
Synthesis and in Silico Evaluation of Novel Compounds for PET-Based Investigations of the Norepinephrine Transporter


 ,
,  and
and
Abstract
:1. Introduction


2. Results and Discussion




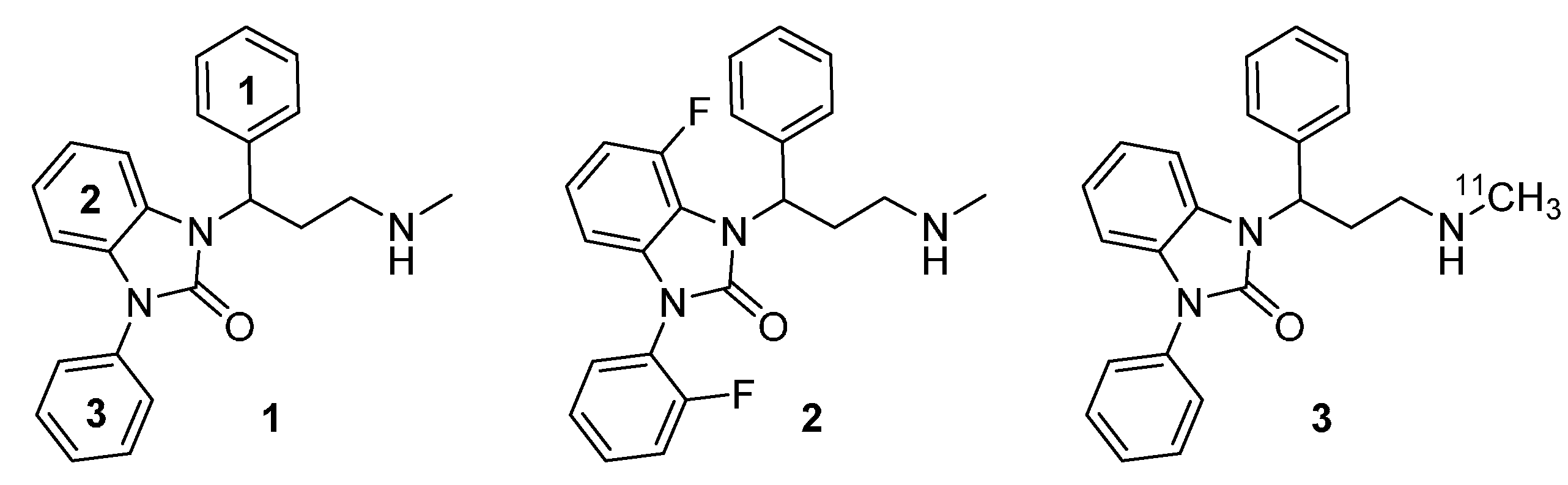{kind=link}
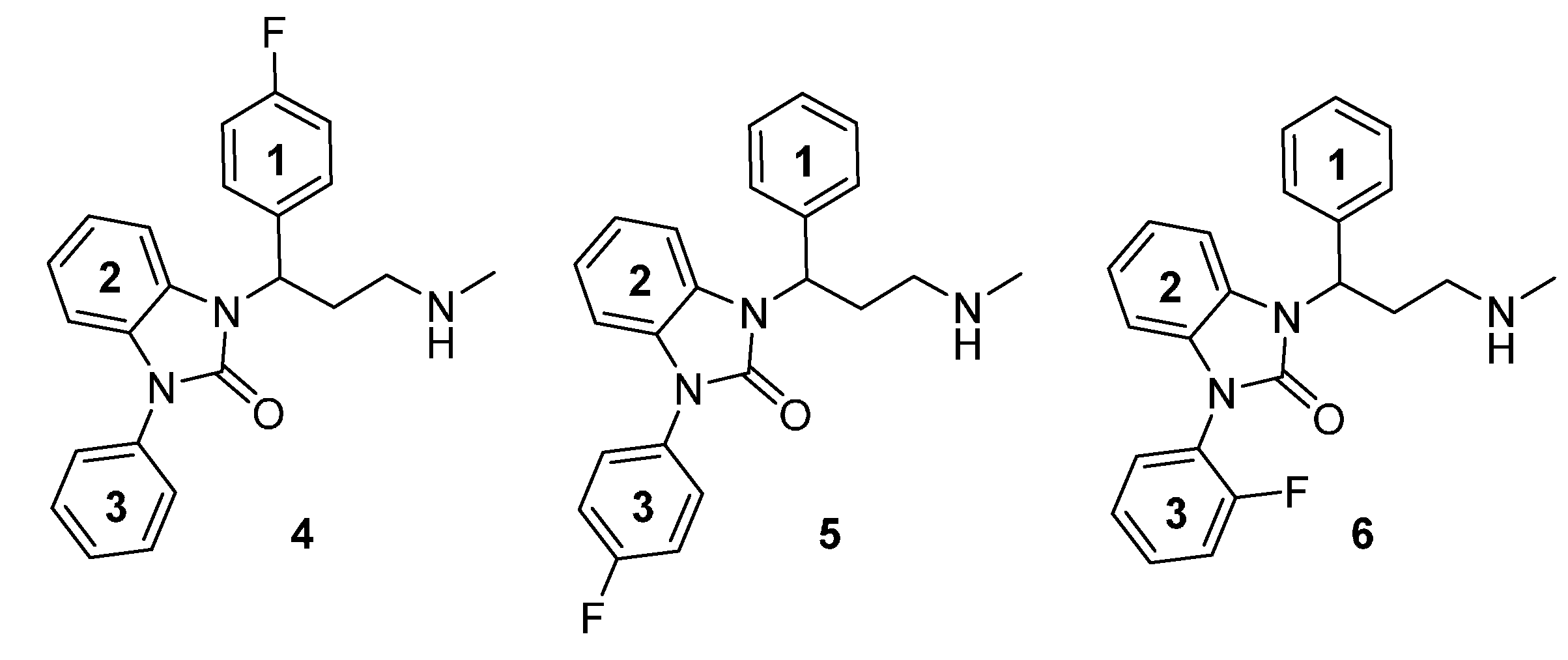{kind=link}
{kind=link}
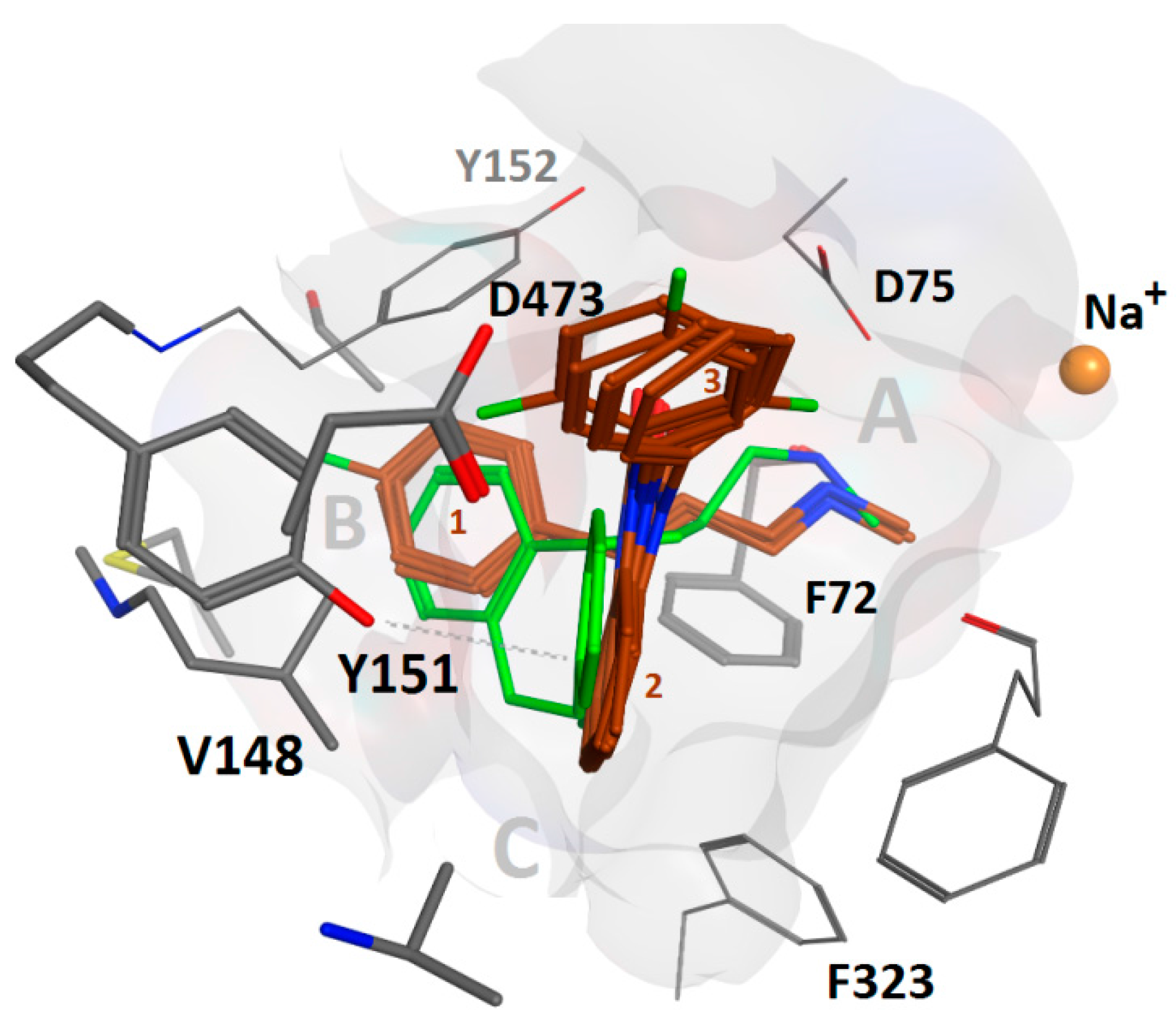{kind=link}
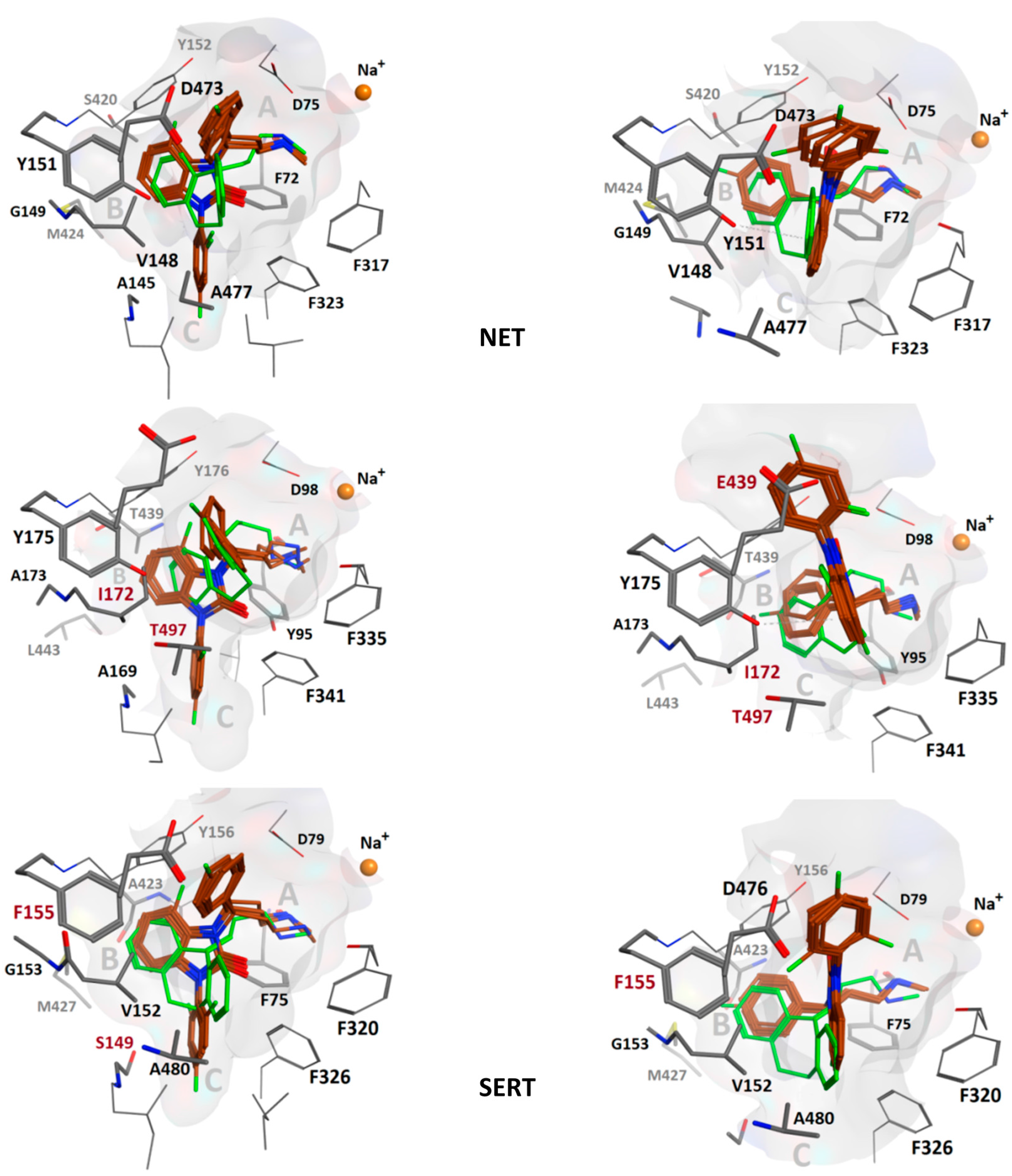{kind=link}
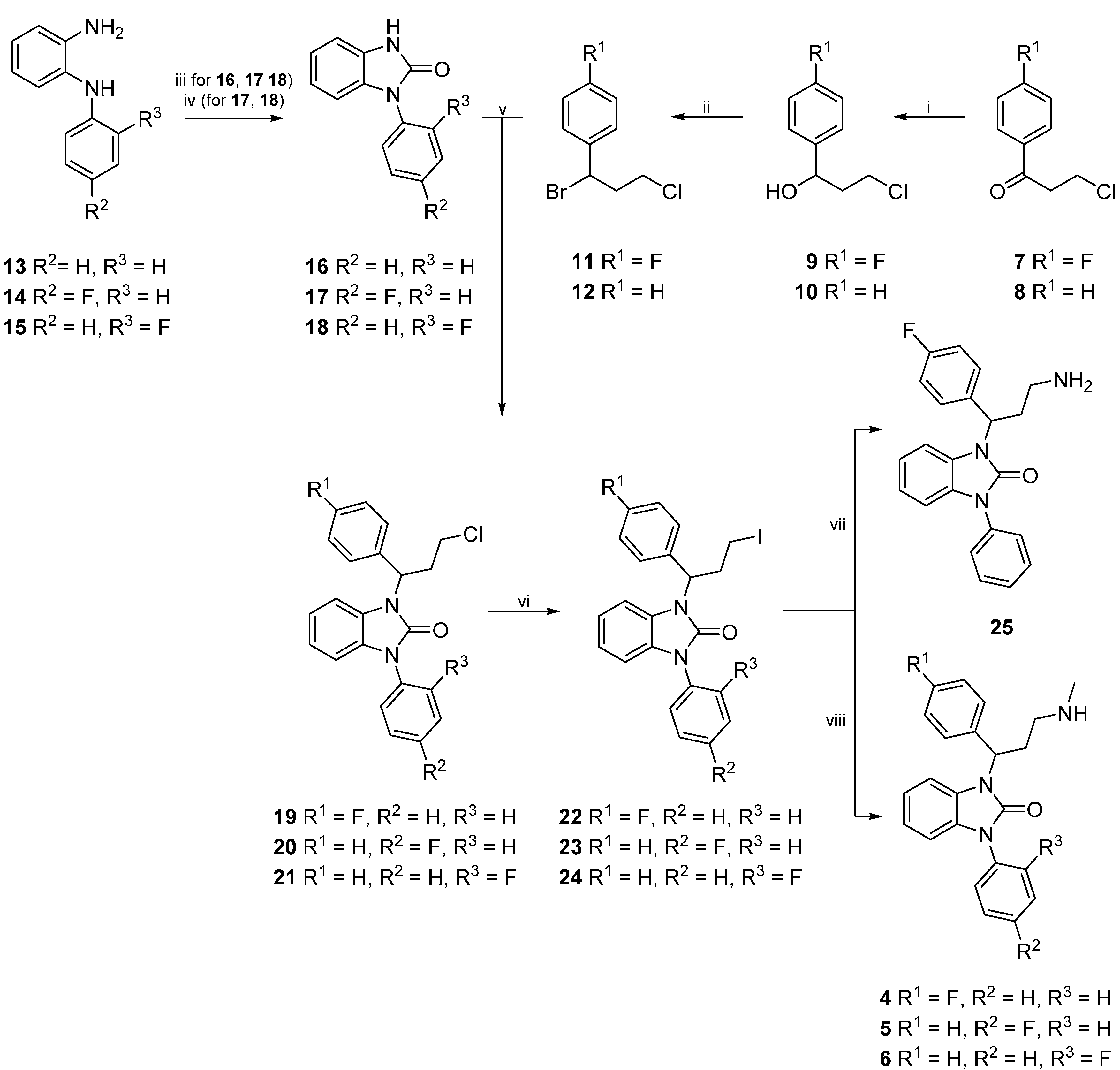{kind=link}
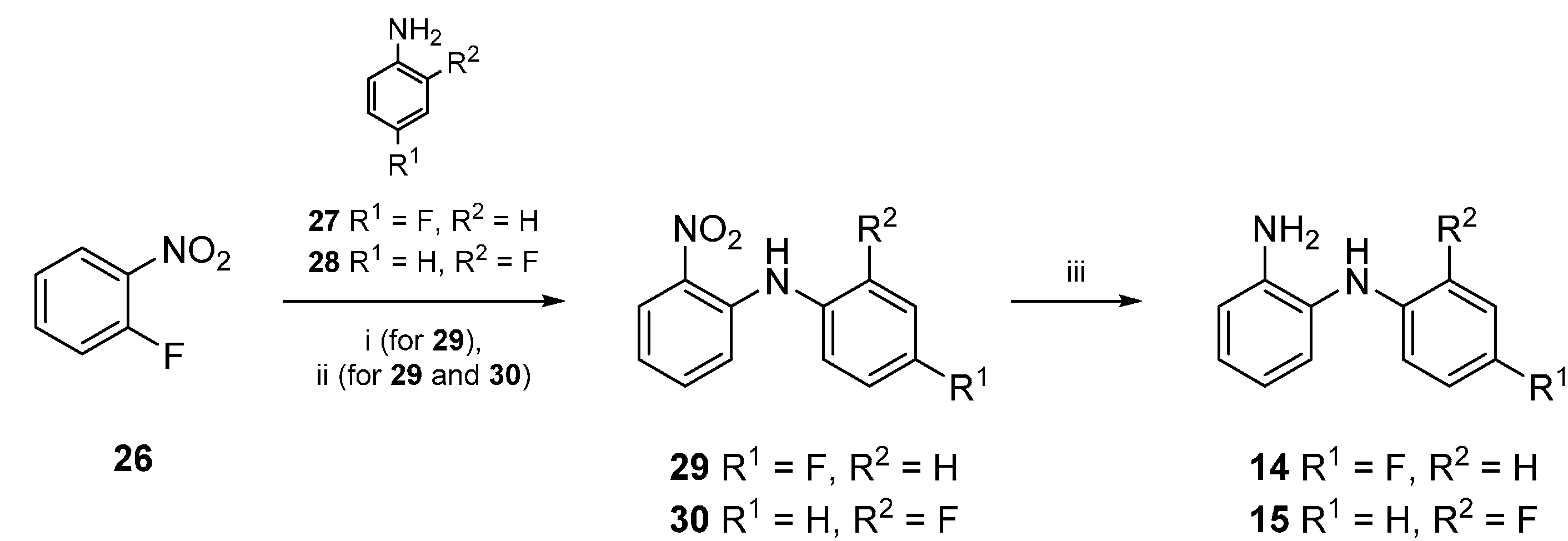{kind=link}
| B Site | C Site | Outer Site | |||||||
|---|---|---|---|---|---|---|---|---|---|
| hNET | S420 | M424 | G149 | V148 | A145 | F72 | D473 | Y151 | A477 |
| hSERT | T439 | L443 | A173 | I172 | A177 | Y95 | E493 | Y175 | T497 |
| hDAT | A423 | M427 | G153 | V152 | S149 | F76 | D476 | F155 | A480 |

3. Experimental Section
3.1. General
3.2. Syntheses
3.2.1. General Procedure for the Synthesis of 9 and 10
3.2.2. General Procedure for the Synthesis of 11 and 12
3.2.3. General Procedure for the Synthesis of 29 and 30
3.2.4. Alternative Procedure for the Synthesis of 29
3.2.5. General Procedure for the Synthesis of 14 and 15
3.2.6. General Procedure for the Synthesis of 16–18
3.2.7. Alternative Procedure for the Synthesis of 17 and 18
3.2.8. General Procedure for the Synthesis of 19–21
3.2.9. General Procedure for the Synthesis of 22–24
3.2.10. General procedure for the synthesis of 1-(3-amino-1-(4-fluorophenyl)propyl)-3-phenyl-1,3- dihydro-2H-benzimidazol-2-one (25)
3.2.11. General Procedure for the Synthesis of 4–6
3.3. Computational Methods

