Figure 1 shows the impact of precursor feed on silica surface area as well as BET equivalent diameter (calculated by d
BET = 6/(SSA × ρ
silica) where SSA is the specific surface area of the prepared particles in cm
2/g and ρ
silica is the density of silica, taken as 2.2 g/cm
3 [
5]) for the 3, 5 and 7 mL/min fed FSP silica, hereafter referred to as 3, 5 and 7-SiO
2, respectively. Increasing precursor feed rate from 3 to 7 mL/min resulted in a decrease in surface area from ~340 to 240 m
2/g. A higher precursor feed rate means that more fuel is being fed to the flame resulting in a hotter flame (evidenced by the increasing flame height from ~6.5 to 9 cm). Additionally, a higher precursor feed rate means a higher concentration of silica within the flame, in turn promoting sintering (by increasing the likelihood of collisions). By increasing the fuel content and silica concentration within the flame the particle growth rate and sintering is encouraged thus resulting in larger particles [
5].
Figure 1.
Impact of precursor feed rate and fuel equivalence ratio (Φ) on specific surface area and Brunauer-Emmett-Teller (BET)-equivalent diameter of flame spray pyrolysis (FSP)-produced silica; as prepared, and after 5% Ni impregnation and calcination. Included are TEM micrographs of FSP (a) 3, (b) 5 and (c) 7-SiO2.
Figure 2.
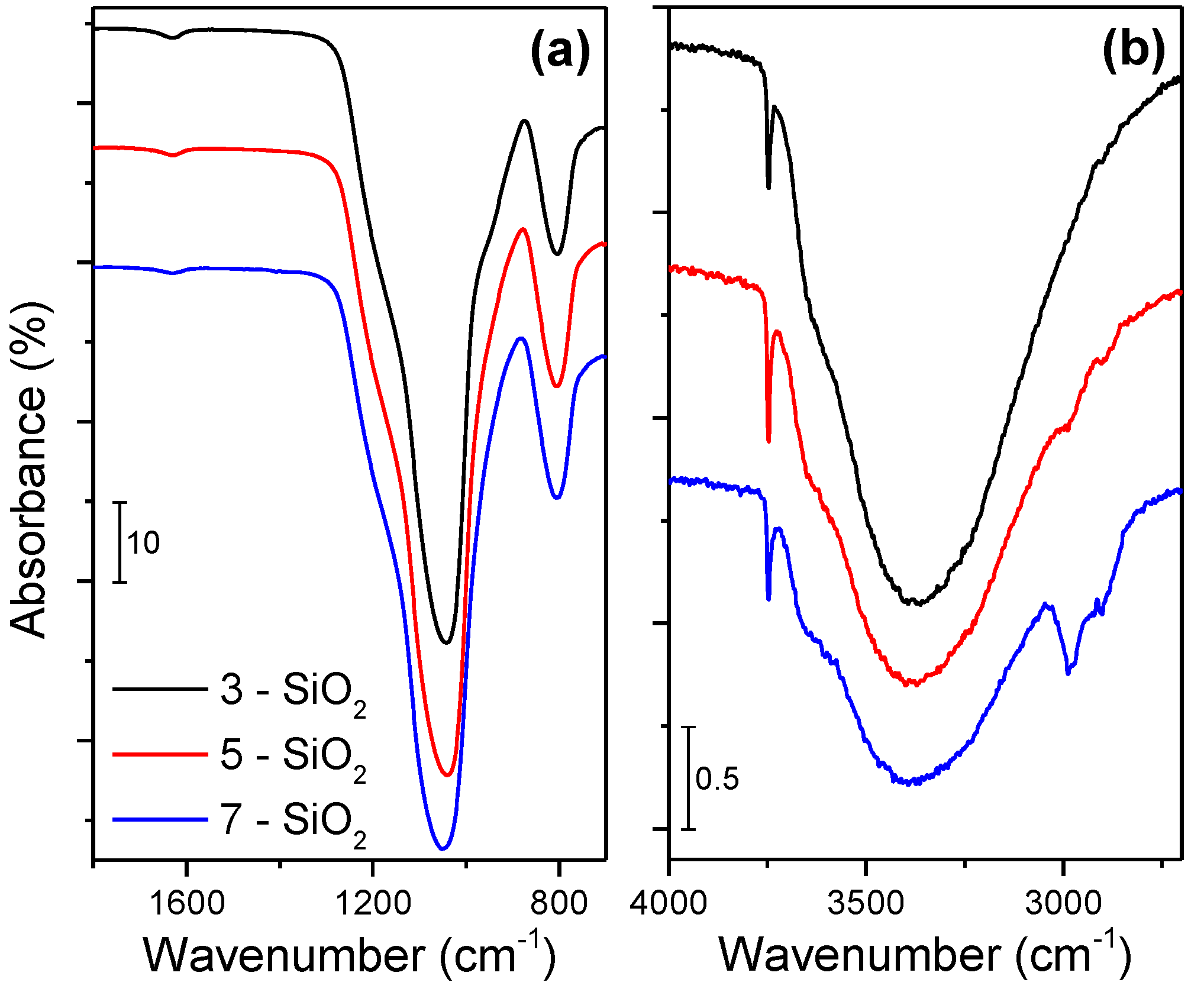
FTIR spectra of as prepared 3, 5 and 7-SiO2 showing the (a) 1800–700 cm−1 and (b) 4000–2700 cm−1 wavenumber regions.
