Abstract
A series of 18 N-alkyl substituted 3-aminopyrazine-2-carboxamides was prepared in this work according to previously experimentally set and proven conditions using microwave assisted synthesis methodology. This approach for the aminodehalogenation reaction was chosen due to higher yields and shorter reaction times compared to organic reactions with conventional heating. Antimycobacterial, antibacterial, antifungal and photosynthetic electron transport (PET) inhibiting in vitro activities of these compounds were investigated. Experiments for the determination of lipophilicity were also performed. Only a small number of substances with alicyclic side chain showed activity against fungi which was the same or higher than standards and the biological efficacy of the compounds increased with rising lipophilicity. Nine pyrazinamide derivatives also inhibited PET in spinach chloroplasts and the IC50 values of these compounds varied in the range from 14.3 to 1590.0 μmol/L. The inhibitory activity was connected not only with the lipophilicity, but also with the presence of secondary amine fragment bounded to the pyrazine ring. Structure-activity relationships are discussed as well.
1. Introduction
Pyrazinamide (PZA) is considered to be one of the most important drugs in the World. It has numerous uses and the pyrazine ring is also contained in many natural compounds. Nevertheless, its main field of use is still the treatment of tuberculosis (TB). The reason is clear—Mycobacterium tuberculosis is the most dreadful microorganisms in the World. Although the incidence of new TB cases has been falling slowly throughout the last decade, the danger is posed by the group of resistant strains [1]. There are currently two types of resistance. The first group consists of multidrug-resistant mycobacteria, which are resistant to all first-line antituberculotic agents (rifampicin/rifabutin, isoniazid, pyrazinamide, ethambutol, streptomycin). The second group is more treacherous because of its resistance to isoniazid and rifampicin combined with resistance to any fluoroquinolone used in the current therapy and to at least one of the three parenteral second-line antituberculotic drugs (amikacin, kanamycin, capreomycin) [2]. There was another category in the past called totally drug-resistant TB that was first observed in India. These strains were resistant to all known first- and second-line antituberculosis agents and posed a threat to epidemiologists all over the World. But they have practically disappeared with the discovery and approval of the new anti-tuberculosis agent bedaquiline for their therapy because it is active against the resistant Mycobacterium tuberculosis [3]. PZA is classified as the first-line antituberculotic agent that is unique for its therapeutic properties. It acts in acidic environment as a bactericidal sterilising drug that is able to kill the dormant forms of tubercle bacilli, especially in combination with rifampicin. These so called persistors are just situated in nidus where an acidic pH is typical [4].
On the contrary, the mechanism of action of PZA is still under the investigation by researchers all over the World. One of the first theories suggested was that it is necessary to activate PZA via the enzyme pyrazinamidase (nicotinamidase) (EC 3.5.1.19) to the active form pyrazinecarboxylic acid (POA). This acid molecule gets into the mycobacterial cell and causes the inner cell compartment acidification during the ionic efflux. These consequences lead to cellular death [5,6,7]. The gene encoding pyrazinamidase is known as pncA gene and its mutation leads to the resistance of mycobacteria to pyrazinamide [8].
Another theory propounded the inhibition of fatty acid synthase I (FAS I) (EC 2.3.1.85). This is the enzyme essential for synthesis of mycolic acids, which are vital for the mycobacterial cell wall integrity. Despite the fact that PZA is effective in this aspect, this mechanism was rejected by Boshoff who has proven that PZA caused only low inhibition [9]. PZA analogues that act as FAS I inhibitors are from the group of 5-chloropyrazinamide, esters of 5-chloropyrazinoic acid or POA and their derivatives [10,11,12].
A recent theory introduced by Zhang et al. proposed the inhibition of trans-translation as the main way of PZA action. This cellular process is essential for survival and virulence of mycobacteria and its interruption leads to fatal blockade of proteosynthesis. All these hypotheses were supported by in vitro experiments [13].
PZA and its derivatives have also many other useful applications, as they show antiviral, antifungal, antibacterial and as well as antineoplastic activities [14,15,16,17,18,19,20]. Several pyrazine derivatives were also found to inhibit photosynthetic electron transport (PET) in plant chloroplasts [21,22,23]. Using EPR spectroscopy it was found that the site of action of pyrazine-derived PET inhibitors is the intermediate D•, i.e., tyrosine radical (TyrD•) situated in the 161st position in D2 protein on the donor side of photosystem (PS) 2 [24,25]. Similar site of action in the photosynthetic apparatus of spinach chloroplasts was confirmed for 2-alkylsulfanylpyridine-4-carbothioamides [26] and substituted benzanilides and thiobenzanilides [27]. Further experiments with artificial electron donor 1,5-diphenylcarbazide (DPC) confirmed that PET between PS2 and PS1 was interrupted by 2,6-disubstituted pyridine-4-thiocarboxamides exclusively on the donor side of PS2 in the section between the primary electron donor of PS2 (H2O) and Z•/D• intermediate and the photosynthetic transport chain from P 680 to plastoquinone QB occurring on the acceptor side of PS2 was not damaged [25], while PET on the acceptor side of PS2 was partially damaged by 5-tert-butyl-6-chloro-N-(3-fluorophenyl)pyrazine-2-carboxamide and 5-tert-butyl-N-(3-hydroxy-4-chlorophenyl)pyrazine-2-carboxamide [25] or by N-benzylpyrazine-2-carboxamides [22]. Many structural variations of pyrazine compounds with herbicidal properties can be found in the patent literature focused on compounds with herbicidal activity that are useful for the control of unwanted vegetation [28,29,30].
A further aspect of this work is the use of microwave-assisted reactions. It is known that microwave-assisted syntheses have become into general awareness during the last decades and their applications are still in the focus of many researchers. These reactions usually provide higher yields, shorter reaction times and better chemical conversions in comparison with conventional heating methods. Moreover, sometimes the desired products can be obtained only by using the microwaves accelerated reactions [31]. If emerging side-products are detected, they can be eliminated by many separation methods such as chromatography or crystallization. The advantages of these reactions are explained by the interaction between microwaves and molecules themselves, neither vessel sides nor any solvents used. There is also the possibility of reaching temperatures higher than the boiling point of solvent, especially in over-pressurized systems. This has opened a new way for drug discovery and development [32]. We focus herein on the synthesis of pyrazine derivatives with aliphatic or alicyclic side chains and their antimycobacterial, antibacterial, antifungal and PET-inhibiting activities.
2. Results and Discussion
2.1. Chemistry
Compounds synthesized in this work were prepared according to the general procedures shown in Scheme 1. The identity and purity were proven by NMR spectra, elemental analysis and melting point.
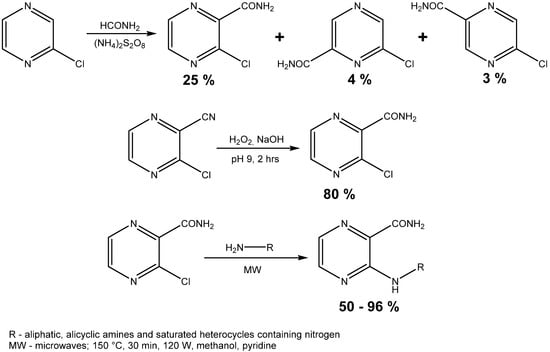
Scheme 1.
Synthesis of starting compound and microwave assisted synthesis of final compounds.
Scheme 1.
Synthesis of starting compound and microwave assisted synthesis of final compounds.
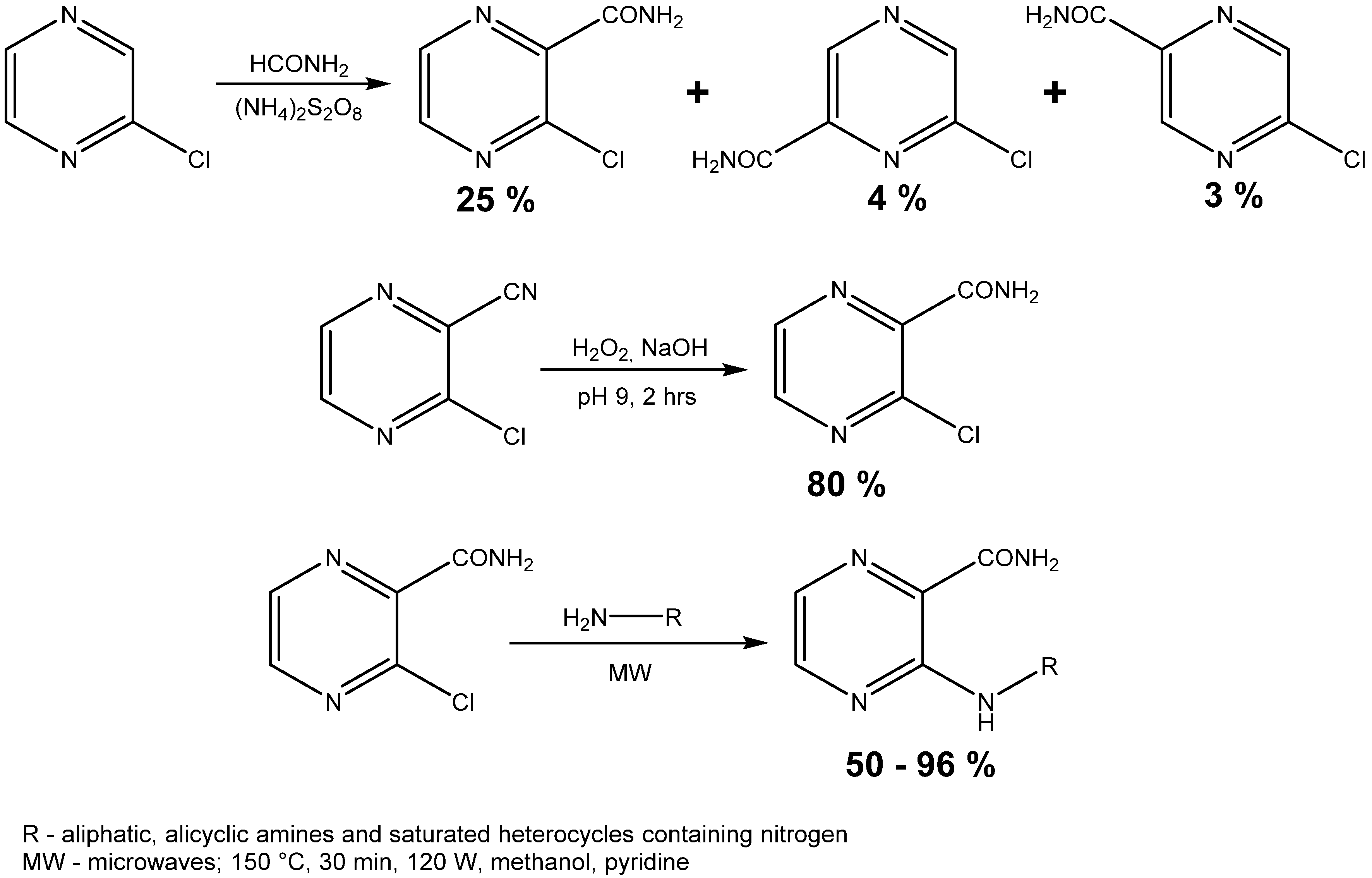
The second method of starting compound preparation was approximately three times more efficacious than the first one comparing the yields of the reactions (80% vs. 25%). The melting point of 3-chloropyrazine-2-carboxamide was compared to the value stated in a previously published paper and the results were practically the same, as our measured value was in the 187.1–189.2 °C range and the reported values were 186.0–186.5 °C [33] and 188.0–190.0 °C [34]. 3-Chloropyrazine-2-carboxamide was then treated with 18 aliphatic or alicyclic amines and saturated heterocycles containing nitrogen. A series of experiments was done at the beginning to compare the efficiency of both methods—conventional and microwave-assisted heating (Table 1).