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sung, U.; Apparsundram, S.; Galli, A.; Kahlig, K.M.; Savchenko, V.; Schroeter, S.; Quick, M.W.; Blakely, R.D. A regulated interaction of syntaxin 1A with the antidepressant-sensitive norepinephrine transporter establishes catecholamine clearance capacity. J. Neurosci. 2003, 23, 1697–1709. [Google Scholar]
- Kim, C.H.; Hahn, M.K.; Joung, Y.; Anderson, S.L.; Steele, A.H.; Mazei-Robinson, M.S.; Gizer, I.; Teicher, M.H.; Cohen, B.M.; Robertson, D.; et al. A polymorphism in the norepinephrine transporter gene alters promoter activity and is associated with attention-deficit hyperactivity disorder. Proc. Natl. Acad. Sci. USA 2006, 103, 19164–19169. [Google Scholar]
- Hahn, M.K.; Robertson, D.; Blakely, R.D. A mutation in the human norepinephrine transporter gene (SLC6A2) associated with orthostatic intolerance disrupts surface expression of mutant and wild-type transporters. J. Neurosci. 2003, 23, 4470–4478. [Google Scholar]
- Mirbolooki, M.R.; Upadhyay, S.K.; Constantinescu, C.C.; Pan, M.L.; Mukherjee, J. Adrenergic pathway activation enhances brown adipose tissue metabolism: A [18F]FDG PET/CT study in mice. Nucl. Med. Biol. 2014, 41, 10–16. [Google Scholar]
- Lin, S.L.; Fan, X.; Yeckel, C.W.; Weinzimmer, D.; Mulnix, T.; Gallezot, J.D.; Carson, R.E.; Sherwin, R.S.; Ding, Y.S. Ex vivo and in vivo Evaluation of the Norepinephrine Transporter Ligand [11C]MRB for Brown Adipose Tissue Imaging. Nucl. Med. Biol. 2012, 39, 1081–1086. [Google Scholar]
- Stöber, G.; Nöthen, M.M.; Pörzgen, P.; Brüss, M.; Bönisch, H.; Knapp, M.; Beckmann, H.; Propping, P. Systematic search for variation in the human norepinephrine transporter gene: Identification of five naturally occurring missense mutations and study of association with major psychiatric disorders. Am. J. Med. Genet. (Neuropsychiatr. Genet.) 1996, 67, 523–532. [Google Scholar]