The absorption bands do not show any significant differences in the nature of the silica when prepared at different feed rates. In contrast, the FTIR spectra in
Figure 2b display some variances in the surface species present on the silica. The broad peak near 3387 cm
−1 corresponds to hydrogen bonded –OH groups (which can be attributed to H–bonded H
2O, H–bonded –OH vibrations of residual alcohol and H–bonded Si–OH [
23]). The sharp band evident at 3749 cm
−1 corresponds to isolated silanol groups [
23]. While the resolution makes it difficult to quantify the ratio of H–bonded to isolated –OH groups on the silica surface there is evidence of some variations. The percentage of isolated –OH groups to H–bonded –OH groups (normalised to the maximum Si–O–Si peak) increases with increasing precursor feed rate from 0.9%, 1.8% to 2.1% with 3, 5 and 7 mL/min, respectively.
The fuel equivalence ratio (Φ), which is a numerical representation of the oxygen supply to the flame [
24], is displayed in
Figure 1 (upper y-axis). A Φ value > 1 indicates that the flame is oxygen deficient. Irrespective of the precursor feed rate, a consistent oxygen flow is supplied to flame, and thus a higher precursor feed rate results in a more oxygen deficient flame. As expected, the 7 mL/min precursor flow rate provides the most oxygen deficient flame with an equivalence ratio of approximately 1.7. The impact of the elevated Φ is illustrated in
Figure 2 with the presence of distinct C–H stretches around 2780–2880 cm
−1 for 7-SiO
2 [
18,
20,
25]. The C–H bonding presence implies incomplete precursor combustion which results in residual C–H groups (2980 cm
−1 is CH
3, 2930 cm
−1 is CH
2 [
20]) residing on the silica surface. There is some evidence of a minor C–H bonding presence on 5-SiO
2 (Φ = 1.3) while the 3-SiO
2 (Φ = 0.95) does not possess any evidence of C–H surface species. It should be noted that FTIR was also performed on all the samples after impregnation and calcination. No C–H adsorption bands were observed in this instance, indicating that calcination is sufficient for oxidising the residual carbon species.
2.1. Nickel Properties
Following impregnation with 5% Ni, and calcination TEM, XRD and H
2-TPR were used to characterise the nature of the NiO deposits. After impregnation and calcination the surface area of the samples decreased by approximately 7%–30% (
Figure 1). As FSP synthesised materials are non-porous the decrease in surface area can be attributed to sintering due to heat treatment of the particles.
Figure 3 displays the TEM micrographs and corresponding NiO deposit size distributions on the varying silica supports with a minimum of 300 measured deposits. The average measured deposit sizes are shown in
Table 1. From the results it is evident that 3-SiO
2 has the smallest NiO deposit size. The 3-SiO
2 fed FSP silica has the smallest SiO
2 particle size and therefore has the largest area for the Ni to disperse across during impregnation, thus resulting in the smallest deposits. Our previous findings have shown the impacts of higher surface area supports during Ni impregnation, whereby a greater surface area support facilitated a higher Ni dispersion [
6]. Interestingly, the increasing average NiO deposit size does not follow the decrease in SiO
2 surface area for the 5 and 7-SiO
2. 7-SiO
2 has a slightly smaller average NiO deposit size than the 5-SiO
2 with the narrowest particle size distribution of all samples. 5-SiO
2 shows the broadest NiO size distribution along with the highest average deposit size.
