
Seven-Membered Rings through Metal-Free Rearrangement Mediated by Hypervalent Iodine

Abstract
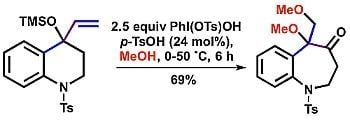
:
1. Introduction

2. Results and Discussion



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Conditions | Product (Yield) |
|---|---|---|
| 1 | 1.0 equiv HTIB, 20 mol% p-TsOH, MeOH, −72 °C to rt, 2 h |  |
| 2 | 1.0 equiv HTIB, MeOH, −72 to 30 °C, 2.5 h |  |
| 3 | (1) 1.0 equiv HTIB, MeOH, −72 to 30 °C, 2.5 h; (2) 2 weeks |  |
| 4 | 2.5 equiv HTIB, MeOH, rt, 2 h |  |



| Entry | Substrate | Products (Isolated Yield) |
|---|---|---|
| 1 |  |  |
| 2 |  |  |
| 3 |  |  |
| 4 |  |  |
| 5 a | 3e | 8e (67%) |
| 6 a |  |  |
| 7 b |  |  |
| 8 |  |  |
| 9 |  |  |
| 10 |  |  |
| 11 |  |  |
| 12 b |  |  |




| Doxorubicin |  |  |  |  |  | |
|---|---|---|---|---|---|---|
| 5f and 8f (1:1) | 8d | 8g | 8l | 12f | ||
| U251 | 0.20 | 4.8 | 8.2 | 43.2 | 50.7 | 5.2 |
| UACC-62 | 0.86 | 18.1 | 34.4 | 59.3 | 91.3 | 8.4 |
| MCF-7 | 1.2 | 14.0 | 25.9 | 52.8 | 70.1 | 6.0 |
| NCI-ADR/RES | 3.5 | 19.8 | 42.0 | 57.9 | 79.9 | 6.5 |
| 786-0 | 0.27 | 15.9 | 27.6 | 72.8 | 102.4 | 6.7 |
| NCI-H460 | 0.61 | 37.2 | 60.3 | 64.6 | 157.7 | 22.9 |
| PC-3 | 0.74 | 3.6 | 8.5 | 44.8 | 88.5 | 10.6 |
| HT29 | 11.4 | 17.5 | 24.4 | 55.7 | 124.6 | 34.6 |
| K562 | 0.96 | >250 | 25.6 | >250 | 167.6 | 46.3 |
| HaCat | 0.16 | 7.5 | 11.8 | 48.1 | 206.0 | 4.1 |
| Mean LogTGI | −0.081 | >1.2 | 1.35 | >1.8 | 2.02 | 1.03 |
3. Experimental Section
General Information
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kleinke, A.S.; Webb, D.; Jamison, T.F. Recent Progress in the Synthesis of Oxepanes and Medium Ring Ethers. Tetrahedron 2012, 68, 6999–7018. [Google Scholar] [CrossRef]
- Sharma, A.; Appukkuttan, P.; van der Eycken, E. Microwave-Assisted Synthesis of Medium-Sized Heterocycles. Chem. Commun. 2012, 48, 1623–1637. [Google Scholar] [CrossRef]
- Majumdar, K.C. Regioselective Formation of Medium-Ring Heterocycles of Biological Relevance by Intramolecular Cyclization. RSC Adv. 2011, 1, 1152–1170. [Google Scholar] [CrossRef]
- Majumdar, K.C.; Chattopadhyay, B. New Synthetic Strategies for Medium-Sized and Macrocyclic Compounds by Palladium-Catalyzed Cyclization. Curr. Org. Chem. 2009, 13, 731–757. [Google Scholar] [CrossRef]
- Chattopadhyay, S.K.; Karmakar, S.; Biswas, T.; Majumdar, K.C.; Rahaman, H.; Roy, B. Formation of Medium-Ring Heterocycles by Diene and Enyne Metathesis. Tetrahedron 2007, 63, 3919–3952. [Google Scholar] [CrossRef]
- Lopez, F.; Mascarenas, J.L. The Oxygen-Bridge Templating Approach to Eight- and Nine-Membered Carbocycles: Recent Developments Based on Catalytic Reactions. Chem. Eur. J. 2007, 13, 2172–2178. [Google Scholar] [CrossRef] [PubMed]
- Snyder, N.L.; Haines, H.M.; Peczuh, M.W. Recent Developments in the Synthesis of Oxepines. Tetrahedron 2006, 62, 9301–9320. [Google Scholar] [CrossRef]
