2.1. Structural Characterizations
Compound
1 crystallizes in the monoclinic space group
C2/c. The atomic numbering and displacement ellipsoid plot are presented in
Figure 2. Crystal data and structural refinement of compound
1 are presented in
Table 1. Intramolecular H-bonds C(10)–H(10A)···O(3) and N(4)–H(4)···O(2) result in the formation of non-planar and planar six-membered pseudo rings, respectively. The dihedral angle formed between aromatic plane and indole ring is 44.20(6)°. The piperazine moiety displays a typical chair conformation, as the torsion angles of N(2)–C(18)–C(10)–N(1) and N(2)–C(16)–C(21)–N(1) are is −58.6(2)° and 58.7(2)°, respectively.
Figure 2.
Crystallographic structure of compound 1. Displacement ellipsoids are drawn at the 30% probability level, and intramolecular hydrogen bonds are presented in dashed lines.
Figure 2.
Crystallographic structure of compound 1. Displacement ellipsoids are drawn at the 30% probability level, and intramolecular hydrogen bonds are presented in dashed lines.
Table 1.
Crystal data and structural refinement of compound 1, 2 and 3.
Table 1.
Crystal data and structural refinement of compound 1, 2 and 3.
| Empirical Formula | C23H27N5O4 | C24H30N4O3 | C23H28N4O2 |
|---|
| Formula weight | 437.49 | 422.52 | 392.49 |
| Temperature (K) | 293(2) | 293(2) | 293(2) |
| Crystal system | monoclinic | Triclinic | orthorhombic |
| Space group | C2/c | P-1 | Pccn |
| a, b, c (Å) | 21.385(4), 7.8960(16), 28.227(6) | 9.998(2), 10.692(2), 13.614(3) | 15.762(3), 35.112(7), 7.8452(16) |
| α, β, γ (°) | 90, 93.13(3), 90 | 100.89(3), 105.05(3), 98.87(3) | 90, 90, 90 |
| Volume (Å3), Z | 4759.2(17), 8 | 1348.0(5), 2 | 4341.9(15), 8 |