Figure 3.
TEM images and corresponding NiO size distribution of (a,b) 3 (c,d) 5 and (e,f) 7-SiO2 after impregnation with 5% Ni and calcination.
Figure 3.
TEM images and corresponding NiO size distribution of (a,b) 3 (c,d) 5 and (e,f) 7-SiO2 after impregnation with 5% Ni and calcination.
Table 1.
Summary of Ni properties of Ni-SiO2 with silica being prepared by FSP at varying feed rates.
Table 1.
Summary of Ni properties of Ni-SiO2 with silica being prepared by FSP at varying feed rates.
| Silica Feed Rate (mL/min) | Actual Ni Loading (%) a | NiO Size (nm) |
|---|
| TEM (σ) b | XRD c |
|---|
| 3 | 4.4 | 21.7 (13.9) | 13.3 |
| 5 | 4.3 | 25.9 (16.1) | 13.8 |
| 7 | 4.4 | 24.9 (12.3) | 13.2 |
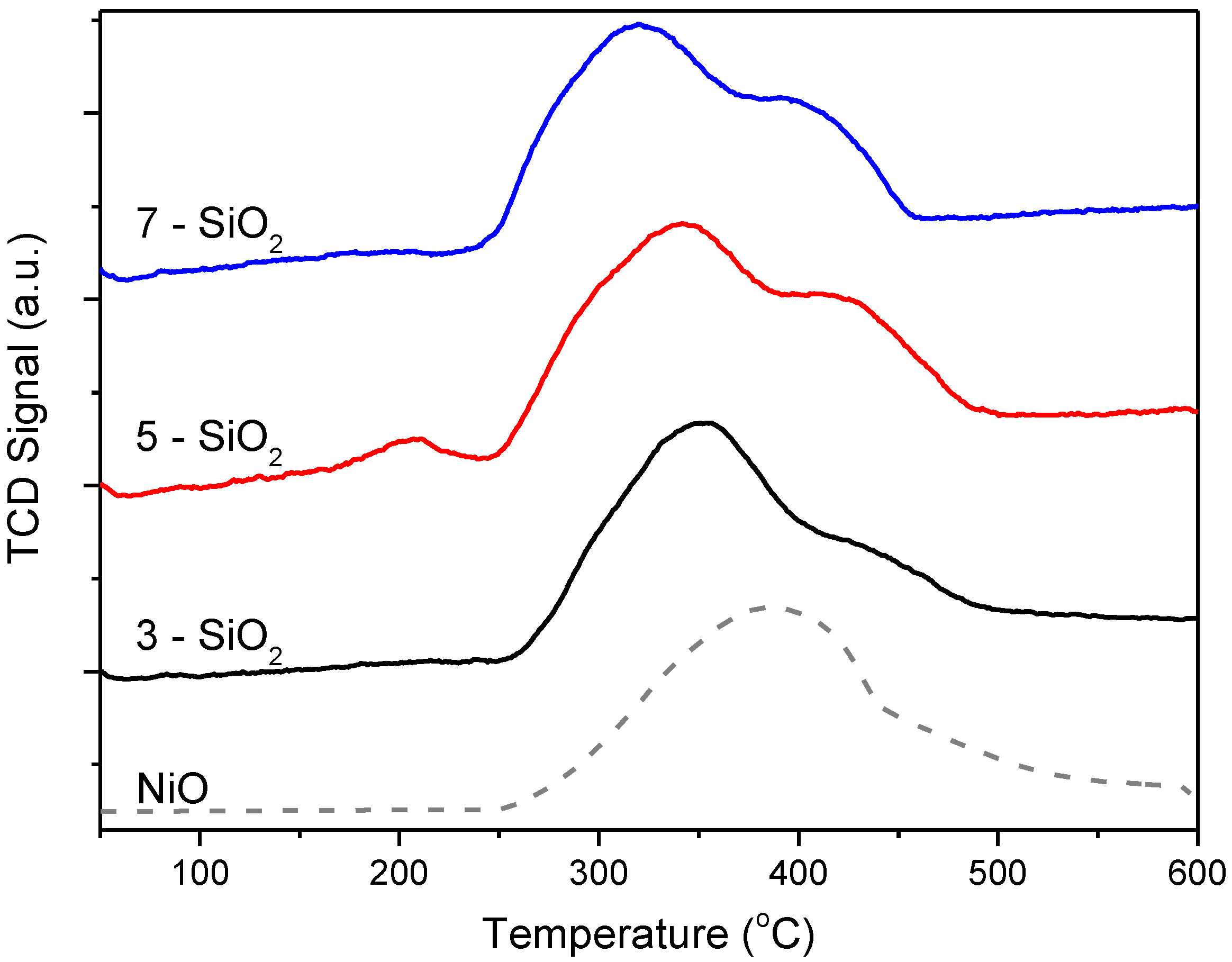
H
2-TPR was used to assess whether there was an impact of varying FSP support properties on NiO-support interaction with the reduction profiles shown in
Figure 4. The H
2-TPR profile for NiO powder (dashed line) was included for comparison. All reduction profiles show one main reduction peak (at approximately 320–350 °C) coupled with a broad, higher temperature shoulder at greater than 400 °C. The shoulder occurs at a similar temperature to the bulk NiO and is therefore assigned to NiO deposits weakly interacting with the silica support. It follows that the more significant, lower temperature peak then corresponds to NiO deposits with stronger metal-support interaction. When comparing the profiles between 3, 5 and 7-SiO
2 it is clear that there are some differences in metal-support interaction, even though all samples show the presence of these two Ni species, in varying proportions. 3-SiO
2 shows the lowest relative quantity of bulk NiO, which is consistent with the TEM particle size distributions. 5-SiO
2, on the other hand, shows a significantly higher amount of bulk NiO. An additional slight hump present at around 200 °C is evident for 5-SiO
2 which is tentatively attributed to trace Ni
2O
3 [
26,
27], however, no peaks were evident for this particular species in XRD (vide infra) and the peak is very small thus the quantity is seemingly insignificant. Compared to 5-SiO
2, 7-SiO
