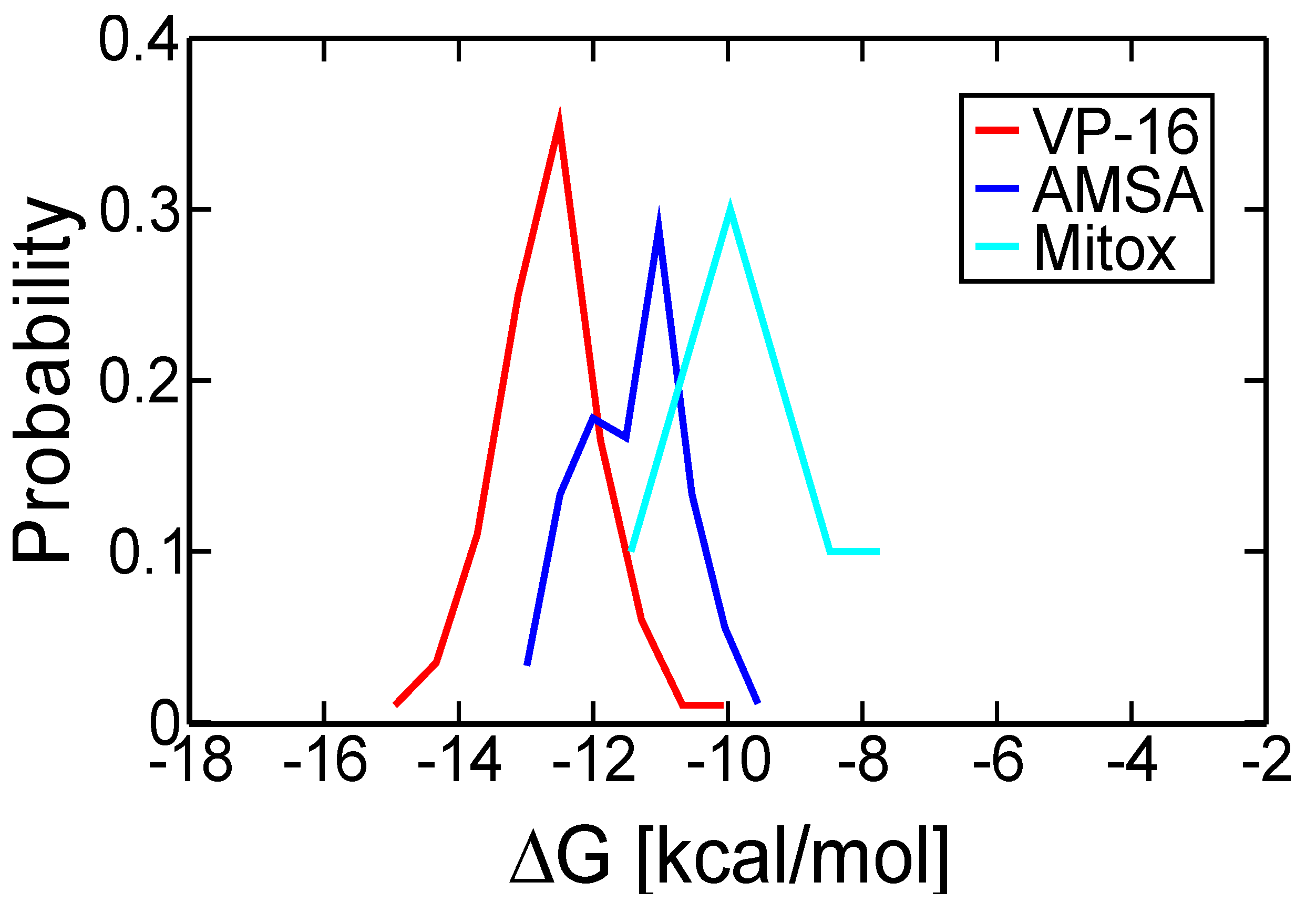
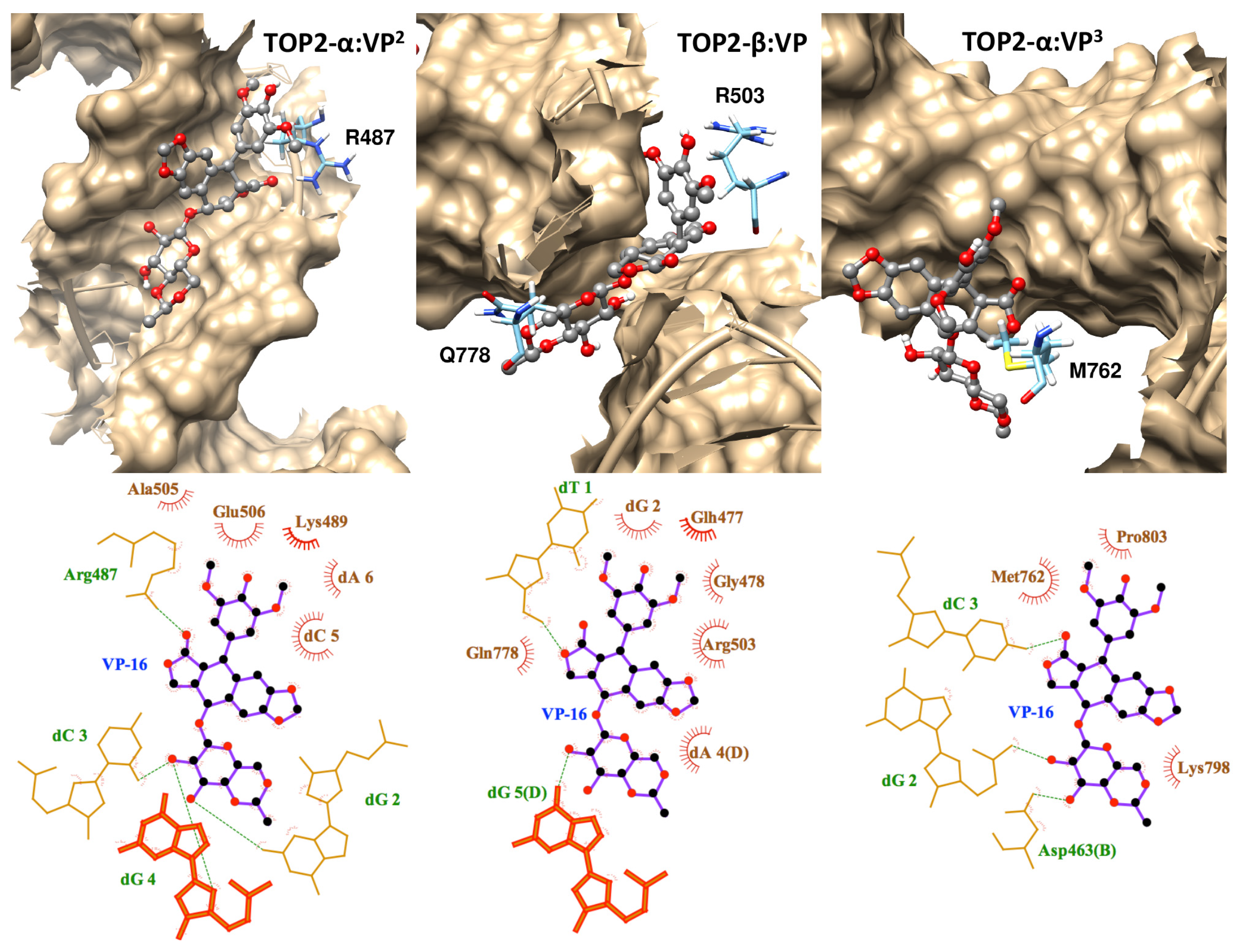
2.1. Molecular Docking Reproduced the Binding Mode of VP-16 in the Cocrystalized Cleavage Complex of TOP2-β


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | VP-16 | m-AMSA | Mitoxantrone |
|---|---|---|---|
| Rescore (kcal/mol) | −14.3 | −2.68 | +1.26 |
| Refined ∆G (kcal/mol) | −14.82 | −3.17 | −1.20 |
| refined RMSD (Å) | 0.62 | 0.64 | 1.07 |
| Docking ∆G (kcal/mol) | −14.79 | −7.53 | −5.59 |
| docking RMSD (Å) | 0.65 | 8.01 | 13.50 |
| Most probable ∆G (kcal/mol) | −12.51 | −11.03 | −10.72 |
| estimated Ki (nM) | 0.68 | 8.21 | 13.98 |
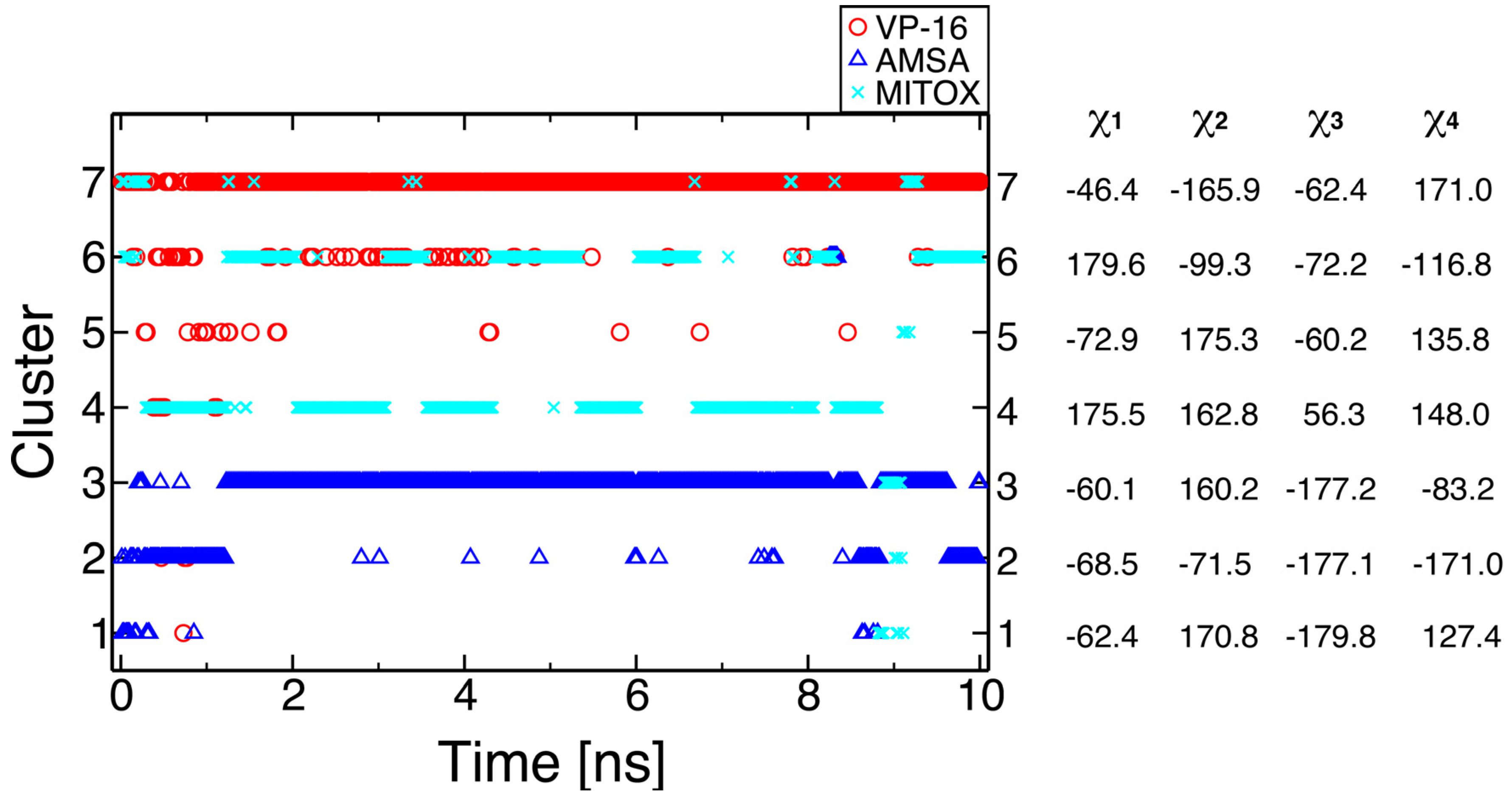
2.2. Molecular Dynamics Simulations Revealed Conformational Changes of the Cleavage Complexes in Response to Ligand Binding


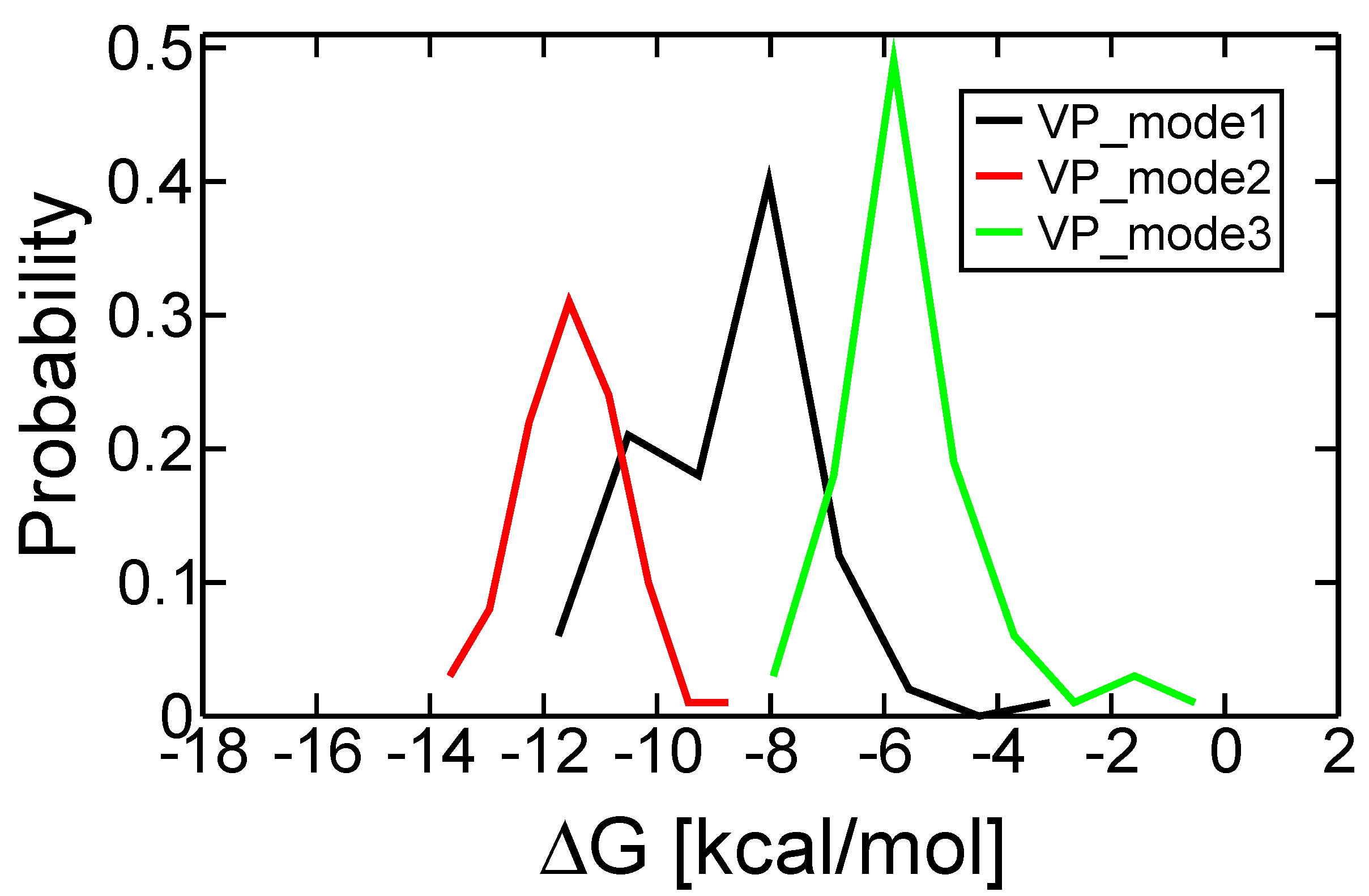
2.3. Discriminating the High-Affinity Binding Mode of VP-16 in the Cleavage Complex of TOP2-α Using Free Energy Spectra
| Initial Binding Mode | 1 | 2 a | 3 |
|---|---|---|---|
| Docking ∆G (kcal/mol) | −10.43 | −10.01 | −9.58 |