- Michaut, A.; Rodriguez, J. Selective Construction of Carbocyclic Eight-Membered Rings by Ring-Closing Metathesis of Acyclic Precursors. Angew. Chem. Int. Ed. 2006, 45, 5740–5750. [Google Scholar] [CrossRef]
- Nakamura, I.; Yamamoto, Y. Transition-metal-catalyzed reactions in heterocyclic synthesis. Chem. Rev. 2004, 104, 2127–2198. [Google Scholar] [CrossRef] [PubMed]
- Yet, L. Metal-Mediated Synthesis of Medium-Sized Rings. Chem. Rev. 2000, 100, 2963–3007. [Google Scholar] [CrossRef] [PubMed]
- Yet, L. Free Radicals in the Synthesis of Medium-Sized Rings. Tetrahedron 1999, 55, 9349–9403. [Google Scholar] [CrossRef]
- Hoberg, J.O. Synthesis of Seven-Membered Oxacycles. Tetrahedron 1998, 54, 12631–12670. [Google Scholar] [CrossRef]
- Bauer, R.A.; Wenderski, T.A.; Tan, D.S. Biomimetic Diversity-Oriented Synthesis of Benzannulated Medium Rings via Ring Expansion. Nat. Chem. Biol. 2013, 9, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Yousufb, S.K.; Mukherjee, D. Importance and Synthesis of Benzannulated Medium-Sized and Macrocyclic Rings (BMRs). RSC Adv. 2014, 4, 43241–43257. [Google Scholar] [CrossRef]
- Illuminati, G.; Mandolini, L. Ring-Closure Reactions of Bifunctional Chain Molecules. Acc. Chem. Res. 1981, 14, 95–102. [Google Scholar] [CrossRef]
- Crimmins, M.T.; Choy, A.L. An Asymmetric Aldol-Ring-Closing Metathesis Strategy for the Enantioselective Construction of Oxygen Heterocycles: An Efficient Approach to the Enantioselective Synthesis of (+)-Laurencin. J. Am. Chem. Soc. 1999, 121, 5653–5660. [Google Scholar] [CrossRef]
- Trost, B.M.; Xie, J. Palladium-Catalyzed Asymmetric Ring Expansion of Allenylcyclobutanols: An Asymmetric Wagner-Meerwein Shift. J. Am. Chem. Soc. 2006, 128, 6044–6045. [Google Scholar] [CrossRef] [PubMed]
- Waters, S.P.; Tian, Y.; Li, Y.M.; Danishefsky, S.J. Total Synthesis of (−)-Scabronine G, an Inducer of Neurotrophic Factor Production. J. Am. Chem. Soc. 2005, 127, 13514–13515. [Google Scholar] [CrossRef] [PubMed]
- Kantorowski, E.J.; Kurth, M.J. Expansion to Seven-Membered Rings. Tetrahedron 2000, 56, 4317–4353. [Google Scholar] [CrossRef]
- Kim, S.; Uh, K.H. Mercurinium Ion Mediated Ring Expansion of 1-Alkenyl-1-Cycloalkanols. Tetrahedron Lett. 1992, 33, 4325–4328. [Google Scholar] [CrossRef]
- Silva, L.F., Jr.; Carneiro, V.M.T. Thallium(III) in Organic Synthesis. Synthesis 2010, 1059–1074. [Google Scholar]
- Kim, S.; Uh, K.H. Thallium Ion Mediated Ring Expansion of 1-Trimethylsilyloxy-1-alkenylcycloalkanes to a-exo-Methylenecycloalkanones. Tetrahedron Lett. 1996, 37, 3865–3866. [Google Scholar] [CrossRef]
- Silva, L.F., Jr.; Olofsson, B. Hypervalent Iodine Reagents in the Total Synthesis of Natural Products. Nat. Prod. Rep. 2011, 28, 1722–1754. [Google Scholar] [CrossRef] [PubMed]
- Zhdankin, V.V.; Stang, P.J. Chemistry of Polyvalent Iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T. Hypervalent Iodine Chemistry in Synthesis: Scope and New Directions. Angew. Chem. Int. Edit. 2005, 44, 3656–3665. [Google Scholar] [CrossRef]