| ρcalc (g·cm−3) | 1.221 | 1.041 | 1.201 |
| μ/mm−1 | 0.7 | 0.561 | 0.624 |
| F(000) | 1856 | 452 | 1680 |
| Crystal size (mm3) | 0.3 × 0.2 × 0.2 | 0.3 × 0.2 × 0.2 | 0.3 × 0.25 × 0.23 |
| θ range for data collection (°) | 3.136–68.242 | 3.463–68.213 | 3.073–62.499 |
| h, k, l | −25–25, −9–9, −33–33 | −12–11, −12–12, −16–16 | −18–18, −42–42, −8–9 |
| Reflections collected | 37,917 | 24,318 | 74,888 |
| Independent reflections, Rint | 4353, 0.0731 | 4851, 0.0574 | 3455, 0.0485 |
| Data/restraints/parameters | 4353/0/290 | 4851/0/282 | 3455/85/294 |
| Goodness-of-fit on F2 | 1.084 | 0.996 | 1.002 |
| R1, wR2 [I ≥ σ (I)] | 0.0550, 0.1426 | 0.0620, 0.1773 | 0.0592, 0.1474 |
| R1, wR2 [all data] | 0.0717, 0.1532 | 0.0806, 0.1935 | 0.0764, 0.1684 |
| Largest diff. peak/hole (e·Å−3) | 0.30/−0.21 | 0.38/−0.20 | 0.24/−0.18 |
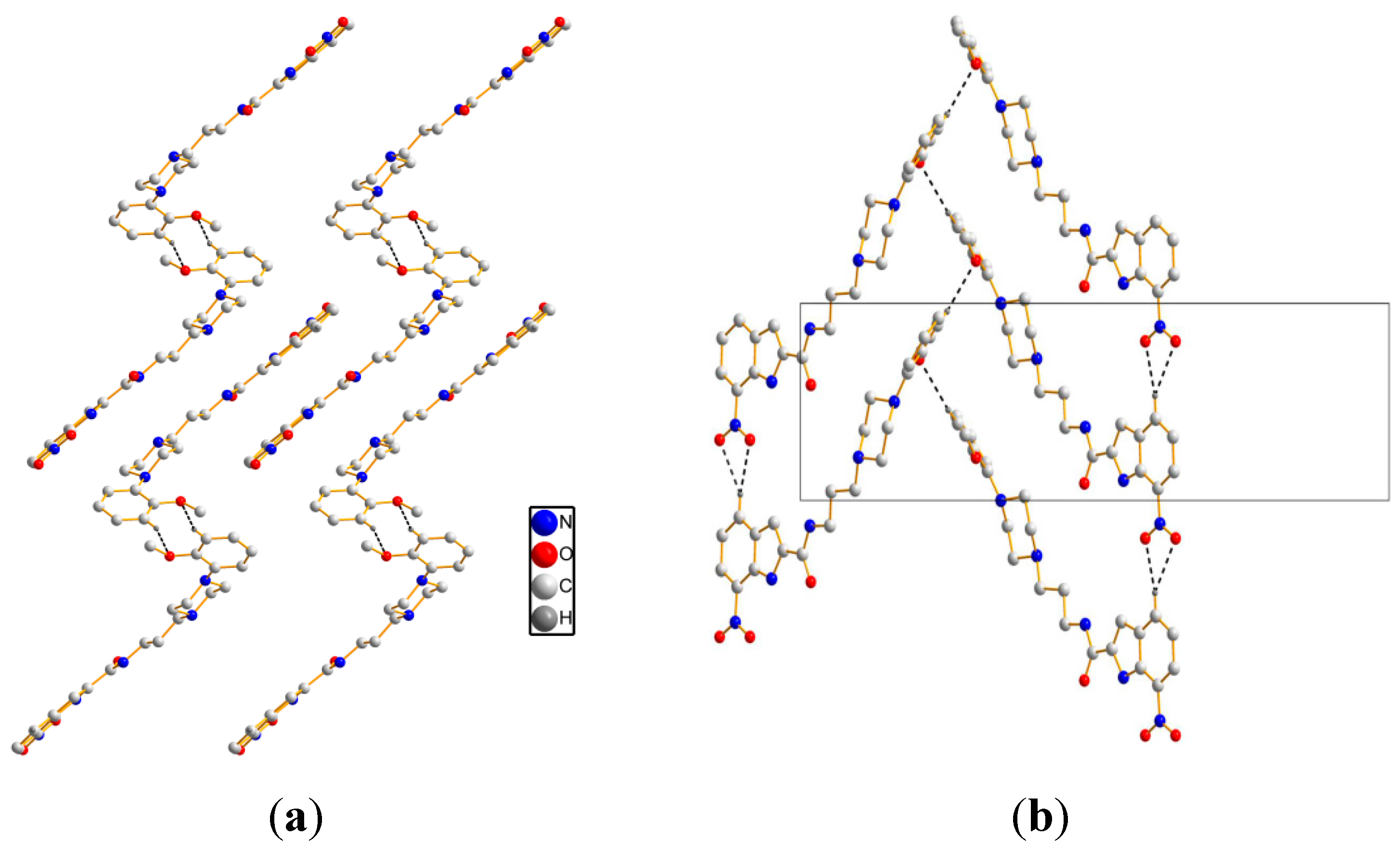
As for crystal packing, two independent molecules form
(8) ring motif along the
b axis, see
Figure 3a. Simultaneously, intermolecular hydrogen bonds [C(24)–H(24)···O(2) and C(24)–H(24)···O(4)] are observed to realize a
(4) ring motif along the
a axis. The framework is further reinforced by zigzag H-bondings [C(27)–H(27)···O(3)], see
Figure 3b. The detailed intermolecular H-bonds data are presented in
Table 2.
Figure 3.
Crystal packing of 1. (a) viewing along the b axis; (b) viewing along the a axis. Black dashed lines show the intermolecular hydrogen bonds.
Figure 3.
Crystal packing of 1. (a) viewing along the b axis; (b) viewing along the a axis. Black dashed lines show the intermolecular hydrogen bonds.
Table 2.
Intermolecular hydrogen bonds for compound 1 (Å, °).
Table 2.