| Most probable ∆G (kcal/mol) | −8.07 | −11.54 | −5.83 |
| Estimated Ki | 1.21 µM | 3.48 nM | 52.88 µM |

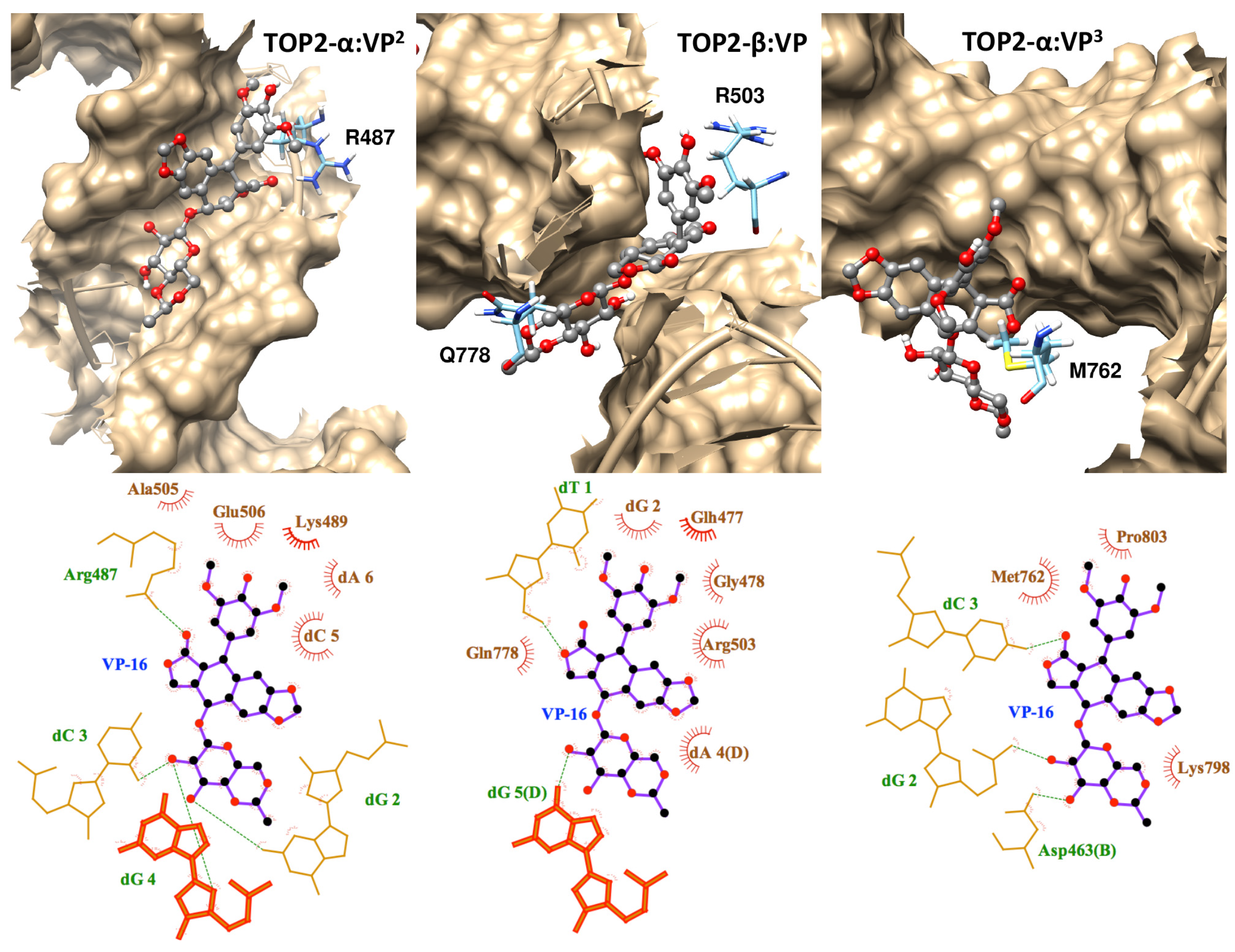
3. Experimental
3.1. Protein Models of TOP2-α and TOP2-β
3.2. Molecular Docking with the Use of AutoDock4RAP
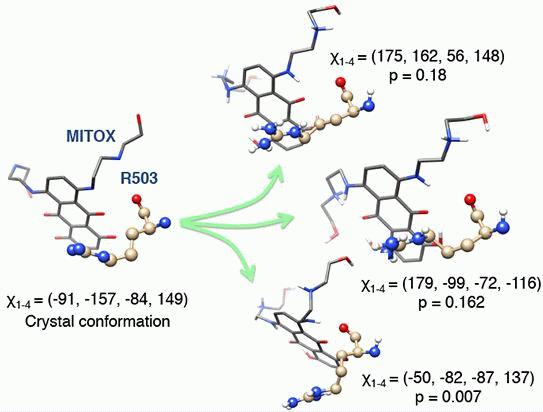
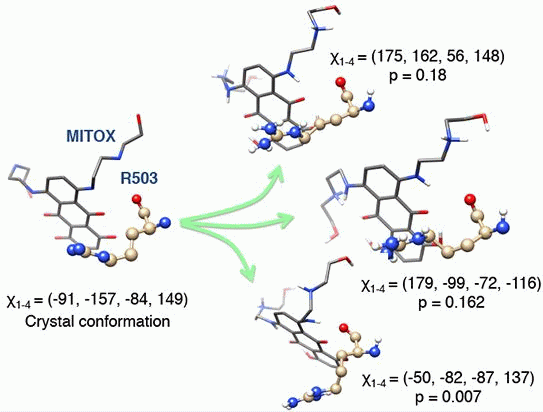
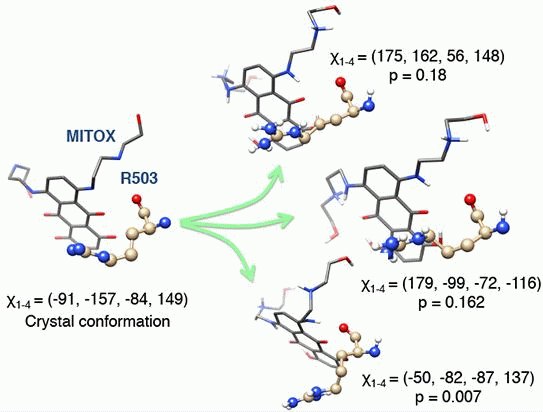
3.3. Molecular Dynamics Simulations, Free Energy Spectra, and Analyses on R503 Rotamer
3.4. Interaction Diagrams
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, J.C. Interaction between DNA and an Escherichia coli protein ω. J. Mol. Biol. 1971, 55, 523–533. [Google Scholar] [CrossRef]
- Roca, J.; Wang, J.C. The capture of a DNA double helix by an ATP-dependent protein clamp—A key step in DNA transport by type-II DNA topoisomerases. Cell 1992, 71, 833–840. [Google Scholar] [CrossRef]
- Howard, M.T.; Neece, S.H.; Matson, S.W.; Kreuzer, K.N. Disruption of a topoisomerase-DNA cleavage complex by a DNA helicase. Proc. Natl. Acad. Sci. USA 1994, 91, 12031–12035. [Google Scholar] [CrossRef]