- Young, J.B.; Landsberg, L. Catecholamines and the adrenal medulla. In Williams Textbook of Endocrinology, 9th ed.; Wilson, J.D., Foster, D.W., Eds.; W.B. Saunders Co.: Philadelphia, PA, USA, 1998; p. 680. [Google Scholar]
- Tellioglu, T.; Robertson, D. Genetic or acquired deficits in the norepinephrine transporter: Current understanding of clinical implications. Exp. Rev. Mol. Med. 2001, 1–10. [Google Scholar]
- Blakely, R.D.; Bauman, A.L. Biogenic amine transporters: regulation in flux. Curr. Opin. Neurobiol. 2000, 10, 328–336. [Google Scholar]
- Zhu, M.Y.; Shamburger, S.; Li, J.; Ordway, G.A. Regulation of the Human Norepinephrine Transporter by Cocaine and Amphetamine. J. Pharmacol. Exp. Ther. 2000, 295, 951–959. [Google Scholar]
- Moron, J.A.; Brockington, A.; Wise, R.A.; Rocha, B.A.; Hope, B.T. Dopamine Uptake through the Norepinephrine Transporter in Brain Regions with Low Levels of the Dopamine Transporter: Evidence from Knock-Out Mouse Lines. J. Neurosci. 2002, 22, 386–395. [Google Scholar]
- Schroeter, S.; Apparsundaram, S.; Wiley, R.G.; Miner, L.H.; Sesack, S.R.; Blakely, R.D. Immunolocalization of the cocaine- and antidepressant-sensitive l-norepinephrine transporter. J. Comp. Neurol. 2000, 420, 211–232. [Google Scholar]
- Torres, G.E.; Gainetdinov, R.R.; Caron, M.G. Plasma membrane monoamine transporters: Structure, regulation and function. Nat. Rev. Neurosci. 2003, 4, 13–25. [Google Scholar]
- Ordway, G.A.; Stockmeier, C.A.; Cason, G.W.; Klimek, V. Pharmacology and Distribution of Norepinephrine Transporters in the Human Locus Coeruleus and Raphe Nuclei. J. Neurosci. 1997, 17, 1710–1719. [Google Scholar]