- Moriarty, R.M. Organohypervalent Iodine: Development, Applications, and Future Directions. J. Org. Chem. 2005, 70, 2893–2903. [Google Scholar] [CrossRef] [PubMed]
- Singh, F.V.; Wirth, T. Oxidative Rearrangements with Hypervalent Iodine Reagents. Synthesis 2013, 45, 2499–2511. [Google Scholar] [CrossRef]
- Silva, L.F., Jr. Hypervalent Iodine-Mediated Ring Contraction Reactions. Molecules 2006, 11, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: London, UK, 1997. [Google Scholar]
- Wirth, T. Topics in Current Chemistry. In Hypervalent Iodine Chemistry—Modern Developments in Organic Synthesis, 1st ed.; Springer: Berlin, Germany, 2003; Volume 224, pp. 1–264. [Google Scholar]
- Justik, M.W.; Koser, G.F. Oxidative Rearrangements of Arylalkenes with Hydroxy(tosyloxy)iodo Benzene in 95% Methanol: A General, Regiospecific Synthesis of α-Aryl Ketones. Tetrahedron Lett. 2004, 45, 6159–6163. [Google Scholar] [CrossRef]
- Justik, M.W.; Koser, G.F. Application of Hydroxy(tosyloxy)iodo Benzene in the Wittig-Ring Expansion Sequence for the Synthesis of β-Benzocycloalkenones from α-Benzocycloalkenones. Molecules 2005, 10, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.P.; Khatod, H.S. Enantioselective Synthesis of the Essential Oil and Pheromonal Component Ar-Himachalene by a Chiral Pool and Chirality Induction Approach. Tetrahedron-Asymmetry 2012, 23, 1410–1415. [Google Scholar] [CrossRef]
- Silva, L.F., Jr.; Vasconcelos, R.S.; Nogueira, M.A. Iodine(III)-Promoted Ring Expansion of 1-Vinylcycloalkanol Derivatives: A Metal-Free Approach Toward Seven-Membered Rings. Org. Lett. 2008, 10, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B. Ruthenium-Catalyzed Olefin Metathesis Double-Bond Isomerization Sequence. J. Org. Chem. 2004, 69, 7672–7687. [Google Scholar] [CrossRef] [PubMed]
- Karimi, B.; Golshani, B. Mild and Highly Efficient Method for the Silylation of Alcohols Using Hexamethyldisilazane Catalyzed by Iodine under Nearly Neutral Reaction Conditions. J. Org. Chem. 2000, 65, 7228–7230. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.A.W.; Rose, M.E. Rapid, Simple, and Mild Procedure for Alkylation of Phenols, Alcohols, Amides and Acids. Tetrahedron 1979, 35, 2169–2173. [Google Scholar] [CrossRef]
- Yusubov, M.S.; Wirth, T. Solvent-Free Reactions with Hypervalent Iodine Reagents. Org. Lett. 2005, 7, 519–521. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.M.; Paknikar, S.K.; Dev, V.; Beauchamp, P.S.; Kamat, S.P. Biogenetic-Type Synthesis of (+)-Cymbodiacetal, a Constituent of Cymbopogon Martinii. J. Nat. Prod. 2004, 67, 700–702. [Google Scholar] [CrossRef] [PubMed]
- Rocha, D.F.O.; Hamilton, K.; Goncalves, C.C.S.; Machado, G.; Marsaioli, A.J. 6-Alkyl-3,4-dihydro-2H-pyrans: Chemical Secretion Compounds in Neotropical Harvestmen. J. Nat. Prod. 2011, 74, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Ferraz, H.M.C.; Bombonato, F.I.; Longo, L.S. Synthetic Approaches to Naturally Occurring Ten-Membered-Ring Lactones. Synthesis 2007, 3261–3285. [Google Scholar]