Intermolecular hydrogen bonds for compound 1 (Å, °).
| D–H···A | D–H | H···A | D···A | D–H···A (°) |
|---|
| C(24)–H(24)···O(2) i | 0.93 | 2.49 | 3.318(3) | 147.9(3) |
| C(24)–H(24)···O(4) i | 0.93 | 2.44 | 3.294(3) | 152.4(2) |
| C(27)–H(27)···O(3) ii | 0.93 | 2.55 | 3.409(3) | 154.4(3) |
The crystal of compound
2 gives triclinic form with space group
P-1, see
Figure 4. The crystal data and structural refinement of compound
2 are presented in
Table 1. The dihedral angle of aromatic plane and indole ring is 11.2(9)°, which is obviously smaller in comparison to that of compound
1. In order to explain the discrepancy of two specific conformations, we performed conformational analysis, see
Table 3. As a comparison to the bond lengths, bond angles and torsion angles between
1 and
2, significant differences are observed to be at the amide alkyl linkage, e.g., the torsion angle [N(2)–C(20)–C(8)–C(19)] of
1 is −179.0(2)° while corresponding angle [N(2)–C(12)–C(13)–C(14)] of
2 is −52.6(3)°. This case is most likely ascribed to the differences of intramolecular and intermolecular hydrogen bonds that promote the formation of specific conformers.
Figure 4.
Crystallographic structure of compound 2. Displacement ellipsoids are drawn at the 30% probability level, and intramolecular hydrogen bond is shown in dashed line.
Figure 4.
Crystallographic structure of compound 2. Displacement ellipsoids are drawn at the 30% probability level, and intramolecular hydrogen bond is shown in dashed line.
Table 3.
Selected geometric parameters of compound 1 and 2 (Å, °).
Table 3.
Selected geometric parameters of compound 1 and 2 (Å, °).
| Compound 1 | Compound 2 |
|---|
| Bonds | Dist. | Bonds | Dist. |
| N(1)–C(17) | 1.419(2) | N(1)–C(3) | 1.413(3) |
| N(1)–C(10) | 1.464(2) | N(1)–C(8) | 1.474(3) |
| N(3)–C(14) | 1.337(2) | N(3)–C(15) | 1.331(3) |
| N(4)–C(11) | 1.393(2) | N(4)–C(16) | 1.370(3) |
| C(8)–C(20) | 1.512(3) | C(12)–C(13) | 1.504 (4) |
| Angle | (°) | Angle | (°) |
| C(20)–C(8)–C(19) | 110.64(17) | C(12)–C(15)–C(14) | 115.9(2) |
| C(19)–N(3)–C(14) | 122.09(18) | C(15)–N(3)–C(14) | 121.7(2) |
| N(2)–C(20)–C(8)–C(19) | −179.0(2) | N(2)–C(12)–C(13)–C(14) | −52.6(3) |
| C(20)–C(8)–C(19)–N(3) | 171.5(2) | C(12)–C(13)–C(14)–N(3) | −56.2(3) |
| C(8)–C(19)–N(3)–C(14) | 171.2(2) | C(13)–C(14)–N(3)–C(15) | 94.9(3) |
Another particular interest for us in studying the crystal structure of
2 is to investigate the stability force comprising intermolecular interactions. As shown in
Figure 5, C–H···π interactions are found to be the critical forces to stabilize the three-dimensional structure [
21]. The geometry parameters of C–H···π interactions are listed in
Table 4. Two molecules form a dimer of head-tail crosslinking through C(24)–H(24B)···π interactions. Additionally, a dimer constructed by two antiparallel molecules plays a significant role in stabilizing the packing structure.
Figure 5.
Crystal packing of 2. Red dashed lines show the intermolecular C–H···π interactions.
Figure 5.
Crystal packing of 2. Red dashed lines show the intermolecular C–H···π interactions.
Table 4.
C–H···π interaction geometry of compound 2 (Å, °).
Table 4.
C–H···π interaction geometry of compound 2 (Å, °).
| ···X–H | Cg * | H···Cg | X···Cg | X–H···Cg (°) |
|---|
| C(10)–H(10A) i | N(4)–C(16)–C(17)–C(18)–C(19) | 2.70 | 3.437(3) | 133 |
| C(10)–H(10A) i | C(18)–C(19)–C(23)–C(22)–C(21)–C(20) | 2.78 | 3.753(3) | 178 |
| C(24)–H(24B) ii | C(2)–C(3)–C(4)–C(5)–C(6)–C(7) | 3.00 | 3.917(4) | 160 |
Crystal structure of compound
3 was herein described clearly. It gives orthorhombic with space group
Pccn. As shown in
Figure 6, the piperazine ring, C(12) and C(13) atoms were disordered. The dihedral angle of aromatic plane and indole ring is 57.6(8)°. With respect to the packing structure, the intermolecular hydrogen bonds (
Table 5) form network structure. The NH [N(3)–H(3), N(4)–H(4)] of a donor molecule realizes H-bond with the O atoms of an adjacent molecule, see
Figure 7. Furthermore, compared to hydrogen bonds (3.294–3.409 Å) in compound
1, the H-bonds (2.953–3.151 Å) in compound
3 have shorter bond lengths, indicating that higher bond energies contribute to the stability of the three-dimensional structure of
3.