- Smith, H.R.; Beveridge, T.J. R.; Porrino, L.J. Distribution of norepinephrine transporters in the non-human primate brain. Neuroscience 2006, 138, 703–714. [Google Scholar]
- Zhou, J. Norepinephrine transporter inhibitors and their therapeutic potential. Drugs Future 2004, 29, 1235–1244. [Google Scholar]
- Curatolo, P.; D’Agati, E.; Moavero, R. The neurobiological basis of ADHD. Ital. J. Pediatr. 2010, 36, 79. [Google Scholar]
- Mash, D.C.; Ouyang, Q.; Qin, Y.; Pablo, J. Norepinephrine transporter immunoblotting and radioligand binding in cocaine abusers. Neurosci. Methods 2005, 143, 79–85. [Google Scholar]
- Barr, C.L.; Kroft, J.; Feng, Y.; Wigg, K.; Roberts, W.; Malone, M.; Ickowicz, A.; Schachar, R.; Tannock, R.; Kennedy, J.L. The Norepinephrine Transporter Gene and Attention-Deficit Hyperactivity Disorder. Am. J. Med. Genet. (Neuropsychiatr. Genet.) 2002, 114, 255–229. [Google Scholar]
- Klimek, V.; Stockmeier, C.; Overholser, J.; Meltzer, H.Y.; Kalka, S.; Dilley, G.; Ordway, G.A. Reduced Levels of Norepinephrine Transporters in the Locus Coeruleus in Major Depression. J. Neurosci. 1997, 17, 8451–8458. [Google Scholar]
- Nedergaard, J.; Cannon, B. The changed metabolic world with human brown adipose tissue: Therapeutic visions. Cell Metab. 2010, 11, 268–272. [Google Scholar]
- Gulyas, B.; Brockschnieder, D.; Nag, S.; Pavlova, E.; Kasa, P.; Beliczai, Z.; Legradi, A.; Gulya, K.; Thiele, A.; Dyrks, T.; et al. The norepinephrine transporter radioligand [18F]FD2MeNER shows significant decreases in NET density in the locus coeruleus and the thalamus in Alzheimer’s disease: A post-mortem autoradiographic study in human brains. Neurochem. Int. 2010, 56, 789–798. [Google Scholar]