- Berger, J.M.; Gamblin, S.J.; Harrison, S.C.; Wang, J.C. Structure and mechanism of DNA topoisomerase II. Nature 1996, 379, 225–232. [Google Scholar] [CrossRef]
- Berger, J.M.; Wang, J.C. Recent developments in DNA topoisomerase II structure and mechanism. Curr. Opin. Struct. Biol. 1996, 6, 84–90. [Google Scholar] [CrossRef]
- Chen, S.H.; Chan, N.L.; Hsieh, T.S. New mechanistic and functional insights into DNA topoisomerases. Annu. Rev. Biochem. 2013, 82, 139–170. [Google Scholar] [CrossRef]
- Bailly, C. Contemporary challenges in the design of topoisomerase II inhibitors for cancer chemotherapy. Chem. Rev. 2012, 112, 3611–3640. [Google Scholar]
- Willmore, E.; Frank, A.J.; Padget, K.; Tilby, M.J.; Austin, C.A. Etoposide targets topoisomerase IIα and IIβ in leukemic cells: Isoform-specific cleavable complexes visualized and quantifiedin situ by a novel immunofluorescence technique. Mol. Pharmacol. 1998, 54, 78–85. [Google Scholar]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Pommier, Y. Drugging topoisomerases: Lessons and challenges. ACS Chem. Biol. 2012, 8, 82–95. [Google Scholar] [CrossRef]