The remaining reactions were performed in microwave reactor and the conditions, which were proven experimentally, were as follows: 140 °C, 30 min, 120 W, methanol as a solvent and pyridine as a base (Scheme 1). Yields obtained from the aminodehalogenation were in a range between 50.0% and 95.8% and their variability can be explained by the steric effects of some substituents (dibutyl-, tert-pentyl-) that lead to worse outcomes.
This reaction yielded N-substituted 3-aminopyrazine-2-carboxamides (Table 2). We prepared a series consisted of 18 compounds of which 16 were novel. They were analytically characterized (NMR and IR spectra, melting point, elemental analysis) and submitted to biological screening. The obtained analytical data were fully consistent with the proposed structures. 3-(Methylamino)pyrazine-2-carboxamide was previously synthesized by Osden et al. (reported melting point was 200–201 °C) and by Albert et al. (reported melting point 198–199 °C) [33,35]. The light yellow crystalline compound we obtained melted at 200.0–201.4 °C. 3-(Cyclohexylamino)pyrazine-2-carboxamide was previously synthesized by Keir et al. and reported melting point was 128–129 °C [36]. Our light yellow crystalline compound melted at 129.0–130.3 °C.

Table 1.
Table comparing the efficiency of conventional and microwave-assisted methodology showing the yields, reaction times and conditions.
| Compound | Type of Synthesis | Time | Conditions | Yield | |
|---|---|---|---|---|---|
| 1 | 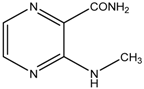 | MW | 30 min | 140 °C, 120 W, MeOH, overpressure, 1 eq. pyridine, 2 eq. amine | 61.4% |
| Conventional | 60 min | 110 °C, toluene, 1 eq. pyridine, 2 eq. amine | 53.0% | ||
| 2 | 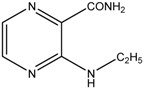 | MW | 30 min | 140 °C, 120 W, MeOH, overpressure, 1 eq. pyridine, 2 eq. amine | 78.7% |
| Conventional | 60 min | 110 °C, toluene, 1 eq. pyridine, 2 eq. amine | 46.3% | ||
| 3 | 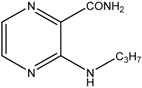 | MW | 30 min | 140 °C, 120 W, MeOH, overpressure, 1 eq. pyridine, 2 eq. amine | 87.0% |
| Conventional | 60 min | 110 °C, toluene, 1 eq. pyridine, 2 eq. amine | 56.1% | ||
| 10 | 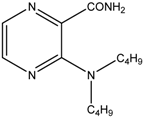 | MW | 30 min | 140 °C, 120 W, MeOH, overpressure, 1 eq. pyridine, 2 eq. amine | 67.2% |
| Conventional | 60 min | 110 °C, toluene, 1 eq. pyridine, 2 eq. amine | 0% | ||
Table 2.
List of prepared compounds and their lipophilicity.
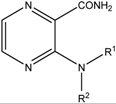 | ||||
|---|---|---|---|---|
| Nr. | R1 | R2 | logP/ClogP | logk |
| 1 | CH3 | H | −1.10/0.20 | −0.4028 |
| 2 | C2H5 | H | −0.76/0.73 | −0.2014 |
| 3 | C3H7 | H | −0.27/1.25 | 0.0038 |
| 4 | C4H9 | H | 0.14/1.78 | 0.2287 |
| 5 | C5H11 | H | 0.56/2.31 | 0.4563 |
| 6 | tert-C5H11 | H | 0.26/1.96 | 0.5468 |
| 7 | C6H13 | H | 0.98/2.84 | 0.6860 |
| 8 | C7H15 | H | 1.40/3.37 | 0.9198 |
| 9 | C8H17 | H | 1.81/3.90 | 1.1533 |
| 10 | C4H9 | C4H9 | 2.17/3.40 | 0.5197 |
| 11 | cyclopentyl | H | 0.03/1.67 | 0.2537 |
| 12 | cyclohexyl | H | 0.45/2.23 | 0.4523 |
| 13 | cycloheptyl | H | 0.87/2.79 | 0.6575 |
| 14 | –(CH2)4– | 0.01/0.34 | −0.4889 | |
| 15 | –(CH2)5– | 0.42/0.90 | −0.2333 | |
| 16 | –(CH2)2O(CH2)2– | −0.71/−0.49 | −0.6238 | |
| 17 | –(CH2)2NCH3(CH2)2– | −0.55/0.08 | −0.4253 | |
| 18 | –(CH2)2NH(CH2)2– | −0.93/−0.50 | −2.3522 | |
2.2. Calculated and Experimentally Determined Lipophilicity
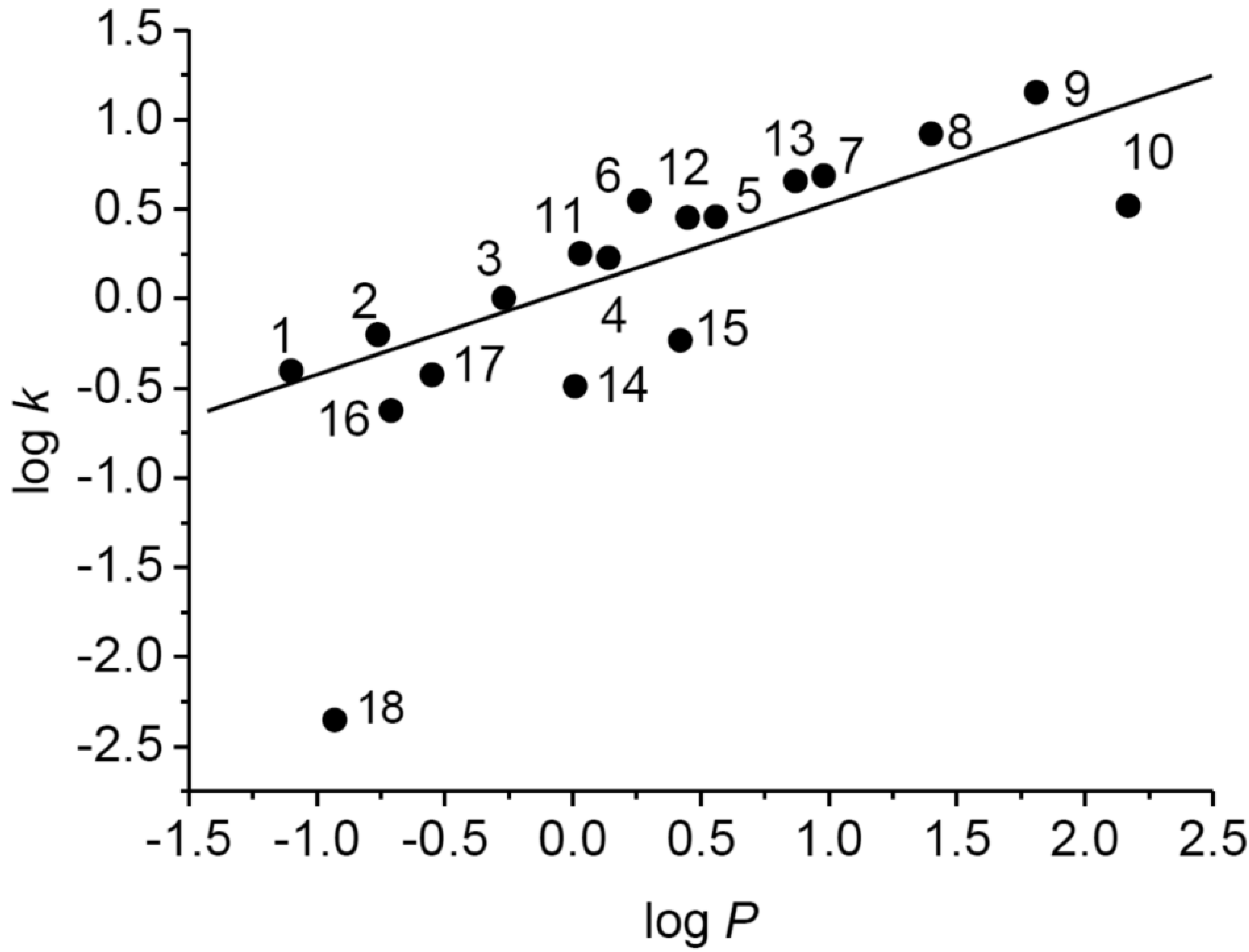
The dependence of experimentally determined logk on calculated logP values of tested compounds is shown in Figure 1. While for compounds 1–17 linear dependence of logk on logP was determined (r = 0.816), the estimated logk value for 3-(piperazin-1-yl)pyrazine-2-carboxamide (18) was considerably lower than expected. This could be connected with the presence of a secondary cyclic amine that can be very easily ionized.
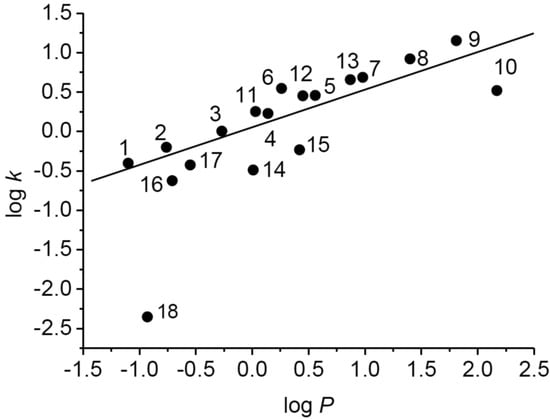
Figure 1.
Dependence of experimentally determined logk on calculated logP values of tested compounds.
Figure 1.
Dependence of experimentally determined logk on calculated logP values of tested compounds.