Figure 6.
Crystallographic structure of compound 3. Displacement ellipsoids are drawn at the 30% probability level.
Figure 6.
Crystallographic structure of compound 3. Displacement ellipsoids are drawn at the 30% probability level.
Figure 7.
Crystal packing of 3 along the c axis. Black dashed lines show the intermolecular H-bonds.
Figure 7.
Crystal packing of 3 along the c axis. Black dashed lines show the intermolecular H-bonds.
Table 5.
Intermolecular hydrogen bonds for compound 3 (Å, °).
Table 5.
Intermolecular hydrogen bonds for compound 3 (Å, °).
| D–H···A | D–H | H···A | D···A | D–H···A (°) |
|---|
| N(3)–H(3)···O(2) i | 0.86 | 2.14 | 2.953 (4) | 157.5 (2) |
| N(4)–H(4)···O(1) ii | 0.86 | 2.37 | 3.151 (1) | 151.7 (2) |
2.2. DFT Calculations
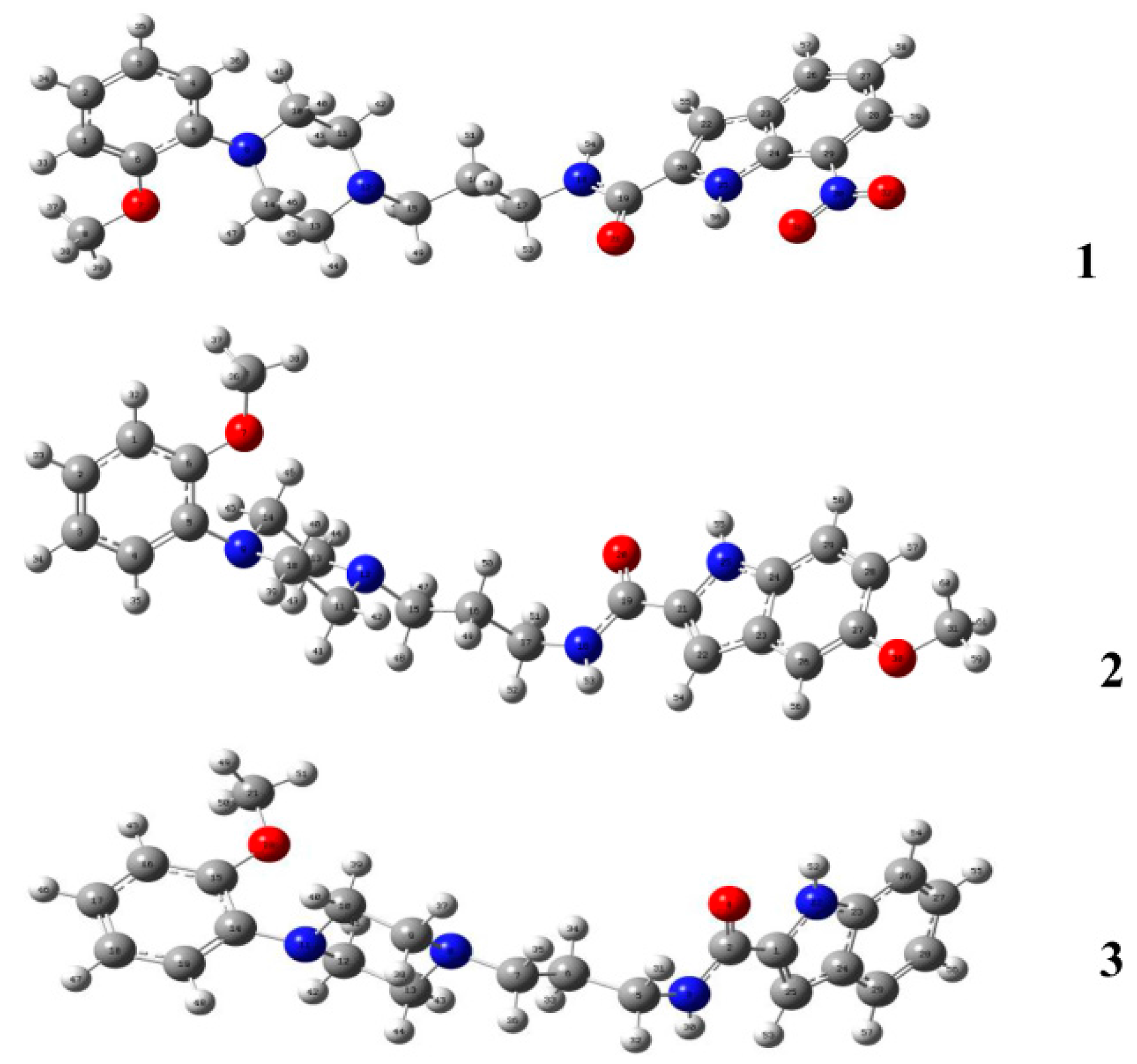
Compounds
1–
3 were subject to theoretical calculations using Gaussian 09 at the B3LYP/6-311G (d, p) level of theory to elucidate their structural properties. The optimized geometries are shown in
Figure 8. The comparison of the calculated bond lengths and bond angles among compounds
1–
3 and the corresponding solid-state values are listed in
Table 6. The bond-length deviations between the calculated and solid-state conformers are 0.001 to 0.021 Å, and the deviations of bond angles range from 0.1 to 1.6°. The results suggest that there is no apparent difference between the theoretically calculated and experimental crystallographic conformations.
Figure 8.
DFT-optimized conformations of compound 1, 2 and 3 at the B3LYP/6-311G (d, p) level of theory.
Figure 8.
DFT-optimized conformations of compound 1, 2 and 3 at the B3LYP/6-311G (d, p) level of theory.
Table 6.
Optimized geometrical parameters of theoretically calculated and solid-state conformers.