- Rousseau, G. Medium Ring Lactones. Tetrahedron 1995, 51, 2777–2849. [Google Scholar] [CrossRef]
- Silva, L.F., Jr.; Sousa, R.M.F.; Ferraz, H.M.C.; Aguilar, A.M. Thallium Trinitrate-Mediated Ring Contraction of 1,2-Dihydronaphthalenes: The Effect of Electron-Donating and Electron-Withdrawing Groups. J. Braz. Chem. Soc. 2005, 16, 1160–1173. [Google Scholar]
- Silva, L.F., Jr.; Siqueira, F.A.; Pedrozo, E.C.; Vieira, F.Y.M.; Doriguetto, A.C. Iodine(III)-Promoted Ring Contraction of 1,2-Dihydronaphthalenes: A Diastereoselective Total Synthesis of (±)-Indatraline. Org. Lett. 2007, 9, 1433–1436. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, F.A.; Ishikawa, E.E.; Fogaca, A.; Faccio, A.T.; Carneiro, V.M.T.; Soares, R.R.S.; Utaka, A.; Tebeka, I.R.M.; Bielawski, M.; Olofsson, B.; et al. Metal-Free Synthesis of Indanes by Iodine(III)-Mediated Ring Contraction of 1,2-Dihydronaphthalenes. J. Braz. Chem. Soc. 2011, 22, 1795–1807. [Google Scholar] [CrossRef]
- Ferraz, H.M.C.; Aguilar, A.M.; Silva, L.F. Model Studies Toward the Synthesis of Natural Indans Utilizing a Thallium(III)-Mediated Ring-Contraction Reaction. Synthesis 2003, 1031–1034. [Google Scholar]
- Dohi, T.; Ito, M.; Yamaoka, N.; Morimoto, K.; Fujioka, H.; Kita, Y. Hypervalent Iodine(III): Selective and Efficient Single-Electron-Transfer (SET) Oxidizing Agent. Tetrahedron 2009, 65, 10797–10815. [Google Scholar] [CrossRef]
- Kita, Y.; Egi, M.; Ohtsubo, M.; Saiki, T.; Okajima, A.; Takada, T.; Tohma, H. Hypervalent Iodine(III)-Induced Intramolecular Cyclization Reaction of Substituted Phenol Ethers with an Alkyl Azido Side-Chain: A Novel and Efficient Synthesis of Quinone Imine Derivatives. Chem. Pharm. Bull. 1999, 47, 241–245. [Google Scholar] [CrossRef]
- Tankard, M.H.; Whitehurst, J.S. Ethoxyethynyl Carbinol-αβ-Unsaturated Ester Conversion—Stereomutation Accompanying a Selenium Dioxide Oxidation. Tetrahedron 1974, 30, 451–454. [Google Scholar] [CrossRef]
- Ahmad, A.; Silva, L.F., Jr. Synthesis of Chromanes and 4H-Chromenes: Exploring the Oxidation of 2H-Chromenes and Dihydro-1-benzoxepines by Hypervalent Iodine(III). Synthesis 2012, 44, 3671–3677. [Google Scholar] [CrossRef]
- Liu, P.; Liu, S.J.; Zhang, J.Z.; Tian, G.R. Selective Oxidation of Sulfides to Sulfoxides with Poly 4-Hydroxy(tosyloxy)iodo Styrene. Synth. Commun. 2005, 35, 3173–3177. [Google Scholar] [CrossRef]
- So, M.; Kotake, T.; Matsuura, K.; Inui, M.; Kamimura, A. Concise Synthesis of 2-Benzazepine Derivatives and Their Biological Activity. J. Org. Chem. 2012, 77, 4017–4028. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsov, V.; Palma, A.; Ewert, C. Synthesis and Applicability of Partially Reduced 2-Benzazepines. Curr. Org. Chem. 2001, 5, 519–551. [Google Scholar] [CrossRef]