2 shows relatively smaller quantities of bulk NiO (represented by the smaller high temperature shoulder) as well as a narrower peak. This is consistent with the TEM micrographs showing on average a smaller size as well as a narrower deposit size distribution.
Figure 4.
H2-TPR results of 5% Ni on 3, 5 and 7-SiO2 and bulk NiO.
Figure 4.
H2-TPR results of 5% Ni on 3, 5 and 7-SiO2 and bulk NiO.
XRD was used to understand the difference in NiO crystallinity for the 5% Ni loaded on 3, 5 and 7-SiO
2 (
Figure 5). All profiles show similar patterns with peaks at 2θ values of 38, 44, 63, 76 and 79° corresponding to NiO(111), (200), (220), (222) and (311) planes, respectively. All XRD patterns also show a broad hump and tailing centered at around 23° which is characteristic of non-crystalline silica [
28,
29,
30]. The Scherrer equation was used to estimate NiO crystal size with the results displayed in
Table 1. The crystal size estimates show similar values between the differing silica supports, with a slight change in size depending on support synthesis conditions. The crystal sizes, consistent with TEM and TPR results, show a slight increase in the following order 3 < 7 < 5-SiO
2. When comparing the average NiO deposit size determined by TEM (22–26 nm) to the crystal size from XRD (~13 nm) it can be concluded that NiO deposits are multi-crystalline for all samples. Typically H
2 or CO chemisorption would be utilised to determine Ni dispersion. In this case, the relatively low Ni content and dispersion relative to the signal-to-noise ratio of the analysis made reliably determining the dispersion problematic. For this study, the XRD, H
2-TPR and TEM results are sufficient in showing the impact of the differing silica support on the nature of the Ni species present.
Figure 5.
XRD patterns of 5% Ni on 3, 5 and 7-SiO2.
Figure 5.
XRD patterns of 5% Ni on 3, 5 and 7-SiO2.
To ensure that the impregnated samples all had consistent Ni loadings, ICP-OES was utilised with the results being displayed in
Table 1. All samples show equivalent Ni loadings, thus the differences in Ni sizes do not arise from variations in Ni loading.