2.3. Biological Assays
2.3.1. Antimycobacterial in Vitro Screening
All prepared compounds were tested against the wild strain Mycobacterium tuberculosis H37Rv. Four substances (compounds 6‒9) showed low activity against this strain with corresponding minimal inhibition concentration (MIC) values equal to 50 μg/mL. However, this activity was negligible comparing to the activity of the standard isoniazid (0.39 μg/mL) and pyrazinamide (12.5 μg/mL).
2.3.2. Antifungal and Antibacterial in Vitro Screening
The results of antibacterial screening have shown only low activities against Staphylococcus epidermidis. The antibacterial activity of compounds 5, 7 and 13 expressed by 80% inhibition concentration (IC80) values was lower than 250 μmol/L and did not reach the values of antibiotic standards.
Antifungal evaluation showed more interesting results. There were seven active substances and two of them (compounds 12 and 13) showed better activity than two standards (voriconazole, fluconazole) (Table 3). The alicyclic side chain with seven carbons was more active than the 6-membered ring. The 80% inhibition of control was observed in the range from 15.62 to 31.25 μmol/L for compound 12 and in the range from 7.81 to 31.25 μmol/L for compound 13 against nearly a whole spectrum of fungi tested after 24 h of incubation. Five substances (compounds 4, 5, 7, 8 and 11) were less active than the standards and their IC80 values were not lower than 250 μmol/L. The activity spectrum of these compounds was also wider and did not include only one fungal strain.
Table 3.
Results of antifungal evaluation for the compounds with alicyclic side chains and corresponding standards.
| IC80 [μmol/L] | ||||||||
|---|---|---|---|---|---|---|---|---|
| 11 | 12 | 13 | AMP | VOR | NYS | FLU | ||
| CA | 24 h | 500 | 31.25 | 15.62 | 0.12 | 0.008 | 0.98 | 0.24 |
| 48 h | >500 | 62.5 | 31.25 | 0.49 | 0.008 | 1.95 | 0.24 | |
| CT | 24 h | >500 | 125 | 62.5 | 1.95 | 250 | 1.95 | >500 |
| 48 h | >500 | 500 | 500 | 1.95 | 250 | 3.9 | >500 | |
| CK | 24 h | 250 | 15.62 | 7.81 | 1.95 | 0.98 | 1.95 | 125 |
| 48 h | 500 | 62.5 | 31.25 | 1.95 | 1.95 | 3.9 | 250 | |
| CG | 24 h | 250 | 15.62 | 7.81 | 0.98 | 250 | 1.95 | 31.25 |
| 48 h | >500 | 31.25 | 15.62 | 1.95 | 250 | 3.9 | 250 | |
| TA | 24 h | 500 | 31.25 | 31.25 | 0.49 | 7.81 | 1.95 | 250 |
| 48 h | >500 | 250 | 125 | 0.98 | 31.25 | 1.95 | 500 | |
| AF | 24 h | 500 | 15.62 | 7.81 | 1.95 | 0.49 | 1.95 | >500 |
| 48 h | >500 | 62,5 | 7.81 | 1.95 | 0.98 | 3.9 | >500 | |
| LC | 24 h | >500 | 250 | 62.5 | 7.81 | 250 | 15.62 | >500 |
| 48 h | >500 | 500 | 250 | 7.81 | 250 | 31.25 | >500 | |
| TM | 72 h | 500 | 15.62 | 15.62 | 1.95 | 0.06 | 3.9 | 7.81 |
| 120 h | 500 | 15.62 | 15.62 | 1.95 | 0.12 | 7.81 | 125 | |
CA: Candida albicans; CT: C. tropicalis; CK: C. krusei; CG: C. glabrata; TA: Trichosporon asahii; AF: Aspergillus fumigatus; LC: Lichtheimia corymbifera; TM: Trichophyton mentagrophytes; AMP: amphotericin B; VOR: voriconazole; NYS: nystatin; FLU: fluconazole.
It is not possible to predict structure-activity relationships for antibacterial and antifungal efficacy of studied compounds in the whole tested group due to small number of active substances and a lack of IC80 values better than standards. However, a saturated ring is probably important for antifungal activity and the activity also increases with increasing lipophilicity of the compounds. Unfortunately, the series is too small (only three compounds) to prove this hypothesis.
2.3.3. Photosynthetic Electron Transport Inhibiting Activity Evaluation
The 3-substituted pyrazine-2-carboxamide derivatives were tested for their PET inhibiting activity. However, the PET-inhibiting activity, expressed by IC50 value (compound concentration in mol/L causing 50% inhibition of PET) in the investigated set, could be determined only for nine compounds. The values varied in a wide concentration range from 14.3 μmol/L (compound 9) to 1590.0 μmol/L (compound 3) (Table 4). The rest of compounds 1, 2, 10, 14, 15, 16, 17, and 18 did not inhibit PET in spinach chloroplasts. Because all tested compounds containing heterocyclic amines as substituent in position 3 on the pyrazine ring, i.e., 14, 15, 16, 17, and 18, were found to be inactive it could be concluded that increased basicity of these compounds caused by the introduction of nitrogen atom(s) into the ring is connected with activity loss. Moreover, these substituents ultimately create a trisubstituted amine while by introduction of amino alkyl or amino cycloalkyl substituents to pyrazine-2-carboxamide disubstituted amine is formed. However, it could be stressed that trisubstituted amine is present also in compound with R = (C4H9)2 (10) which did not show PET inhibiting activity as well.
Table 4.
IC50 values [μmol/L] of tested compounds related to PET inhibition in spinach chloroplasts in comparison with 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU) standard.
| Compound | IC50 [μmol/L] |
|---|---|
| 3 | 1590 |
| 4 | 203 |
| 5 | 38.6 |
| 6 | 480 |
| 7 | 32.9 |
| 9 | 14.3 |
| 11 | 25.8 |
| 12 | 83.0 |
| 13 | 19.3 |
| DCMU | 1.9 |
The PET-inhibiting activity was strongly affected by lipophilic properties of the compounds (Figure 2) expressed as logP or logk. The PET-inhibiting activity increased with increasing lipophilicity of the compound and the dependence of log (1/IC50) vs. logP showed quasi-parabolic course (Figure 2A). The only exception was compound 11 (R = cyclopentyl with logP = 0.03) activity of which was considerably higher (IC50 = 25.8 μmol/L) than that of compound 4 with logP = 0.14 (IC50 = 203 μmol/L). Similarly, quasi-parabolic course was also observed for the dependence of PET inhibiting activity on logk. However, with respect to the quasi-parabolic course of this dependence, compound 11 (logk = 0.2537) showed higher and compound 6 (logk = 0.5468) lower PET inhibiting activity (Figure 2B).
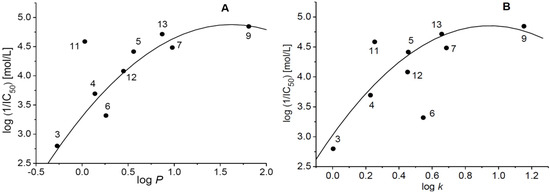
Figure 2.
Dependence of PET-inhibiting activity on lipophilicity of studied compounds expressed as logP (A) or logk (B).
Figure 2.
Dependence of PET-inhibiting activity on lipophilicity of studied compounds expressed as logP (A) or logk (B).
The dependence of log (1/IC50) on logP can be expressed by following equation after excluding the 3-(cyclopentylamino)pyrazine-2-carboxamide (11):
log(1/IC50) = 3.302 (±0.137) + 1.943 (±0.366) logP − 0.599 (±0.211) (logP)2
r = 0.955, s = 0.253, F = 25.79, n = 8
r = 0.955, s = 0.253, F = 25.79, n = 8
On the other hand, after excluding the 3-(tert-pentylamino)pyrazine-2-carboxamide (6) the dependence of log (1/IC50) on logk can be expressed by following equation:
log (1/IC50) = 3.028 (±0.329) + 0.386 (±0.126) logk − 2.040 (±1.028) (logk)2
r = 0.881, s = 0.378, F = 8.68, n = 8
r = 0.881, s = 0.378, F = 8.68, n = 8
The obtained results indicated that IC50 could not be determined for compounds with too low lipophilicity, namely 1 (logP = −1.3), 2 (logP = −0.76).
As mentioned above, the ineffectiveness of compound 10 (R = (C4H9)2) as well as compounds 14‒18 with heterocyclic substituents could be connected mainly with the absence of a secondary amine fragment (-NH-) bounded to the pyrazine ring. Consequently, possible interactions of these compounds with constituents of the photosynthetic apparatus are limited. On the other side, a 2-carboxamino substituent CONH2 on the pyrazine ring plays an important role in the interactions of the tested pyrazine derivatives with constituents of the photosynthetic apparatus. Moreover, increasing length of the alkyl substituent contributes not only leads to better transport of the compound to its site of action but incorporation of longer alkyl chains into the thylakoid membrane that results in membrane damage and consecutive increase of PET inhibiting activity. Similar results were also obtained with 2-alkylthio-4-pyridinecarbothioamides [26].
Artificial electron donors could be used to specify the site of inhibitory action of PET inhibitors in the photosynthetic apparatus because they can restore PET activity in chloroplasts of which was previously inhibited. For example, 2,5-diphenylcarbazide (DPC) can supply electrons in the site of Z•/D• intermediate which is situated on the donor side of PS2 [37]. Consequently, if PET was inhibited in the section between the oxygen evolving complex and Z•/D• intermediate, DPC can restore PET between the core of PS2 (P680) and secondary quinone acceptor QB which is situated on the acceptor side of PS2 and 2,6-dichlorophenol-indophenol (DCPIP) photoreduction occurs again. However, if the site of PET inhibitor is located between P680 and QB, photoreduction of DCPIP (an artificial electron acceptor used for monitoring the PET through PS2 from H2O to QB) remains inhibited. Activity of chloroplasts, which was restricted by studied pyrazine-2-carboxamides, was practically completely restored after DPC addition. Thus, it can be concluded that the site of action of tested compounds in the electron transport chain is situated exclusively on the donor side of PS2, between oxygen evolving complex and Z•/D• intermediate, while the section of the electron transport chain on the acceptor side of PS2 between P680 and QB is not damaged. Similar results were also obtained with N-substituted 5-amino-6-methylpyrazine-2,3-dicarbonitriles [23], 2-benzylsulphanylbenzimidazoles [38] and ring-substituted 1-hydroxynaphthalene-2-carboxanilides [39]. On the other hand, addition of DPC restored PET in chloroplasts, which was inhibited by N-benzylpyrazine-2-carboxamides only to 77%–88% indicating that the site of PET inhibition is situated not only on the donor but also on the acceptor side of PS2 [22]. Similar sites of action on both sides of PS2 were determined previously for 5-tert-butyl-N-(3-hydroxy-4-chlorophenyl)-pyrazine-2-carboxamide and 5-tert-butyl-6-chloro-N-(3-fluorophenyl)pyrazine-2-carboxamide [24].