Table 6.
Optimized geometrical parameters of theoretically calculated and solid-state conformers.
| Compound 1 |
|---|
| Bond lengths (Å) | DFT | X-ray |
| O(7)–C(6) | 1.362 | 1.373(3) |
| N(25)–C(24) | 1.354 | 1.362(2) |
| C(15)–C(16) | 1.525 | 1.511(3) |
| C(27)–C(28) | 1.396 | 1.385(3) |
| Bond angles (°) | | |
| C(20)–C(19)–N(18) | 116.5 | 115.8(2) |
| C(15)–C(16)–C(17) | 111.5 | 110.8(2) |
| N(12)–C(15)–C(16) | 113.3 | 114.9(2) |
| Compound 2 |
| Bond lengths (Å) | DFT | X-ray |
| O(7)–C(6) | 1.36 | 1.380(3) |
| N(25)–C(24) | 1.371 | 1.370(3) |
| C(15)–C(16) | 1.527 | 1.515(4) |
| C(27)–C(28) | 1.409 | 1.394(4) |
| Bond angles (°) | | |
| O(21)–C(19)–C(18) | 123.4 | 122.6(2) |
| C(15)–C(16)–C(17) | 111.5 | 115.9(2) |
| N(12)–C(15)–C(16) | 113.5 | 114.5(2) |
| Compound 3 |
| Bond lengths (Å) | DFT | X-ray |
| O(4)–C(2) | 1.221 | 1.242(3) |
| N(22)–C(23) | 1.366 | 1.371(3) |
| C(5)–C(7) | 1.524 | 1.506(9) |
| C(14)–C(19) | 1.385 | 1.376(4) |
| Bond angles (°) | | |
| O(4)–C(2)–N(3) | 123.4 | 122.3(2) |
| O(20)–C(15)–C(14) | 116.0 | 115.0(2) |
| N(22)–C(23)–C(24) | 107.4 | 107.5(2) |
Molecular orbitals are very useful for physicists and chemists because the energy gap between HOMO and LUMO characterizes the chemical reactivity and kinetic stability of molecule [
22]. As shown in
Figure 9, the HOMO of
1 is localized on the arylpiperazine moiety, whereas the LUMO is mainly concentrated on the 7-nitro-1
H-indole moiety. The HOMO and LUMO orbitals of
2 and
3 are localizes over the amide and indole moieties. The HOMO-LUMO energy gaps are 2.728, 4.324 and 4.610 eV for compound
1,
2 and
3, respectively (
Table 7).
Figure 9.
HOMO, LUMO surfaces of compound 1, 2 and 3 simulated at the B3LYP/6-311G (d, p) level of theory.