- Azarova, A.M.; Lyu, Y.L.; Lin, C.P.; Tsai, Y.C.; Lau, J.Y.; Wang, J.C.; Liu, L.F. Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc. Natl. Acad. Sci. USA 2007, 104, 11014–11019. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar]
- Raschi, E.; Vasina, V.; Poluzzi, E.; de Ponti, F. The herg k+ channel: Target and antitarget strategies in drug development. Pharmacol. Res. 2008, 57, 181–195. [Google Scholar] [CrossRef]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef]
- Wendorff, T.J.; Schmidt, B.H.; Heslop, P.; Austin, C.A.; Berger, J.M. The structure of DNA-bound human topoisomerase II alpha: Conformational mechanisms for coordinating inter-subunit interactions with DNA cleavage. J. Mol. Biol. 2012, 424, 109–124. [Google Scholar] [CrossRef]
- Wu, C.C.; Li, Y.C.; Wang, Y.R.; Li, T.K.; Chan, N.L. On the structural basis and design guidelines for type II topoisomerase-targeting anticancer drugs. Nucleic Acids Res. 2013, 41, 10630–10640. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, J.-G.; Wang, B.; Li, Z.-M. Integrating molecular docking, DFT and CoMFA/CoMSIA approaches for a series of naphthoquinone fused cyclic α-aminophosphonates that act as novel topoisomerase II inhibitors. J. Mol. Model. 2011, 17, 1899–1909. [Google Scholar] [CrossRef]
- Wei, H.; Ruthenburg, A.J.; Bechis, S.K.; Verdine, G.L. Nucleotide-dependent domain movement in the atpase domain of a human type IIA DNA topoisomerase. J. Biol. Chem. 2005, 280, 37041–37047. [Google Scholar] [CrossRef]
- Wang, J.C.; Lin, J.H.; Chen, C.M.; Perryman, A.L.; Olson, A.J. Robust scoring functions for protein-ligand interactions with quantum chemical charge models. J. Chem. Inf. Model. 2011, 51, 2528–2537. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar]
- Jakalian, A.; Bush, B.L.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 2000, 21, 132–146. [Google Scholar]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Weiner, S.J.; Kollman, P.A.; Case, D.A.; Singh, U.C.; Ghio, C.; Alagona, G.; Profeta, S.; Weiner, P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984, 106, 765–784. [Google Scholar]