Interaction of substituted pyrazine-2-carboxamides with residues of aromatic amino acids (AAA), mainly tryptophan and tyrosine occurring in photosynthetic proteins situated mainly in PS2, which was documented by the quenching of AAA fluorescence at 334 nm contributed to PET inhibition. Figure 3 presents fluorescence emission spectra of AAA of untreated spinach chloroplasts and of chloroplasts treated with increasing concentrations of compound 7. As shown in Figure 3, the quenching of the fluorescence of aromatic amino acids at 334 nm increased with increasing concentration of pyrazine derivative. This finding is in accordance with the results obtained in the above mentioned study of PET inhibition in spinach chloroplasts by tested pyrazine-2-carboxamides using artificial electron acceptor DCPIP.
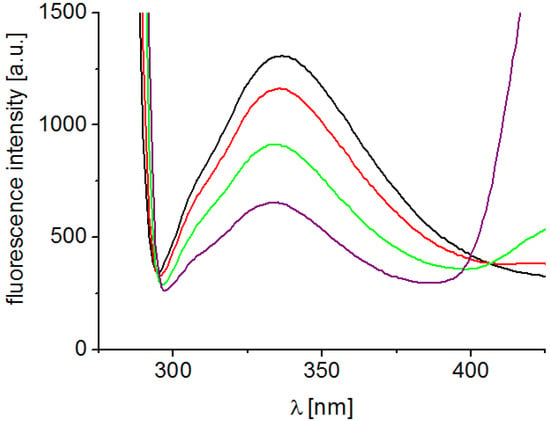
Figure 3.
Fluorescence emission spectra of aromatic amino acids in suspension of spinach chloroplasts without and with compound 7 (c = 0, 6, 24 and 48 μmol/L; the curves from top to bottom); excitation wave length λ = 275 nm; chlorophyll concentration 10 mg/L.
Figure 3.
Fluorescence emission spectra of aromatic amino acids in suspension of spinach chloroplasts without and with compound 7 (c = 0, 6, 24 and 48 μmol/L; the curves from top to bottom); excitation wave length λ = 275 nm; chlorophyll concentration 10 mg/L.
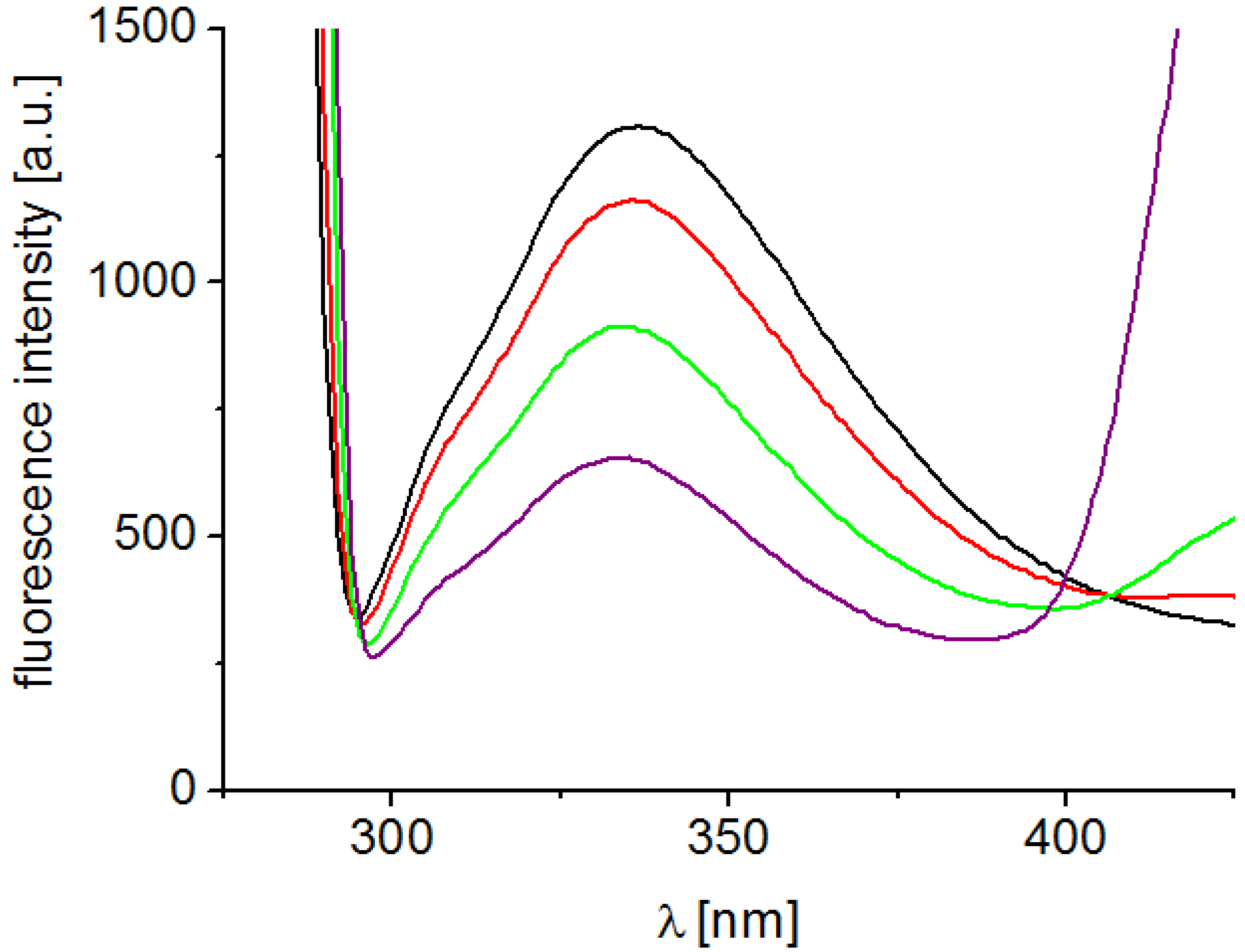
The quenching of the fluorescence of aromatic amino acids in the presence of 5-bromo- and 3,5-dibromo-2-hydroxy-N-phenylbenzamides and ring-substituted 2-hydroxynaphthalene-1-carboxanilides was observed previously [40,41].
3. Experimental
3.1. General
All chemicals were reagent or higher grade of purity and were purchased from Sigma-Aldrich (Steinheim, Germany) or Fluorochem Ltd. (Hadfield, Derbyshire, UK), unless stated otherwise.
The starting compound was prepared according to proven conventional organic synthesis methodology. The final aminodehalogenation reaction was performed in a CEM Discover microwave reactor with a focused field (CEM Corporation, Matthews, NC, USA) connected to an Explorer 24 autosampler (CEM Corporation) and this equipment was running under CEM’s SynergyTM software for setting and monitoring the conditions of reactions. The temperature of the reaction mixture was monitored by internal infrared sensor. The progress of the reaction was checked by Thin Layer Chromatography (TLC) (Alugram® Sil G/UV254, Machery-Nagel, Postfach, Germany) with UV detection using wavelength 254 nm.
All obtained products were purified by crystallization or by preparative flash chromatograph CombiFlash® Rf (Teledyne Isco Inc., Lincoln, NE, USA). The type of elution was gradient, using the mixture of hexane (LachNer, Neratovice, Czech Republic) and ethyl acetate (Penta, Prague, Czech Republic) as mobile phase. Silica gel (0.040–0.063 nm, Merck, Darmstadt, Germany) was used as the stationary phase.
NMR spectra were taken with spectrometers Varian Mercury-VxBB 300 with frequencies 299.95 MHz for 1H and 75.43 MHz for 13C or Varian VNMR S500 (499.87 MHz for 1H and 125.71 MHz for 13C) (Varian Corporation, Palo Alto, CA, USA). Chemical shifts were reported in ppm (δ) and were applied indirectly to tetramethylsilane as a signal of solvent (2.49 for 1H and 39.7 for 13C in DMSO-d6). Infrared spectra were recorded with spectrometer FT-IR Nicolet 6700 (Thermo Scientific, Waltham, MA, USA) using attenuated total reflectance (ATR) methodology. Elemental analyses were measured with EA 1110 CHNS Analyzer (Fisons Instruments S. p. A., Carlo Erba, Milano, Italy). Melting points were assessed by SMP3 Stuart Scientific (Bibby Sterling Ltd., Staffordshire, UK) and were uncorrected. LogP and ClogP were calculated with PC program CS ChemBioDraw Ultra 13.0 (CambridgeSoft, Cambridge, MA, USA).
3.2. Synthesis of Starting Compound and Final Products
The starting compound 3-chloropyrazine-2-carboxamide was synthesized using two published procedures. The first method was classified as less effective and was based on the homolytic amidation of 2-chloropyrazine. Thus, 2-chloropyrazine (0.17 mol) was dissolved in formamide (3.7 mol), heated to 90 °C and ammonium peroxodisulphate (0.18 mol) was added portionwise over one hour period. This mixture reacted for another one hour at 90 °C and then it was left to stand for 24 h at laboratory temperature. Dilution with 100 mL of water was followed by filtration and this filtrate was extracted continuously with chloroform for 16 h [34,42]. The mixture of three positional isomers was separated by flash chromatography using silica gel as stationary phase. The second process used 3-chloropyrazine-2-carbonitrile, which was submitted to partial hydrolysis of the nitrile group. The powdered carbonitrile (0.104 mol) was added little by little into the reaction mixture of concentrated hydrogen peroxide (0.95 mol) and water (195 mL) heated to 50 °C. The pH was adjusted and regulated around a value of 9 using an 8% solution of sodium hydroxide and the temperature of the reaction was regulated between 55 and 60 °C. The reaction was stopped after 2.5 h and was cooled to 5 °C. Newly-emerged crystals were removed by suction and recrystallized from ethanol [42].
The starting compound (1.27 mmol) was treated with 18 aliphatic amines, alicyclic amines or saturated heterocycles containing at least one nitrogen atom (2.54 mmol). Four reactions were completed by conventional heating methods. The conditions were 110 °C, toluene as a solvent and pyridine (1.27 mmol) as a base. The reaction time was set to one hour. Then the reactions were completed using the microwave reactor with focused field and conditions used for syntheses were 140 °C, 30 min, 120 W, methanol used as a solvent and pyridine (1.27 mmol) as a base. They were set experimentally with respect to prior experience. The progress of reaction was monitored with TLC in system hexane/ethyl acetate (1:1). Then the mixture was separated by flash column chromatograph using gradient elution. Mobile phases were hexane and ethyl acetate again.