2.2. Discussion of Ni-SiO2 Properties
As was expected, increasing the FSP precursor feed rate decreased the SiO
2 specific surface area over the range of 240–340 m
2/g. As the SiO
2 specific surface area increased it was anticipated that NiO deposit size would then decrease (
i.e., dispersion would increase) although this was not found to be the case. Instead, the average NiO deposit size increases in the following order; 3 < 7 < 5-SiO
2. While the silica specific surface area was a significant factor which governed NiO deposit size, FTIR demonstrated that changing the precursor feed rate also impacted on the silica surface chemistry. In particular, increasing the FSP feed rate altered the ratio of H–bonded to isolated silanol groups on the silica surface as well introduced residual carbon species. Recent research has demonstrated the importance of silica surface properties during impregnation [
7,
17,
18]. It is well established that there are two types of silanol groups on silica surfaces; H–bonded Si–OH and isolated (non H–bonded) Si–OH [
14,
15,
20]. As mentioned earlier, upon impregnating SiO
2 with Ag it was found that varying silanol groups by calcination significantly altered the active site (Ag) dispersion and metal-support interaction [
17]. It has also been reported by others that modifying the surface of a silica support (by ethylene glycol pre-treatment) resulted in more dispersed Ni species giving greater activity for the DRM [
18]. The studies concluded that during impregnation the Ni
2+ interacts preferentially with H–bonded –OH groups which results in larger Ni species. Additionally, Lv
et al., postulated that residual C–H groups and a reduction in total surface hydroxyl groups (comparative to Si–O–Si) contributed to increased Ni dispersion [
18]. It is difficult to ascertain whether the increase in Ni dispersion arises from the variation of silanol groups on the silica surface or introduction of residual C–H groups or a combination of the two. However is clear is that the difference in surface properties of the FSP-prepared silica has an impact on the dispersion and Ni-support interaction, although to a lesser extent than the impact of varying surface area. These findings demonstrate FSP is suitable for producing silica as a support for NiO catalysts with synthesis conditions able to be effectively tuned.
2.3. Activity Results
The impact of FSP SiO
2 support precursor feed rate on the performance of 5% Ni-SiO