- Wilson, A.A.; Johnson, D.P.; Mozley, D.; Hussey, D.; Ginovart, N.; Nobrega, J.; Garcia, A.; Meyer, J.; Houle, S. Synthesis and in vivo evaluation of novel radiotracers for the in vivo imaging of the norepinephrine transporter. Nucl. Med. Biol. 2003, 30, 85–92. [Google Scholar]
- Takano, A.; Gulyas, B.; Varrone, A.; Halldin, C. Saturated norepinephrine transporter occupancy by atomoxetine relevant to clinical doses: a rhesus monkey study with (S,S)-[(18)F]FMeNER-D (2). Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1308–1314. [Google Scholar]
- Schou, M.; Zoghbi, S.S.; Shetty, H.U.; Shchukin, E.; Liow, J.S.; Hong, J.; Andrée, B.A.; Gulyás, B.; Farde, L.; Innis, R.B.; et al. Investigation of the metabolites of [11C](S,S)-MeNER in humans, monkeys and rats. Mol. Imaging Biol. 2009, 11, 23–30. [Google Scholar]
- Zhang, P.; Terefenko, E.A.; McComas, C.C.; Mahaney, P.E.; Vu, A.; Trybulski, E.; Koury, E.; Johnston, G.; Bray, J.; Deecher, D. Synthesis and activity of novel 1- or 3-(3-amino-1-phenyl propyl)-1,3-dihydro-2H-benzimidazol-2-ones as selective norepinephrine reuptake inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 6067–6070. [Google Scholar]
- Mark, C.; Bornatowicz, B.; Mitterhauser, M.; Hendl, M.; Nics, L.; Haeusler, D.; Lanzenberger, R.; Berger, M.L.; Spreitzer, H.; Wadsak, W. Development and automation of a novel NET-PET tracer: [C-11]Me@APPI. Nucl. Med. Biol. 2013, 40, 295–303. [Google Scholar]
- Varney, M.D.; Romines, W.H.; Boritzki, T.; Margosiak, S.A.; Barlett, C.; Howland, E.J. Synthesis and biological evaluation of -n[4-(2-trans-[([2,6-diamino-4(3H)-oxopyrimidin-5-yl]methyl)thio]cyclobutyl)benzoyl] -l-glutamic acid a novel 5-thiapyrimidinone inhibitor of dihydrofolate reductase. J. Heterocycl. Chem. 1995, 32, 1493–1498. [Google Scholar]