3.3. Analytical Data of the Prepared Compounds
3-(Methylamino)pyrazine-2-carboxamide (1). Light yellow crystalline solid. Yield 61.4%; m.p. 200.0–201.4 °C (described in the literature 200–201 °C [35] or 198–199 °C [33]); IR (ATR-Ge, cm−1): 3413m (-NH-), 3293m, 3179m (-CONH2), 1686vs (-CONH2), 1589s, 1513s, 1436m, 1414s, 1194vs (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.65–8.55 (1H, m, NH), 8.23 (1H, d, J = 2.3 Hz, Ar), 8.11 (1H, bs, NH2), 7.72 (1H, d, J = 2.3 Hz, Ar), 7.63 (1H, bs, NH2), 2.90 (3H, d, J = 5.3 Hz, CH3); 13C-NMR (75 MHz, DMSO-d6) δ 168.9, 155.1, 146.6, 129.5, 126.9, 27.3; Elemental analysis: calc. for C6H8N4O (MW 152.15): 47.36% C, 5.30% H, 36.82% N; found 47.43% C, 5.45% H, 36.68% N.
3-(Ethylamino)pyrazine-2-carboxamide (2). Light yellow crystalline solid. Yield 78.7%; m.p. 117.4–118.5 °C; IR (ATR-Ge, cm−1): 3381m (-NH-), 3294m, 3209m (-CONH2), 1683vs (-CONH2), 1582s, 1533m, 1500vs, 1419m, 1192vs (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.68 (1H, t, J = 4.7 Hz, NH), 8.22 (1H, d, J = 2.3 Hz, Ar), 8.11 (1H, bs, NH2), 7.72 (1H, d, J = 2.3 Hz, Ar), 7.64 (1H, bs, NH2), 3.47–3.32 (2H, m, NCH2), 1.14 (3H, t, J = 7.0 Hz, CH3); 13C-NMR (75 MHz, DMSO-d6) δ 169.0, 154.4, 146.6, 129.6, 126.5, 34.8, 14.8; Elemental analysis: calc. for C7H10N4O (MW 166.18): 50.59% C, 6.07% H, 33.71% N; found 50.66% C, 5.98% H, 33.51% N.
3-(Propylamino)pyrazine-2-carboxamide (3). Yellow crystalline solid. Yield 87.0%; m.p. 93.9–94.9 °C; IR (ATR-Ge, cm−1): 3402m (-NH-), 3337m, 3186m (-CONH2), 1671vs (-CONH2), 1578s, 1533m, 1514vs, 1423s, 1189vs (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.77 (1H, t, J = 6.5 Hz, NH), 8.21 (1H, d, J = 2.3 Hz, Ar), 8.12 (1H, bs, NH2), 7.72 (1H, d, J = 2.3 Hz, Ar), 7.64 (1H, bs, NH2), 3.34 (2H, q, J = 6.5 Hz, NCH2), 1.63–1.46 (2H, m, CH2), 0.89 (3H, t, J = 6.5 Hz, CH3); 13C-NMR (75 MHz, DMSO-d6) δ 169.1, 154.6, 146.6, 129.6, 126.5, 41.7, 22.2, 11.6; Elemental analysis: calc. for C8H12N4O (MW 180.21): 53.32% C, 6.71% H, 31.09% N; found 53.45% C, 6.79% H, 30.93% N.
3-(Butylamino)pyrazine-2-carboxamide (4). Yellow crystalline solid. Yield 92.3%; m.p. 83.1–84.4 °C; IR (ATR-Ge, cm−1): 3409m (-NH-), 3326m, 3177m (-CONH2), 1671vs (-CONH2), 1578s, 1533m, 1518vs, 1423s, 1187vs (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.75 (1H, t, J = 6.6 Hz, NH), 8.22 (1H, d, J = 2.3 Hz, Ar), 8.12 (1H, bs, NH2), 7.73 (1H, d, J = 2.3 Hz, Ar), 7.64 (1H, bs, NH2), 3.38 (2H, q, J = 6.6 Hz, NCH2), 1.59–1.46 (2H, m, CH2), 1.41–1.25 (2H, m, CH2), 0.88 (3H, t, J = 6.6 Hz, CH3); 13C-NMR (75 MHz, DMSO-d6) δ 169.1, 154.6, 146.6, 129.6, 126.5, 40.5, 31.1, 19.9, 13.9; Elemental analysis: calc. for C9H14N4O (MW 194.23): 55.65% C, 7.27% H, 28.85% N; found 55.77% C, 7.27% H, 28.93% N.
3-(Pentylamino)pyrazine-2-carboxamide (5). Light yellow crystalline solid. Yield 92.8%; m.p. 105.0–106.3 °C; IR (ATR-Ge, cm−1): 3379m (-NH-), 3321m, 3145m (-CONH2), 1663vs (-CONH2), 1587s, 1531m, 1509vs, 1418s, 1185vs (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.76 (1H, t, J = 6.5 Hz, NH), 8.22 (1H, d, J = 2.3 Hz, Ar), 8.12 (1H, bs, NH2), 7.73 (1H, d, J = 2.3 Hz, Ar), 7.64 (1H, bs, NH2), 3.38 (2H, q, J = 6.5 Hz, NCH2), 1.62–1.46 (2H, m, CH2), 1.37–1.19 (4H, m, CH2), 0.85 (3H, t, J = 6.5 Hz, CH3); 13C-NMR (75 MHz, DMSO-d6) δ 169.1, 154.6, 146.6, 129.6, 126.4, 40.5, 28.9, 28.7, 22.1, 14.1; Elemental analysis: calc. for C10H16N4O (MW 208.26): 57.67% C, 7.74% H, 26.90% N; found 57.63% C, 7.67% H, 27.06% N.
3-(tert-Pentylamino)pyrazine-2-carboxamide (6). Yellow crystalline solid. Yield 51.3%; m.p. 65.0–66.7 °C; IR (ATR-Ge, cm−1): 3396m (-NH-), 3265m, 3185m (-CONH2), 1668vs (-CONH2), 1581vs, 1504vs, 1415s, 1183vs (pyrazine ring); 1H-NMR (300 MHz, CDCl3) δ 8.65 (1H, bs, NH), 8.13 (1H, d, J = 2.3 Hz, Ar), 7.74 (1H, bs, NH2), 7.61 (1H, d, J = 2.3 Hz, Ar), 5.56 (1H, bs, NH2), 1.86 (2H, q, J = 7.6 Hz, CH2), 1.42 (6H, s, CH3), 0.87 (3H, t, J = 7.6 Hz, CH3); 13C-NMR (75 MHz, CDCl3) δ 169.7, 154.9, 146.3, 128.9, 125.5, 54.2, 33.0, 26.4, 8.4; Elemental analysis: calc. for C10H16N4O (MW 208.26): 57.67% C, 7.74% H, 26.90% N; found 57.80% C, 7.89% H, 26.70% N.
3-(Hexylamino)pyrazine-2-carboxamide (7). Light yellow crystalline solid. Yield 93.6%; m.p. 64.3–65.6 °C; IR (ATR-Ge, cm−1): 3404m (-NH-), 3314m, 3192m (-CONH2), 1659vs (-CONH2), 1587vs, 1536m, 1509vs, 1424m, 1190vs (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.76 (1H, t, J = 6.2 Hz, NH), 8.22 (1H, d, J = 2.3 Hz, Ar), 8.12 (1H, bs, NH2), 7.73 (1H, d, J = 2.3 Hz, Ar), 7.64 (1H, bs, NH2), 3.38 (2H, q, J = 6.2 Hz, NCH2), 1.60–1.44 (2H, m, CH2), 1.39–1.16 (6H, m, CH2), 0.84 (3H, t, J = 6.2 Hz, CH3); 13C-NMR (75 MHz, DMSO-d6) δ 169.1, 154.6, 146.6, 129.6, 126.4, 40.5, 31.2, 28.9, 26.4, 22.2, 14.1; Elemental analysis: calc. for C11H18N4O (MW 222.29): 59.44% C, 8.16% H, 25.20% N; found 59.41% C, 8.24% H, 25.10% N.
3-(Heptylamino)pyrazine-2-carboxamide (8). Yellow crystalline solid. Yield 91.5%; m.p. 66.0–67.3 °C; IR (ATR-Ge, cm−1): 3407m (-NH-), 3316m, 3176m (-CONH2), 1674vs (-CONH2), 1580vs, 1532m, 1514vs, 1421m, 1184s (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.76 (1H, t, J = 6.3 Hz, NH), 8.22 (1H, d, J = 2.3 Hz, Ar), 8.12 (1H, bs, NH2), 7.72 (1H, d, J = 2.3 Hz, Ar), 7.64 (1H, bs, NH2), 3.38 (2H, q, J = 6.3 Hz, NCH2), 1.60–1.45 (2H, m, CH2), 1.37–1.15 (8H, m, CH2), 0.83 (3H, t, J = 6.3 Hz, CH3); 13C-NMR (75 MHz, DMSO-d6) δ 169.1, 154.6, 146.6, 129.6, 126.4, 40.5, 31.4, 29.0, 28.6, 26.7, 22.2, 14.1; Elemental analysis: calc. for C12H20N4O (MW 236.31): 60.99% C, 8.53% H, 23.71% N; found 61.12% C, 8.57% H, 23.64% N.
3-(Octylamino)pyrazine-2-carboxamide (9). Light yellow crystalline solid. Yield 95.8%; m.p. 67.2–68.3 °C; IR (ATR-Ge, cm−1): 3407m (-NH-), 3321m, 3211m (-CONH2), 1664vs (-CONH2), 1594vs, 1539m, 1510vs, 1423m, 1197vs (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.76 (1H, t, J = 6.4 Hz, NH), 8.22 (1H, d, J = 2.3 Hz, Ar), 8.11 (1H, bs, NH2), 7.73 (1H, d, J = 2.3 Hz, Ar), 7.64 (1H, bs, NH2), 3.38 (2H, q, J = 6.4 Hz, NCH2), 1.61–1.45 (2H, m, CH2), 1.37–1.14 (10H, m, CH2), 0.83 (3H, t, J = 6.4 Hz, CH3); 13C-NMR (75 MHz, DMSO-d6) δ 169.1, 154.6, 146.6, 129.6, 126.5, 40.5, 31.4, 29.0, 28.9, 28.8, 26.7, 22.3, 14.1; Elemental analysis: calc. for C13H22N4O (MW 250.34): 62.37% C, 8.86% H, 22.38% N; found 62.42% C, 8.81% H, 22.25% N.