- Grunewald, G.L.; Dahanukar, V.H.; Criscione, K.R. Effects of a 3-Alkyl-, 4-Hydroxy- and/or 8-Aromatic-Substituent on the Phenylethanolamine N-Methyltransferase Inhibitor Potency and α2-Adrenoceptor Affinity of 2,3,4,5-Tetrahydro-1H-2-Benzazepines. Bioorg. Med. Chem. 2001, 9, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.D.; Aristoff, P.A.; Zurenko, G.E.; Schaadt, R.D.; Yagi, B.H.; Ford, C.W.; Hamel, J.C.; Stapert, D.; Moerman, J.K. Synthesis and Biological Evaluation of Benzazepine Oxazolidinone Antibacterials. Bioorg. Med. Chem. Lett. 2003, 13, 4197–4200. [Google Scholar] [CrossRef] [PubMed]
- Kukla, M.J.; Breslin, H.J.; Diamond, C.J.; Grous, P.P.; Ho, C.Y.; Miranda, M.; Rodgers, J.D.; Sherrill, R.G.; de Clercq, E.; Pauwels, R.; et al. Synthesis and Anti-HIV-1 Activity of 4,5,6,7-Tetrahydro-5-Methylimidazo[4,5,1-jk][1,4]benzodiazepin-2(1H)-one (TIBO) Derivatives. J. Med. Chem. 1991, 34, 3187–3197. [Google Scholar] [CrossRef] [PubMed]
- Qadir, M.; Cobb, J.; Sheldrake, P.W.; Whittall, N.; White, A.J.P.; Hii, K.K.; Horton, P.N.; Hursthouse, M.B. Conformation Analyses, Dynamic Behavior and Amide Bond Distortions of Medium-Sized Heterocycles. 1. Partially and Fully Reduced 1-Benzazepines. J. Org. Chem. 2005, 70, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Boeglin, D.; Bonnet, D.; Hibert, M. Solid-Phase Preparation of a Pilot Library Derived from the 2,3,4,5-Tetrahydro-1H-benzo[b]Azepin-5-amine Scaffold. J. Comb. Chem. 2007, 9, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Seto, M.; Aikawa, K.; Miyamoto, N.; Aramaki, Y.; Kanzaki, N.; Takashima, K.; Kuze, Y.; Iizawa, Y.; Baba, M.; Shiraishi, M. Highly Potent and Orally Active CCR5 Antagonists as Anti-HIV-1 Agents: Synthesis and Biological Activities of 1-Benzazocine Derivatives Containing a Sulfoxide Moiety. J. Med. Chem. 2006, 49, 2037–2048. [Google Scholar] [CrossRef] [PubMed]
- Abonia, R.; Cuervo, P.; Insuasty, B.; Quiroga, J.; Nogueras, M.; Cobo, J. A Simple Two-Step Sequence for the Synthesis of Novel 4-Aryl-4,5-dihydro-6H-[1,3]dioxolo[4,5-h]pyrrolo[1,2-a][1]benzazepin-6-ones from 6-Amino-3,4-methylenedioxyacetophenone. Eur. J. Org. Chem. 2008, 4684–4689. [Google Scholar]
- Iqbal, N.; Fiksdahl, A. Gold(I)-Catalyzed Benz[c]azepin-4-ol Synthesis by Intermolecular 5+2 Cycloaddition. J. Org. Chem. 2013, 78, 7885–7895. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Harada, T.; Ohmiya, H.; Sawamura, M. Intramolecular Hydroamination of Alkynic Sulfonamides Catalyzed by a Gold-Triethynylphosphine Complex: Construction of Azepine Frameworks by 7-Exo-Dig Cyclization. Beilstein J. Org. Chem. 2011, 7, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigrowolff, A.; et al. Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor-Cell Lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.M. DTP—Developmental Therapeutics Program NCI/NIH. Compare Methodology 2000. Available online: http://dtp.nci.nih.gov/docs/compare/compare_methodology.html (accessed on 12 January 2015).