- Weiner, S.J.; Kollman, P.A.; Nguyen, D.T.; Case, D.A. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 1986, 7, 230–252. [Google Scholar] [CrossRef]
- Huang, N.L.; Lin, J.H. Prediction of Binding Poses and Binding Affinities for Glycans and Their Binding Proteins Using a Robust Scoring Function for General Protein-Ligand Interactions. In Proceedings of the International Beilstein Symposium on Glyco-Bioinformatics, Potsdam, Germany, 10–14 June 2013; Discovering the Subtleties of Sugars.
- Goddard, T.D.; Huang, C.C.; Ferrin, T.E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 2007, 157, 281–287. [Google Scholar] [CrossRef]
- Lin, J.H.; Perryman, A.L.; Schames, J.R.; McCammon, J.A. The relaxed complex method: Accommodating receptor flexibility for drug design with an improved scoring scheme. Biopolymers 2003, 68, 47–62. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. Ligplot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Chan, N.L. National Taiwan University: Taipei, Taiwan, Unpulished work, 2014.
- O’Boyle, N.; Banck, M.; James, C.; Morley, C.; Vandermeersch, T.; Hutchison, G. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. In Advances in Protein Chemistry; Valerie, D., Ed.; Academic Press: San Diego, CA, USA, 2003; volume 66, pp. 27–85. [Google Scholar]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Roe, D.R.; Cheatham, T.E. PTRAJ and Cpptraj: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, N.-L.; Lin, J.-H. Drug-Induced Conformational Population Shifts in Topoisomerase-DNA Ternary Complexes. Molecules 2014, 19, 7415-7428. https://doi.org/10.3390/molecules19067415
Huang N-L, Lin J-H. Drug-Induced Conformational Population Shifts in Topoisomerase-DNA Ternary Complexes. Molecules. 2014; 19(6):7415-7428. https://doi.org/10.3390/molecules19067415
Chicago/Turabian StyleHuang, Nan-Lan, and Jung-Hsin Lin. 2014. "Drug-Induced Conformational Population Shifts in Topoisomerase-DNA Ternary Complexes" Molecules 19, no. 6: 7415-7428. https://doi.org/10.3390/molecules19067415
APA StyleHuang, N.-L., & Lin, J.-H. (2014). Drug-Induced Conformational Population Shifts in Topoisomerase-DNA Ternary Complexes. Molecules, 19(6), 7415-7428. https://doi.org/10.3390/molecules19067415