3-(Dibutylamino)pyrazine-2-carboxamide (10). Light yellow crystalline solid. Yield 67.2%; m.p. 129.3–130.8 °C; IR (ATR-Ge, cm−1): 3372m, 3197m (-CONH2), 1647vs (-CONH2), 1616m, 1558m, 1507m, 1457m, 1179s (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.08 (1H, d, J = 2.3 Hz, Ar), 7.96 (1H, bs, NH2), 7.73 (1H, d, J = 2.3 Hz, Ar), 7.52 (1H, bs, NH2), 3.40 (4H, q, J = 7.5 Hz, NCH2), 1.56–1.42 (4H, m, CH2), 1.31–1.13 (4H, m, CH2), 0.85 (6H, t, J = 7.5 Hz, CH3); 13C-NMR (75 MHz, DMSO-d6) δ 169.5, 151.7, 141.8, 135.5, 129.7, 48.8, 29.3, 19.8, 14.0; Elemental analysis: calc. for C13H22N4O (MW 250.34): 62.37% C, 8.86% H, 22.38% N; found 62.27% C, 8.66% H, 22.32% N.
3-(Cyclopentylamino)pyrazine-2-carboxamide (11). Yellow crystalline solid. Yield 90.5%; m.p. 107.8–108.8 °C; IR (ATR-Ge, cm−1): 3399m (-NH-), 3285m (-CONH2), 1655vs (-CONH2), 1580vs, 1525m, 1505vs, 1419m, 1172vs (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.79 (1H, d, J = 7.0 Hz, NH), 8.23 (1H, d, J = 2.3 Hz, Ar), 8.12 (1H, bs, NH2), 7.73 (1H, d, J = 2.3 Hz, Ar), 7.64 (1H, bs, NH2), 4.33–4.12 (1H, m, NCH), 2.05–1.88 (2H, m, CH2), 1.75–1.49 (4H, m, CH2), 1.49–1.33 (2H, m, CH2); 13C-NMR (75 MHz, DMSO-d6) δ 169.1, 154.2, 146.6, 129.7, 126.4, 51.5, 32.9, 23.5; Elemental analysis: calc. for C10H14N4O (MW 206.24): 58.24% C, 6.84% H, 27.17% N; found 58.23% C, 6.85% H, 26.98% N.
3-(Cyclohexylamino)pyrazine-2-carboxamide (12). Light yellow crystalline solid. Yield 92.5%; m.p. 129.0–130.3 °C (described in the literature 128–129 °C [36]); IR (ATR-Ge, cm−1): 3455m (-NH-), 3254m (-CONH2), 1683vs (-CONH2), 1583s, 1518vs, 1473m, 1418m, 1182s (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.78 (1H, d, J = 7.6 Hz, NH), 8.21 (1H, d, J = 2.3 Hz, Ar), 8.12 (1H, bs, NH2), 7.72 (1H, d, J = 2.3 Hz, Ar), 7.64 (1H, bs, NH2), 3.96–3.80 (1H, m, NCH), 1.96–1.82 (2H, m, CH2), 1.73–1.60 (2H, m, CH2), 1.60–1.47 (1H, m, CH2), 1.43–1.14 (5H, m, CH2); 13C-NMR (75 MHz, DMSO-d6) δ 169.1, 153.8, 146.6, 129.6, 126.2, 48.0, 32.4, 25.5, 24.4; Elemental analysis: calc. for C11H16N4O (MW 220.27): 59.98% C, 7.32% H, 25.44% N; found 59.78% C, 7.38% H, 25.33% N.
3-(Cycloheptylamino)pyrazine-2-carboxamide (13). Light yellow crystalline solid. Yield 87.4%; m.p. 102.7–104.1 °C; IR (ATR-Ge, cm−1): 3450m (-NH-), 3293m, 3180m (-CONH2), 1674vs (-CONH2), 1581vs, 1506vs, 1465m, 1416s, 1177vs (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.84 (1H, d, J = 7.6 Hz, NH), 8.22 (1H, d, J = 2.3 Hz, Ar), 8.12 (1H, bs, NH2), 7.71 (1H, d, J = 2.3 Hz, Ar), 7.64 (1H, bs, NH2), 4.15–3.99 (1H, m, NCH), 1.95–1.79 (2H, m, CH2), 1.65–1.37 (10H, m, CH2); 13C-NMR (75 MHz, DMSO-d6) δ 169.1, 153.7, 146.6, 129.5, 126.3, 50.2, 34.2, 27.7, 23.7; Elemental analysis: calc. for C12H18N4O (MW 234.30): 61.52% C, 7.74% H, 23.91% N; found 61.50% C, 7.94% H, 24.05% N.
3-(Pyrrolidin-1-ylamino)pyrazine-2-carboxamide (14). Yellow crystalline solid. Yield 82.7%; m.p. 179.6–180.0 °C; IR (ATR-Ge, cm−1): 3384m, 3189m (-CONH2), 1638vs (-CONH2), 1551s, 1517s, 1462s, 1437s, 1191s (pyrazine ring); 1H-NMR (300 MHz, CDCl3) δ 8.14 (1H, d, J = 2.3 Hz, Ar), 7.75 (1H, d, J = 2.3 Hz, Ar), 7.18 (1H, bs, NH2), 5.84 (1H, bs, NH2), 3.54–3.39 (4H, m, NCH2), 2.00–1.85 (4H, m, CH2); 13C-NMR (75 MHz, CDCl3) δ 168.8, 151.5, 143.6, 129.4, 129.2, 49.5, 25.4; Elemental analysis: calc. for C9H12N4O (MW 192.22): 56.24% C, 6.29% H, 29.15% N; found 56.16% C, 6.23% H, 29.11% N.
3-(Piperidin-1-ylamino)pyrazine-2-carboxamide (15). Light yellow crystalline solid. Yield 93.0%; m.p. 126.4–127.7 °C; IR (ATR-Ge, cm−1): 3385m, 3183m (-CONH2), 1646vs (-CONH2), 1552s, 1518s, 1484vs, 1440vs, 1183vs (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.13 (1H, d, J = 2.1 Hz, Ar), 7.92 (1H, bs, NH2), 7.82 (1H, d, J = 2.3 Hz, Ar), 7.51 (1H, bs, NH2), 3.45–3.33 (4H, m, NCH2), 1.64–1.42 (6H, m, CH2); 13C-NMR (75 MHz, DMSO-d6) δ 168.9, 153.0, 142.2, 135.9, 131.1, 48.2, 25.4, 24.1; Elemental analysis: calc. for C10H14N4O (MW 206.24): 58.24% C, 6.84% H, 27.17% N; found 58.29% C, 6.77% H, 27.37% N.
3-(Morpholin-4-yl)pyrazine-2-carboxamide (16). Light yellow crystalline solid. Yield 91.3%; m.p. 146.1–147.5 °C; IR (ATR-Ge, cm−1): 3330m, 3184m (-CONH2), 1655vs (-CONH2), 1609m, 1554s, 1519m, 1484vs, 1436s, 1109vs (pyrazine ring); 1H-NMR (300 MHz, CDCl3) δ 8.20 (1H, d, J = 2.3 Hz, Ar), 7.91 (1H, d, J = 2.3 Hz, Ar), 7.49 (1H, bs, NH2), 5.87 (1H, bs, NH2), 3.82 (4H, t, J = 4.7 Hz, OCH2), 3.54 (4H, t, J = 4.7 Hz, NCH2); 13C-NMR (75 MHz, CDCl3) δ 167.8, 154.4, 143.6, 132.3, 131.3, 66.7, 49.0; Elemental analysis: calc. for C9H12N4O2 (MW 208.22): 51.92% C, 5.81% H, 26.91% N; found 51.80% C, 5.91% H, 26.83% N.
3-(4-Methylpiperazin-1-yl)pyrazine-2-carboxamide (17). Sandy yellow crystalline solid. Yield 50.0%; m.p. 168.8–170.5 °C (decomp.); IR (ATR-Ge, cm−1): 3374m, 3190m (-CONH2), 1647vs (-CONH2), 1552s, 1519m, 1492vs, 1440s, 1186s (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 8.16 (1H, d, J = 2.3 Hz, Ar), 7.97 (1H, bs, NH2), 7.88 (1H, d, J = 2.3 Hz, Ar), 7.56 (1H, bs, NH2), 3.42 (4H, t, J = 5.0 Hz, NCH2), 2.35 (4H, t, J = 5.0 Hz, NCH2), 2.18 (3H, s, CH3); 13C-NMR (75 MHz, DMSO-d6) δ 168.8, 152.8, 142.3, 136.1, 131.8, 54.5, 47.0, 46.0; Elemental analysis: calc. for C10H15N5O (MW 221.26): 54.28% C, 6.83% H, 31.65% N; found 54.19% C, 6.84% H, 31.48% N.
3-(Piperazin-1-yl)pyrazine-2-carboxamide (18). Yellow crystalline solid. Yield 65.8%; m.p. decomp.; IR (ATR-Ge, cm−1): 3389m (-NH-), 3293m, 3179m (-CONH2), 1678vs (-CONH2), 1593s, 1559s, 1527m, 1464s, 1433s, 1159s (pyrazine ring); 1H-NMR (300 MHz, DMSO-d6) δ 9.75 (1H, bs, NH), 8.26 (1H, bs, Ar), 8.08 (1H, bs, NH2), 8.02 (1H, bs, Ar), 7.67 (1H, bs, NH2), 3.68–3.59 (4H, m, NCH2), 3.16–3.07 (4H, m, NCH2); 13C-NMR (75 MHz, DMSO-d6) δ 168.2, 152.6, 142.8, 136.0, 133.4, 44.5, 42.5; Elemental analysis: calc. for C9H13N5O (MW 207.23): 52.16% C, 6.32% H, 33.79% N; found 52.19% C, 6.31% H, 33.59% N.
3.4. Lipophilicity HPLC Determination and Calculations
Experimental lipophilicity parameter logk was ascertained using an Agilent Technologies 1200 SL liquid chromatography HPLC system with a SL G1315C Diode-array Detector, chromatographic pre-column ZORBAX XDB-C18 5 μm, 4 × 4 mm, Part No. 7995118-504 and column ZORBAX Eclipse XDB-C18 5 μm, 4.6 × 250 mm, Part No. 7995118-585 (Agilent Technologies Inc., Colorado Springs, CO, USA) were used. The separation process was controlled by Agilent ChemStation, version B.04.02 extended by spectral module (Agilent Technologies Inc.). A solution of MeOH (HPLC grade, 70%) and H2O (HPLC-Milli-Q Grade, 30%) was used as mobile phase. The total flow of the column was 1.0 mL/min, injection 20 μL, column temperature 30 °C. 210 nm as detection wavelength and 270 nm as monitor wavelength were chosen. The KI methanol solution was used for the dead time (TD) determination. Retention times (TR) of synthesized compounds were measured in minutes. The capacity factors k were calculated using Microsoft Excel according to formula k = (TR− TD)/TD, where TR is the retention time of the solute and TD denotes the dead time obtained via an unretained analyte. Logk, calculated from the capacity factor k, is used as the lipophilicity index converted to logP scale.