Figure 9.
HOMO, LUMO surfaces of compound 1, 2 and 3 simulated at the B3LYP/6-311G (d, p) level of theory.
Table 7.
Frontier molecular orbital energies of 1, 2 and 3 (eV).
Table 7.
Frontier molecular orbital energies of 1, 2 and 3 (eV).
| Compound | EHOMO | ELUMO | ∆ELUMO–HOMO |
|---|
| 1 | −5.395 | −2.667 | 2.728 |
| 2 | −5.508 | −1.184 | 4.324 |
| 3 | −5.807 | −1.197 | 4.610 |
Chemical hardness is a useful concept for understanding the behavior of chemical systems and is associated with the stability and reactivity of a chemical system. The chemical hardness (ƞ) can be expressed as: ƞ = (–
EHOMO +
ELUMO)/2. Using HOMO and LUMO orbital energies, we determined that the values of ƞ
1, ƞ
2 and ƞ
3 are 1.364, 2.162 and 2.305 eV, respectively. The results suggest that
1 is more reactive and less stable than
2 and
3. Electronic chemical potential (µ) is defined as the escaping tendency of electrons from an equilibrium system and is given by µ= (
EHOMO +
ELUMO)/2. The µ values increase in the order of
1 (–4.031 eV) <
3 (–3.502 eV) <
2 (–3.346 eV). Generally, hard molecules will have their electron density changed more hardly than a soft molecule [
23]. In this case, the chemical hardness of
3 is larger than that of
2, but the electronic chemical potential of the former is slightly smaller than the latter. It may be ascribed to the intermolecular hydrogen bonds of
3 that facilitate the electronic transfers. The global electrophilicity power of a ligand, expressed as ω = µ
2/2ƞ, is a measure of the stabilization in energy achieved when the system acquires an additional electronic charge from the environment. The corresponding ω values of
1,
2 and
3 are 5.956, 2.589 and 2.660, respectively.
Figure 10.
MEP surfaces of compound 1, 2 and 3.
Figure 10.
MEP surfaces of compound 1, 2 and 3.
The molecular electrostatic potential (MEP) maps were evaluated using the B3LYP method with the basis set of 6-311G (d, p) to investigate the reactive sites of electrophilic and nucleophilic attacks for the title compounds. The positive (red) regions of MEP were related to nucleophilic reactivity, and the negative (blue) regions referred to electrophilic reactivity. Determining electrostatic potential is a suitable process for analysis based on “recognition” of one molecule by another, similar to that in drug receptor and enzyme substrate interactions, because the two species detect each other through their potentials [
24,
25]. As shown by the MEP map of
1 (
Figure 10), the negative regions are mainly localized over the anisole ring. The maximum positive region is over the NH portion of the amide group (+50.01 kcal/mol). Similar to that of compound
1, the maximum positive region of
2 is localized to its own NH group (+42.69 kcal/mol), and the maximum negative site is concentrated on the O atom of the amide group (–39.34 kcal/mol). The N atom in the piperazine ring (–32.65 kcal/mol) also displays significant electrophilic reactivity. Nevertheless, the NH of indole ring exhibits a strong nucleophilic reactivity (+32.83 kcal/mol). These findings suggest that the above-mentioned sites are involved in the hydrogen-bonding interactions, which is validated by molecular model of the
2-α
1A complex (
Figure 11b). The maximum positive region for
3 is also localized to the NH group (+43.75 kcal/mol).
Figure 11.
The docking complexes of ligands 1 (a); 2 (b); and 3 (c) with α1A-AR. These compounds are shown in stick representation. The elements are coloured as follows: oxygen, red; nitrogen, blue; carbon, yellow. Dashed lines represent the hydrogen bonds or electrostatic interactions.
Figure 11.
The docking complexes of ligands 1 (a); 2 (b); and 3 (c) with α1A-AR. These compounds are shown in stick representation. The elements are coloured as follows: oxygen, red; nitrogen, blue; carbon, yellow. Dashed lines represent the hydrogen bonds or electrostatic interactions.