- Xu, Z.B.; Lu, Y.; Guo, Z.R. An Efficient and Fast Procedure for the Preparation of 2-Nitrophenylamines under Microwave Conditions. Synlett 2003, 4, 564–566. [Google Scholar]
- Wang, X.J.; Xi, M.Y.; Fu, J.H.; Zhang, F.R.; Cheng, G.F.; Yin, D.L.; You, Q.D. Synthesis, biological evaluation and SAR studies of benzimidazole derivatives as H1-antihistamine agents. Chin. Chem. Lett. 2012, 23, 707–710. [Google Scholar]
- Jona, H.; Shibata, J.; Asai, M.; Goto, Y.; Arai, S.; Nakajima, S.; Okamoto, O.; Kawamoto, H.; Iwasawa, Y. Efficient and practical asymmetric synthesis of 1-tert-butyl 3-methyl (3R,4R)-4-(2-oxo-2,3-dihydro-1H-benzimidazol-1-yl)piperidine-1,3-dicarboxylate, a useful intermediate for the synthesis of nociceptin antagonists. Tetrahedron: Asymmetry 2009, 20, 2439–2446. [Google Scholar]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 2013, 503, 85–90. [Google Scholar]
- Wang, H.; Goehring, A.; Wang, K.H.; Penmatsa, A.; Ressler, R.; Gouaux, E. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature 2013, 503, 141–145. [Google Scholar]
- Tatsumi, M.; Groshan, K.; Blakely, R.D.; Richelson, E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur. J. Pharmacol. 1997, 340, 249–258. [Google Scholar]
- Richter, L.; de Graaf, C.; Sieghart, W.; Varagic, Z.; Morzinger, M.; de Esch, I.J.; Ecker, G.F.; Ernst, M. Diazepam-bound GABAA receptor models identify new benzodiazepine binding-site ligands. Nat. Chem. Biol. 2012, 8, 455–464. [Google Scholar]