- Fouche, G.; Cragg, G.M.; Pillay, P.; Kolesnikova, N.; Maharaj, V.J.; Senabe, J. In Vitro Anticancer Screening of South African Plants. J. Ethnopharm. 2008, 119, 455–461. [Google Scholar] [CrossRef]
- Seyferth, D. Di-n-butyldivinyltin [Tin, dibutyldivinyl-]. Org. Synth. Coll. 1963, 4, 258. [Google Scholar]
- Pagliero, R.J.; Pierini, A.B.; Brun, R.; Mazzieri, M.R. Design, Synthesis and 3-D Characterization of 1-Benzenesulfonyl-1,2,3,4-Tetrahydroquinolinesas Lead Scaffold for Antiparasitic Drug. Lett. Drug Des. Discov. 2010, 7, 461–470. [Google Scholar] [CrossRef]
- Catino, A.J.; Nichols, J.M.; Choi, H.; Gottipamula, S.; Doyle, M.P. Benzylic Oxidation Catalyzed by Dirhodium(II,III) Caprolactamate. Org. Lett. 2005, 7, 5167–5170. [Google Scholar] [CrossRef] [PubMed]
- Braunholtz, J.T.; Mann, F.G. Cyclic Keto-Amines. Part II. The Preparation and Spectroscopic Characteristics of Substituted 1:2:3:4-Tetrahydro-4-oxoquinolines. J. Chem. Soc. 1957, 4166–4173. [Google Scholar]
- Shoemaker, R.H. The NCI60 Human Tumour Cell Line Anticancer Drug Screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, S.B.L.; Torre, A.D.; De Carvalho, J.E.; Ruiz, A.L.T.G.; Silva, L.F., Jr. Seven-Membered Rings through Metal-Free Rearrangement Mediated by Hypervalent Iodine. Molecules 2015, 20, 1475-1494. https://doi.org/10.3390/molecules20011475
Silva SBL, Torre AD, De Carvalho JE, Ruiz ALTG, Silva LF Jr. Seven-Membered Rings through Metal-Free Rearrangement Mediated by Hypervalent Iodine. Molecules. 2015; 20(1):1475-1494. https://doi.org/10.3390/molecules20011475
Chicago/Turabian StyleSilva, Siguara Bastos Lemos, Adriana Della Torre, João Ernesto De Carvalho, Ana Lúcia Tasca Gois Ruiz, and Luiz F. Silva, Jr. 2015. "Seven-Membered Rings through Metal-Free Rearrangement Mediated by Hypervalent Iodine" Molecules 20, no. 1: 1475-1494. https://doi.org/10.3390/molecules20011475
APA StyleSilva, S. B. L., Torre, A. D., De Carvalho, J. E., Ruiz, A. L. T. G., & Silva, L. F., Jr. (2015). Seven-Membered Rings through Metal-Free Rearrangement Mediated by Hypervalent Iodine. Molecules, 20(1), 1475-1494. https://doi.org/10.3390/molecules20011475