2.3. Binding Mechanisms of the Ligand-α1A Complex
G protein-coupled receptors (GPCRs) are the largest class of membrane receptors in eukaryotes, and are characterized by seven transmembrane (TM) helices, and N- and C-terminal fragments. The TM helices are connected by alternating intra- and extracellular loop (ECL) regions that are very flexible and important for a wide range of biological functions. When agonists or antagonists bind to GPCRs, the hosts act as molecular switches that modulate downstream effector proteins when turned on [
26]. α
1A-Adrenoceptor is a member of the GPCR family. The accurate crystal structure of α
1A at the atomic resolution cannot be obtained, thereby severely impeding the development of therapeutic BPH medicines through structure-based drug design. Nevertheless, homology-modeling procedures with continuous progresses provide viable tools for structure-based GPCR ligand design.
Functional experiments involving animal tissues validated that compound
1,
2 and
3 exhibit 11.0-, 18.6- and 4.7-fold increases in selectivity, respectively, for α
1A over than the α
1B subtype [
13]. The minimum-energy structures of the ligands were subject to molecular docking using a homology model of the α
1A receptor to determine the structure-activity-binding mechanism relationship. As shown in
Figure 10a, NH in the indole ring as a donor forms H-bond (2.1 Å) with Cys176 residue. The NH of amino group realizes a hydrogen bond (2.8 Å) with the carbonyl oxygen atom of Gln177 in the ECL2 region that has been proved to be essential for GPCR activation [
27]. The indole and benzene rings mainly engage in hydrophobic interactions with Trp102, Trp92, Phe86, Phe308, Phe288 and Phe312. The residues including Asp106, Glu87 and Leu29 participate in
1 binding to the α
1A receptor with van der Waals interactions. In the
2-α
1A-adrenoceptor complex, the NH in indole ring and the carbonyl oxygen atom are involved in two hydrogen bonds with the nitrogen and oxygen atoms of Gln177 (2.2 and 2.5 Å). In addition,
2 contacts via hydrophobic interactions with residues Phe86, Tyr316 and Phe288. The indole ring forms aromatic π-stacking against Phe308 of TM7 (Figure 10b). The cationic nitrogen atom in the piperazine ring for the electrostatic binding to Glu87 of TM2 is also presented. As observed from
3-α
1A complex (Figure 10c), the NH of amino group can realize a H-bond (2.4 Å) with Glu87 of ECL1. The indole and benzene rings for hydrophobic interactions with the region formed by Tyr316, Trp313, Phe308 of TM7 and Phe288 of TM6 are found. The arylpiperazine moiety is in weak van der waals contact with residues Ile175 and Ile178 of ECL2. The protonated piperazine moiety forms a weak electrostatic interaction with Gln177 (4.1 Å between the nitrogen atom of piperazine ring and the amide oxygen atom of Gln177).