- Andersen, J.; Olsen, L.; Hansen, K.B.; Taboureau, O.; Jorgensen, F.S.; Jorgensen, A.M.; Bang-Andersen, B.; Egebjerg, J.; Stromgaard, K.; Kristensen, A.S. Mutational Mapping and Modeling of the Binding Site for (S)-Citalopram in the Human Serotonin Transporter. J. Biol. Chem. 2010, 285, 2051–2063. [Google Scholar]
- Chelli, R.; Gervasio, F.L.; Procacci, P.; Schettino, V. Stacking and T-shape Competition in Aromatic–Aromatic Amino Acid Interactions. J. Am. Chem. Soc. 2002, 124, 6133–6143. [Google Scholar]
- Yu, F.; Zhou, J.N.; Zhang, X.C.; Sui, Y.Z.; Wu, F.F.; Xie, L.J.; Chan, A.S.C.; Wu, J. Copper(II)-Catalyzed Hydrosilylation of Ketones Using Chiral Dipyridylphosphane Ligands: Highly Enantioselective Synthesis of Valuable Alcohols. Chemistry 2011, 17, 14234–14240. [Google Scholar]
- La Regina, G.; Diodata D’Auria, F.; Tafi, A.; Piscitelli, F.; Olla, S.; Caporuscio, F.; Nencioni, L.; Cirilli, R.; La Torre, F.; Rodrigues De Melo, N.; et al. 1-[(3-Aryloxy-3-aryl)propyl]-1H-imidazoles, new imidazoles with potent activity against Candida albicans and dermatophytes. Synthesis, structure-activity relationship, and molecular modeling studies. J. Med. Chem. 2008, 51, 3841–3855. [Google Scholar]
- Panagopoulos, A.M.; Steinman, D.; Goncharenko, A.; Geary, K.; Schleisman, C.; Spaargaren, E.; Zeller, M.; Becker, D.P. Apparent Alkyl Transfer and Phenazine Formation via an Aryne Intermediate. J. Org. Chem. 2013, 78, 3532–3540. [Google Scholar]
- Liu, P.; Wang, Z.; Hu, X. Highly Efficient Synthesis of Ureas and Carbamates from Amides by Iodosylbenzene-Induced Hofmann Rearrangement. Eur. J. Org. Chem. 2012, 10, 1994–2000. [Google Scholar]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2013.