3.5. Biological Assays
3.5.1. Antimycobacterial in Vitro Screening
Mycobacterial screening was performed against M. tuberculosis H37Rv CNCTC My 331/88 (Czech National Collection of Type Cultures, National Institute of Public Health, Prague, Czech Republic) using isoniazid and pyrazinamide (Sigma-Aldrich) as standards. Culturing medium was Middlebrook 7H9 broth (Sigma-Aldrich) with the addition of glycerol (Sigma-Aldrich) and OADC supplement (Himedia, Mumbai, India). Tested compounds were dissolved in dimethylsulfoxide (DMSO) and diluted with medium to final concentrations 100, 50, 25, 12.5, 6.25, 3.125 and 1.5625 μg/mL. The method used for this assay was microdilution broth panel method. The final concentration of DMSO did not exceed 1% (v/v) and did not affect the growth of mycobacteria. The cultures were grown in Middlebrook 7H9 medium at 37 °C in humid dark atmosphere. The antimycobacterial activity was determined using Alamar Blue colouring after 14 days of incubation as MIC (μg/mL). This evaluation was done in cooperation with Department of Clinical Microbiology, University Hospital in Hradec Kralove (Hradec Kralove, Czech Republic).
3.5.2. Antifungal and Antibacterial in Vitro Screenings
Antibacterial evaluation was made using the microdilution broth method in plates M27A-M1 (200 + 10) against 8 bacterial stems from Czech Collection of Microorganisms (Brno, Czech Republic) or clinical isolates from Department of Clinical Microbiology, University Hospital in Hradec Kralove (Staphylococcus aureus CCM 4516/08, Staphylococcus aureus H 5996/08 methicillin resistant, Staphylococcus epidermidis H 6966/08, Enterococcus sp. J 14365/08, Escherichia coli CCM 4517, Klebsiella pneumoniae D 11750/08, Klebsiella pneumoniae J 14368/05 ESBL positive, Pseudomonas aeruginosa CCM 1961). Mueller Hinton broth was used for the cultivation that was done in humid atmosphere and by 35 °C. The readings were made after 24 and 48 h and MIC was set as 80% inhibition of control. The standards were neomycin, bacitracin, penicillin G, ciprofloxacin and phenoxymethylpenicillin [43].
Antifungal evaluation was also accomplished with microdilution broth method. On the contrary, there was used RPMI 1640 broth with glutamine as medium and conditions were humid and dark atmosphere, pH 7.0 (adjusted with 0.165 M morpholinepropanesulfonic acid, MOPS) and 35 °C. 8 fungal strains were used (Candida albicans ATCC 44859, Candida tropicalis 156, Candida krusei E28, Candida glabrata 20/I, Trichosporon asahii 1188, Aspergillus fumigatus 231, Lichtheimia corymbifera 272, Trichophyton mentagrophytes 445) together with four antimycotic standards amphotericin B, voriconazole, nystatin and fluconazole. The MIC was set as 80% inhibition of control and readings were made after 24 and 48 h (50% IC, 72 and 120 h for filament fungi) [44].
3.5.3. Study of Photosynthetic Electron Transport Inhibition
The inhibition of photosynthetic electron transport (PET) in spinach chloroplasts was determined spectrophotometrically (Genesys 6, Thermo Scientific, Madison, WI, USA) using an artificial electron acceptor 2,6-dichlorophenol-indophenol (DCPIP) [45]. The rate of photosynthetic electron transport was monitored as a photoreduction of DCPIP. Chloroplasts were prepared from Spinacia oleracea L. [46]. The measurements were carried out in phosphate buffer (0.02 mol/L, pH 7.2) containing sucrose (0.4 mol/L), MgCl2 (0.005 mol/L) and NaCl (0.015 mol/L). The chlorophyll concentration was 30 mg/L and the samples were irradiated (~100 W/m2 with 10 cm distance) with a halogen lamp (250 W). The 4 cm wide water filter was used to prevent warming of the suspension so the temperature did not exceed 22 °C. The studied compound were dissolved in DMSO due to their limited water solubility. The applied DMSO concentration (up to 4%) did not affect the photochemical activity in spinach chloroplasts. The efficiency of studied compounds was expressed as 50% inhibition concentration relative to the untreated control. The standard for these measurements was DCMU (Diurone®).
The emission fluorescence spectra of aromatic amino acids were recorded on the fluorescence spectrophotometer F-2000 (Hitachi, Tokyo, Japan). The samples of chloroplast suspensions with and without studied inhibitor were excited at wavelength of 275 nm, excitation slit 20 nm and emission slit 10 nm. The samples were kept in the dark for 2 min prior to the measurement. The phosphate buffer used for dilution of the chloroplast suspension was the same as described above. The compounds were added to chloroplast suspension in DMSO solution due to low aqueous solubility. The DMSO concentration in all samples was the same as in the control (10%). The chlorophyll concentration in chloroplast suspension was 10 mg/L.
4. Conclusions
Eighteen N-substituted 3-aminopyrazine-2-carboxamides were synthesized in this work with the application of focused field microwave technology. The reaction conditions, which were set experimentally, lead to higher yields and shorter reaction times compared to the conventional heating method. The difference between these conditions is obvious. The temperature reached during the reactions is much higher than the boiling point of methanol and toluene under normal conditions. Also the time is at least two times shorter. All the compounds were purified and then their structure was confirmed by NMR and IR spectra and they were then tested for their potential biological activities.
The experimentally measured lipophilicity was compared to the CS ChemBioDraw Ultra 13.0 program predicted values. The dependence between logk and logP was linear with one exception where the effect of ionization is appreciable.
The majority of compounds did not show antimycobacterial activity against M. tuberculosis. There were only four compounds (6‒9) with very low activity that were characterized by long or ramified side aliphatic side chain which is probably better for penetration into mycobacterial cell. But the values were significantly lower than the values of standards so it is not possible to predict any structure-activity relationships.
Quite interesting results were found in antifungal assays. Three compounds (11‒13) showed activities against whole tested fungal spectrum and the activity of compounds 12 and 13 was also better than that of a fluconazole standard. Although there were not enough active compounds in the antibacterial and antifungal screenings, we can propose a hypothesis that the side alicyclic chain is necessary for antifungal activity and that the efficacy rises with increasing number of carbons in the alicyclic ring, i.e., increasing lipophilicity.
PET inhibition screening showed some interesting consequences. There were nine compounds active and activity of the most active substance (compound 9) expressed by IC50 value was 14.3 μmol/L. A quasi-parabolic course was observed for the dependence of PET inhibiting activity on the lipophilic properties of substituents and side chain of the most effective compounds had seven or eight carbons. The active compounds were found to be PS2 inhibitors acting on the donor side of photosystem 2.
Based on the obtained results it can be concluded that the biological activity of the compounds is affected by the presence of secondary amine group bounded to pyrazine ring. If this -NH- fragment is missing, the efficacy would also disappear. This phenomenon can be seen in both evaluations.
Acknowledgments
The publication is co-financed by the European Social Fund and the state budget of the Czech Republic. Project No. CZ.1.07/2.3.00/20.0235, the title of the project: TEAB and by the European Social Fund and the state budget of the Czech Republic. Project No. CZ.1.07/2.3.00/30.0022. This study was also supported by the Ministry of Health of Czech Republic (IGA NZ 13346), Grant Agency of Charles University B-CH/710312 and SVV 260 062 as well as by the Slovak Grant Agency VEGA, Grant No. 1/0612/11 and by the Project APVV-0061-11. The authors also wish to thank Ida Dufkova for performing and evaluating the antifungal and antibacterial assays.