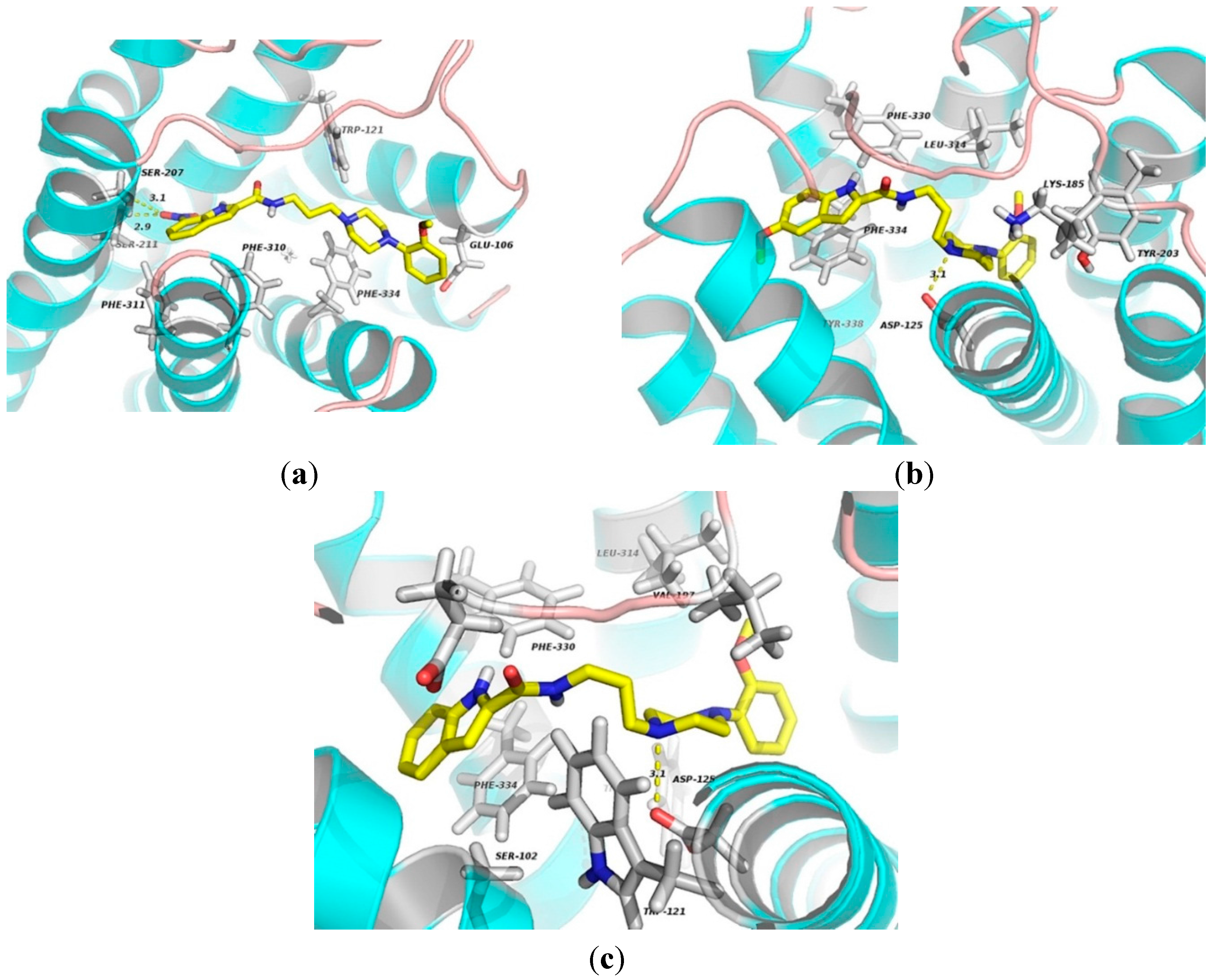
To shed some light on the selectivity for α
1A over than α
1B subtype, molecular dockings using a homology of α
1B receptor were performed. The results show that the oxygen atom of nitrate group of
1 forms two hydrogen bonds with residues Ser207 and Ser211 (3.1 and 2.9 Å) of TM5. The indole and benzene rings mainly engage in hydrophobic interactions with Phe310, Phe311, Phe334 and Trp121 residues (Figure 12a). As for
2-α
1B complex, the ligand contacts via hydrophobic interactions with Phe330, Phe334 and Tyr203. Other residues, including Leu134 and Lys185, involve in ligand binding by van der Waal’s forces. Additionally, the cationic nitrogen atom in the piperazine moiety for electrostatic binding to Asp125 (3.1 Å) is also observed as shown in
Figure 11b. One residue Phe334 in TM7 may be an important binding site for
3-α
1A complex by π-stacking interaction. Compound
3 contacts via van der Waals interactions with residues Ser102, Glu194, Val197 and Leu134. The indole moiety involves in hydrophobic interactions with Phe330 and Trp307. In addition, the electronegative atom oxygen in Asp125 of TM3 makes an electrostatic interaction with the nitrogen atom of the piperazine ring.
Studies on the binding mechanism indicate that residues Gln177, Phe86, Phe308, Phe288 and Glu87 are identified as the major sites for indole-arylpiperazine derivatives (1, 2 and 3) binding to α1A receptor. On the other hand, the important sites for ligands binding with α1B subtype are involved in Asp125, Leu134, Phe334, Phe330, Ser207 and Ser211 residues. We also noticed that the Gln177 residue in ECL2 seems to play a significant role in improving the selectivity of α1A against α1B subtype. The ligands binding to residue Asp125 by electrostatic interactions and contacting Phe334 via hydrophobic interactions or π-stacking interactions contribute to increased affinity for α1B receptor.
Figure 12.
The docking complexes of ligands 1 (a); 2 (b); and 3 (c) with α1B-AR. These compounds are shown in stick representation. The elements are coloured as follows: oxygen, red; nitrogen, blue; carbon, yellow. Dashed lines represent the hydrogen bonds or electrostatic interactions.
Figure 12.
The docking complexes of ligands 1 (a); 2 (b); and 3 (c) with α1B-AR. These compounds are shown in stick representation. The elements are coloured as follows: oxygen, red; nitrogen, blue; carbon, yellow. Dashed lines represent the hydrogen bonds or electrostatic interactions.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