2 for the DRM was evaluated using two approaches; (1) light-off curves over the range 500–800 °C and (2) 24 h stability tests at 700 °C.
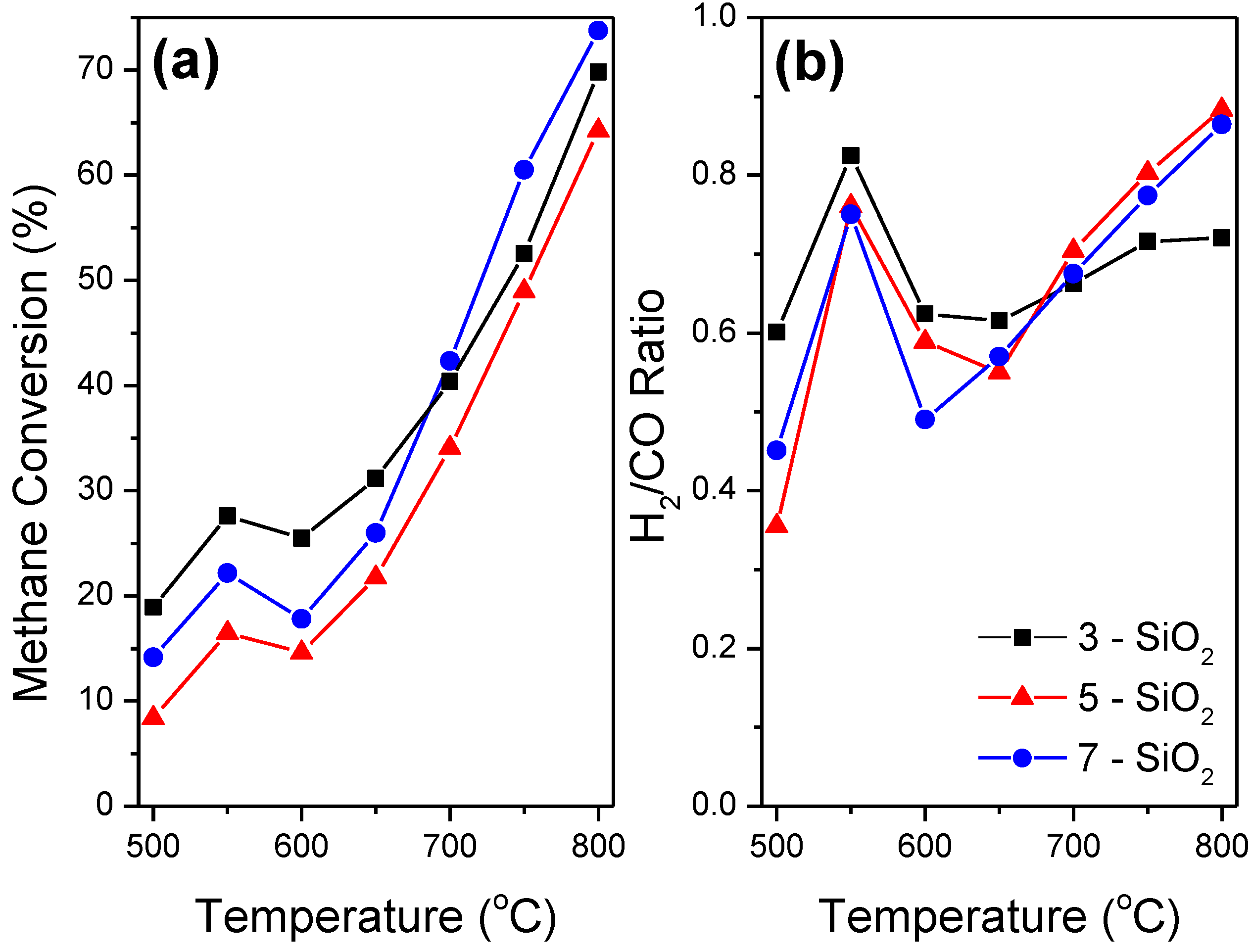
Figure 6 shows CH
4 conversion along with the H
2/CO ratio of each sample with respect to temperature for the light-off profiles. The H
2/CO profiles are of most importance when considering catalysis for the DRM as they provide vital insight into the side reactions occurring within the system. The Boudouard reaction (Equation (2)) at lower temperatures (<650 °C) and methane decomposition (Equation (3)) at higher temperatures (>650 °C) result in carbon formation as well as an increase in H
2/CO ratio. The reverse water gas shift (RWGS) reaction (Equation (4)) results in a decrease in H
2/CO ratio, and increases in terms of thermodynamic favourability with increasing temperature [
8]. A ratio of one indicates a balance of side reactions occurring within the system.
The governing trends evident from the light-off curves (
Figure 6) are consistent with the thermodynamics of the side reactions within the DRM system. With the exception of temperature region 550–600 °C, all samples show an overall increase in CH
4 conversion and H
2/CO ratio conversion as temperature increases. CO
2 consumption (not shown) exhibited a similar profile to the CH
4 light of curve except it remained approximately 10%–20% higher at all temperatures. The CH
4 conversion and H
2/CO ratio plateau in the 600–650 °C range representing a transition between side reaction dominance. At this temperature, the side reactions occurring in the system are at a thermodynamic minimum. Specifically the Boudouard reaction becomes thermodynamically unfavourable whilst both methane decomposition along with the RWGS reaction increases in likelihood with increasing temperature. In addition, the higher CO
2 conversion relative to CH
4 conversion at all temperatures provides further evidence the RWGS reaction and/or CO
2 decomposition are occurring.
Figure 6.
Methane light-off curves (a) and accompanying H2/CO production ratio (b) during dry reforming of methane over 5% Ni supported by 3, 5 and 7-SiO2. Conditions: WHSV = 144 L/(h·gcat), catalyst loading = 0.025 g, N2/CH4/CO2 = 1:1:1.
Figure 6.
Methane light-off curves (a) and accompanying H2/CO production ratio (b) during dry reforming of methane over 5% Ni supported by 3, 5 and 7-SiO2. Conditions: WHSV = 144 L/(h·gcat), catalyst loading = 0.025 g, N2/CH4/CO2 = 1:1:1.