Author Contributions
Ondrej Jandourek, Jan Zitko and Martin Dolezal designed the compounds and the overall study. Ondrej Jandourek carried out the syntheses of final products. Vladimir Kubicek measured the lipophilicity of compounds. Jiri Kunes recorded and interpreted the NMR spectra. Pavla Paterova and Vladimir Buchta carried out the antimycobacterial evaluation. Matus Pesko and Katarina Kralova tested compounds for PET inhibition activity and interpreted the results.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. Global Tuberculosis Report 2013; World Health Organization: Lyon, France, 2013; pp. 1–27. [Google Scholar]
- Zhang, Y.; Chiu Chang, K.; Leung, C.; Yew, W.W.; Gicquel, B.; Fallows, D.; Kaplan, G.; Chaisson, R.E.; Zhang, W. “ZS-MDR-TB” versus “ZR-MDR-TB”: Improving treatment of MDR-TB by identifying pyrazinamide susceptibility. Emerg. Microbes Infect. 2012, 1, e5. [Google Scholar] [CrossRef]
- Velayati, A.A.; Masjedi, M.R.; Farnia, P.; Tabarsi, P.; Ghanavi, J.; Ziazarifi, A.H.; Hoffner, S.E. Emergence of new forms of totally drug-resistant tuberculosis bacilli: Super extensively drug-resistant tuberculosis or totally drug-resistant strains in Iran. Chest 2009, 136, 420–425. [Google Scholar] [CrossRef]
- Lima, C.H.S.; Bispo, M.L.F.; de Souza, M.V.N. Pirazinamida: Um fàrmaco essencial no tratamento da tuberculose. Rev. Virtual Quim. 2011, 3, 159–180. [Google Scholar]
- Zhang, Y.; Wade, M.M.; Scorpio, A. Mode of action of pyrazinamide: Disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 2003, 52, 790–795. [Google Scholar] [CrossRef]
- Konno, K.; Feldmann, F.M.; McDermott, W. Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am. Rev. Respir. Dis. 1967, 95, 461–469. [Google Scholar]
- Zhang, Y.; Scorpio, A.; Nikaido, H.; Sun, Z. Role of acid pH and deficient efflux of pyrazinoic acid in unique susceptibility of Mycobacterium tuberculosis to pyrazinamide. J. Bacteriol. 1999, 181, 2044–2049. [Google Scholar]
- Scorpio, A.; Zhang, Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef]
- Boshoff, H.I.; Mizrahi, V.; Barry, C.E., III. Effects of pyrazinamide on fatty acid synthesis by Whole mycobacterial cells and purified fatty acid synthase I. J. Bacteriol. 2002, 184, 2167–2172. [Google Scholar] [CrossRef]
- Zimhony, O.; Cox, J.S.; Welch, J.T.; Vilcheze, C.; Jacobs, W.R. Pyrazinamide inhibits the eukaryotic-like fatty acid synthetase I (FAS-I) of Mycobacterium tuberculosis. Nat. Med. 2000, 6, 1043–1047. [Google Scholar] [CrossRef]
- Zimhony, O.; Vilcheze, C.; Arai, M.; Welch, J.T.; Jacobs, W.R. Pyrazinoic acid and its n-propyl ester inhibit fatty acid synthase type I in replicating tubercle bacilli. Antimicrob. Agents Chemother. 2007, 51, 752–754. [Google Scholar] [CrossRef]
- Ngo, S.C.; Zimhony, O.; Chung, W.J.; Sayahi, H.; Jacobs, W.R., Jr.; Welch, J.T. Inhibition of isolated mycobacterium tuberculosis fatty acid synthase I by pyrazinamide analogs. Antimicrob. Agents Chemother. 2007, 51, 2430–2435. [Google Scholar] [CrossRef]
- Shi, W.; Zhang, X.; Jiang, X.; Yuan, H.; Lee, J.S.; Barry, C.E.; Wang, H.; Zhang, W.; Zhang, Y. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 2011, 333, 1630–1632. [Google Scholar] [CrossRef]
- Dolezal, M.; Hartl, J.; Miletin, M.; Machacek, M.; Kralova, K. Synthesis and photosynthesis-inhibiting activity of some anilides of substituted pyrazine-2-carboxylic acids. Chem. Pap. 1999, 53, 126–130. [Google Scholar]
- Dolezal, M. Biologically active pyrazines of natural and synthetic origin. Chem. Listy 2006, 100, 959–966. [Google Scholar]
- Chaluvaraju, K.C.; Ishwar, B.K. Synthesis and antimicrobial activities of amino benzylated mannich bases of pyrazinamide. Int. J. ChemTech Res. 2010, 2, 1368–1371. [Google Scholar]
- Dolezal, M.; Zitko, J.; Osicka, Z.; Kunes, J.; Buchta, V.; Vejsova, M.; Dohnal, J.; Jampilek, J.; Kralova, K. Synthesis, antimycobacterial, antifungal and photosynthesis-inhibiting activity of chlorinated N-phenylpyrazine-2-carboxamides. Molecules 2010, 15, 8567–8581. [Google Scholar] [CrossRef]
- Whitehead, R.P.; Unger, J.M.; Flaherty, L.E.; Kraut, E.H.; Mills, G.M.; Klein, C.E.; Chapman, R.A.; Doolittle, G.C.; Hammond, N.; Sondak, V.K. A phase II trial of pyrazine diazohydroxide in patients with disseminated malignant melanoma and no prior chemotherapy—Southwest Oncology Group Study. Invest. New Drugs 2002, 20, 105–111. [Google Scholar]
- Furuta, Y.; Takahashi, K.; Fukuda, Y.; Kuno, M.; Kamiyama, T.; Kozaki, K.; Nomura, N.; Egawa, H.; Minami, S.; Watanabe, Y.; et al. In vitro and in vivo activities of anti-influenza virus compound T-705. Antimicrob. Agents Chemother. 2002, 46, 977–981. [Google Scholar] [CrossRef]
- A-Rahim, Y.I.; Beyer, K.H., Jr.; Vesell, E.S. Studies on pyrazinoylguanidine. 3. Downregulation of lipolysis in isolated adipocytes. Pharmacology 1996, 53, 197–210. [Google Scholar] [CrossRef]
- Dolezal, M.; Kralova, K. Synthesis and evaluation of pyrazine derivatives with herbicidal activity. In Herbicides, Theory and Applications; Soloneski, S., Larramendy, M.L., Eds.; InTech: Vienna, Austria, 2011; pp. 581–610. [Google Scholar]
- Servusova, B.; Eibinova, D.; Dolezal, M.; Kubicek, V.; Paterova, P.; Pesko, M.; Kralova, K. Substituted N-benzylpyrazine-2-carboxamides: Synthesis and biological evaluation. Molecules 2012, 17, 13183–13198. [Google Scholar] [CrossRef]
- Jandourek, O.; Dolezal, M.; Paterova, P.; Kubicek, V.; Pesko, M.; Kunes, J.; Coffey, A.; Guo, J.; Kralova, K. N-Substituted 5-amino-6-methylpyrazine-2,3-dicarbonitriles: Microwave-assisted synthesis and biological properties. Molecules 2014, 19, 651–671. [Google Scholar] [CrossRef]
- Dolezal, M.; Kralova, K.; Sersen, F.; Miletin, M. The site of action of pyrazine-2-carboxylic acids in the photosynthetic apparatus. Folia Pharm. Univ. Carol. 2001, 26, 13–20. [Google Scholar]
- Kralova, K.; Sersen, F.; Miletin, M.; Dolezal, M. Inhibition of photosynthetic electron transport in spinach chloroplasts by 2,6-disubstituted pyridine-4-thiocarboxamides. Chem. Pap. 2002, 56, 214–217. [Google Scholar]
- Kralova, K.; Sersen, F.; Klimesova, V.; Waisser, K. Effect of 2-alkylthio-4-pyridinecarbothioamides on photosynthetic electron transport in spinach chloroplasts. Collect. Czechoslov. Chem. Commun. 1997, 62, 516–520. [Google Scholar] [CrossRef]
- Kralova, K.; Sersen, F.; Kubicova, L.; Waisser, K. Inhibitory effects of substituted benzanilides on photosynthetic electron transport in spinach chloroplasts. Chem. Pap. 1997, 53, 328–331. [Google Scholar]
- Tamai, R.; Ito, M.; Kobayashi, M.; Mitsunari, T.; Nakano, Y. Oxopyrazine Derivative and Herbicide. U.S. Patent Application 2013/0137577, 30 May 2013. [Google Scholar]
- Reingruber, R.; Kraus, H.; Hutzler., J.; Newton, T.W.; Witschel, M.; Moberg, W.K.; Rapado, L.P.; Besong, G; Rack, N.; van der Kloet, A.; et al. Substituted pyrazines having herbicidal activity. U.S. Patent Application 2013/0274109BA1, 17 October 2013. [Google Scholar]
- Nakamura, A.; Ataka, T.; Segawa, H.; Takeuchi, Y.; Takematsu, T. Studies on herbicidal 2,3-dicyanopyrazines. 2. Structure-activity relationships of herbicidal 5-ethylamino- and 5-propylamino-2,3-dicyanopyrazines. Agric. Biol. Chem. 1983, 47, 1561–1567. [Google Scholar] [CrossRef]
- Hayes, B.L. Microwave Synthesis: Chemistry at the Speed of Light; CEM Pub.: Matthews, NC, USA, 2002. [Google Scholar]
- De La Hoz, A.; Diaz-Ortiz, A.; Moreno, A. Microwaves in organic synthesis. Thermal and non-thermal microwave effects. Chem. Soc. Rev. 2005, 34, 164–178. [Google Scholar] [CrossRef]
- Albert, A.; Brown, D.J.; Wood, H.C.S. 406. Pteridine studies. Part VIII. The degradation of pteridine. Methylation of the hydroxypteridines and degradation of the products. J. Chem. Soc. 1956. [Google Scholar] [CrossRef]
- Dlabal, K.; Palat, K.; Lycka, A.; Odlerova, Z. Synthesis and 1H- and 13C-NMR spectra of sulfur derivatives of pyrazine derived from amidation product of 2-chloropyrazine and 6-chloro-2-pyrazinecarbonitrile. Tuberculostatic activity. Collect. Czechoslov. Chem. Commun. 1990, 55, 2493–2500. [Google Scholar] [CrossRef]
- Osdene, T.S.; Taylor, E.C. A new synthetic approach to pteridines*. J. Am. Chem. Soc. 1956, 78, 5451–5452. [Google Scholar] [CrossRef]
- Keir, W.F.; MacLennan, A.H.; Wood, H.C. Amidinoacetamides in the synthesis of pyrazines and pteridines. J. Chem. Soc. 1977, 11, 1321–1325. [Google Scholar]
- Izawa, S. Acceptors and donors for chloroplast electron transport. In Methods in Enzymology; Part, C., Colowick, P., Kaplan, N.O., Eds.; Academic Press: New York, NY, USA; London, UK, 1980; Volume 69, pp. 413–434. [Google Scholar]
- Kralova, K.; Sersen, F.; Pesko, M.; Klimesova, V.; Waisser, K. Photosynthesis-inhibiting effects of 2-benzylsulphanylbenzimidazoles in spinach chloroplasts. Chem. Pap. 2012, 66, 795–799. [Google Scholar] [CrossRef]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Keltosova, S.; Tengler, J.; Bobal, P.; Kollar, P.; Cizek, A.; Kralova, K.; et al. Antimycobacterial and herbicidal activity of ring-substituted 1-hydroxynaphthalene-2-carboxanilides. Bioorg. Med. Chem. 2013, 21, 6531–6541. [Google Scholar]
- Kralova, K.; Sersen, F.; Pesko, M.; Waisser, K.; Kubicova, L. 5-Bromo- and 3,5-dibromo-2-hydroxy-N-phenylbenzamides - inhibitors of photosynthesis. Chem. Pap. 2014, 68, 46–52. [Google Scholar] [CrossRef]
- Gonec, T.; Kos, J.; Zadrazilova, I.; Pesko, M.; Govender, R.; Keltosova, S.; Chambel, B.; Pereira, D.; Kollar, P.; Imramovsky, A.; et al. Antibacterial and herbicidal activity of ring-substituted 2-hydroxynaphthalene-1-carboxanilides. Molecules 2013, 18, 9397–9419. [Google Scholar] [CrossRef]
- Jampilek, J.; Dolezal, M.; Kunes, J.; Satinsky, D.; Raich, I. Novel regioselective preparation of 5-chloropyrazine-2-carbonitrile, pyrazine-2-carboxamide and coupling study of substituted phenylsulfanylpyrazine-2-carboxylic acid derivatives. Curr. Org. Chem. 2005, 9, 49–60. [Google Scholar]
- Jones, R.N.; Barry, A.L. Optimal dilution susceptibility testing conditions, recommendations for MIC interpretation, and quality control guidelines for the ampicillin-sulbactam combination. J. Clin. Microbiol. 1987, 25, 1920–1925. [Google Scholar]
- National Committee for Clinical Laboratory Standards. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts: Proposed Standard M 27-P; National Committee for Clinical Laboratory Standards: Villanova, PA, USA, 1992. [Google Scholar]
- Kralova, K.; Sersen, F.; Sidoova, E. Photosynthesis inhibition produced by 2-alkylthio-6-R-benzothiazoles. Chem. Pap. 1992, 46, 348–350. [Google Scholar]
- Masarovicova, E.; Kralova, K. Approaches to measuring plant photosynthesis activity. In Handbook of Photosynthesis, 2nd ed.; Pessarakli, M., Ed.; Taylor & Francis Group: Boca Raton, FL, USA, 2005; pp. 617–656. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).