- Sali, A.; Potterton, L.; Yuan, F.; van Vlijmen, H.; Karplus, M. Evaluation of comparative protein modeling by MODELLER. Proteins 1995, 23, 318–326. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar]
- XLStat; Addinsoft Inc.: New York, NY, USA, 2009.
- Sample Availability: Samples of the compounds 4–6, 9–25, and 29 and 30 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neudorfer, C.; Seddik, A.; Shanab, K.; Jurik, A.; Rami-Mark, C.; Holzer, W.; Ecker, G.; Mitterhauser, M.; Wadsak, W.; Spreitzer, H. Synthesis and in Silico Evaluation of Novel Compounds for PET-Based Investigations of the Norepinephrine Transporter. Molecules 2015, 20, 1712-1730. https://doi.org/10.3390/molecules20011712
Neudorfer C, Seddik A, Shanab K, Jurik A, Rami-Mark C, Holzer W, Ecker G, Mitterhauser M, Wadsak W, Spreitzer H. Synthesis and in Silico Evaluation of Novel Compounds for PET-Based Investigations of the Norepinephrine Transporter. Molecules. 2015; 20(1):1712-1730. https://doi.org/10.3390/molecules20011712
Chicago/Turabian StyleNeudorfer, Catharina, Amir Seddik, Karem Shanab, Andreas Jurik, Christina Rami-Mark, Wolfgang Holzer, Gerhard Ecker, Markus Mitterhauser, Wolfgang Wadsak, and Helmut Spreitzer. 2015. "Synthesis and in Silico Evaluation of Novel Compounds for PET-Based Investigations of the Norepinephrine Transporter" Molecules 20, no. 1: 1712-1730. https://doi.org/10.3390/molecules20011712
APA StyleNeudorfer, C., Seddik, A., Shanab, K., Jurik, A., Rami-Mark, C., Holzer, W., Ecker, G., Mitterhauser, M., Wadsak, W., & Spreitzer, H. (2015). Synthesis and in Silico Evaluation of Novel Compounds for PET-Based Investigations of the Norepinephrine Transporter. Molecules, 20(1), 1712-1730. https://doi.org/10.3390/molecules20011712