5-SiO
2 shows the lowest CH
4 conversion over the entire temperature range which can be attributed to the larger Ni deposit size. A larger deposit size means less surface area of active sites per mass of Ni present which intrinsically results in a less active catalyst. 5-SiO
2 also has the highest H
2/CO ratio at higher temperature, showing the most significant increase in the 650–800 °C temperature range. H
2-TPR, supported by TEM and XRD, show that 5-SiO
2 has the most bulk NiO present. Bulk NiO with minimal metal-support interaction has a tendency to promote methane decomposition [
6,
31] and therefore will result in higher carbon formation at higher temperatures. Although the high H
2/CO ratio implies that 5-SiO
2 will produce the largest quantity of carbon from methane decomposition, all samples contained approximately the same quantity of carbon present after reaction. TGA indicated that the quantity of formed carbon during the DRM was 56.4, 56.6 and 57.7% for the 3, 5 and 7-SiO
2 supports, respectively. However, the relatively higher H
2/CO ratio at lower temperatures exhibited by 3-SiO
2 coupled with the lower H
2/CO ratio at higher temperature imply that there are differing dominant side reactions within the system despite the equivalent carbon formation. 3-SiO
2 outperforms all samples at lower temperatures however, above 700 °C 7-SiO
2 is the most active. This indicates that 3-SiO
2 has deactivated to a greater extent at lower temperatures. The lower H
2/CO ratio at higher temperatures and equivalent carbon formation over the full temperature range implies that 3-SiO
2 is more prone to carbon formation, and thus elevated deactivation, through the Boudouard reaction. The implication from the results is that 5-SiO
2 is more susceptible to carbon formation via methane decomposition whilst 3-SiO
2 is more inclined to form carbon through the Boudouard reaction. 7-SiO
2 seems to lie in between the 3 and 5-SiO
2 in terms of the prominence of side reactions.
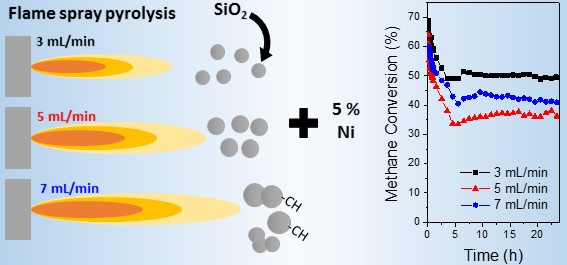
The stability of the prepared catalysts was assessed by 24 h stability tests run at 700 °C in order to minimise the impact of the Boudouard reaction (
Figure 7). The results indicate that, as expected, 3-SiO
2 facilitates the highest methane conversion. The silica shows approximately 30% loss in activity over the 24 h period, with the most significant drop evident within the first 5 h. The carbon formation is significantly lower than all other samples at 700 °C, which further supports the findings drawn from the light-off curves (
Figure 6), namely that 3-SiO
2 results in a reduction in methane decomposition. TEM images (
Figure 7c) show the main species of carbon formed were nanotubes, typical of Ni-catalysts for the DRM [
32]. The TGA curves (not shown) did not indicate significant differences in the carbon species across the different supports.
Figure 7.
Twenty four hour stability tests with methane conversion (a) and accompanying H2/CO production ratio during dry reforming of methane over 5% Ni supported by 3, 5 and 7-SiO2 with (b) showing carbon formation determined by TGA and (c) TEM of spent 3-SiO2. Conditions: WHSV = 144 L/(h·gcat), catalyst loading = 0.025 g, N2/CH4/CO2 = 1:1:1 at a temperature of 700 °C.
Figure 7.
Twenty four hour stability tests with methane conversion (a) and accompanying H2/CO production ratio during dry reforming of methane over 5% Ni supported by 3, 5 and 7-SiO2 with (b) showing carbon formation determined by TGA and (c) TEM of spent 3-SiO2. Conditions: WHSV = 144 L/(h·gcat), catalyst loading = 0.025 g, N2/CH4/CO2 = 1:1:1 at a temperature of 700 °C.
Comparatively, 5-SiO
2 shows the greatest deactivation, losing approximately 43% of its activity, most significantly within the first 5 h. This coupled with the highest carbon formation shows that the larger quantity of bulk Ni (as well as the largest average Ni size) present on 5-SiO
2 results in methane decomposition thus leading to the most unstable catalyst. Interestingly, whilst the deactivation and carbon formation suggest that 5-SiO
2 has the highest quantity of methane decomposition the H
2/CO ratio suggests there are further side reactions occurring in the system. The implication is that 5-SiO
2 is most prone to both the RWGS reaction as well as methane decomposition. This tendency toward the RWGS reaction results in an overall lower H
2/CO ratio, despite the evidence of methane decomposition. 7-SiO
2 shows an intermediate behavior in terms of deactivation (31%) and carbon formation (14.4%). This follows strongly with the measured Ni size (
Section 2.2).
The differences in the activity, stability and selectivity of the produced catalysts likely originate from the varying nature of the Ni deposits on the silica surface. As the silica is a relatively inert support without significant oxygen mobility and acidity/basicity, the impacts of the varying support synthesis conditions on the activity of the prepared catalysts can be attributed to the change in Ni properties. Larger Ni deposits result in decreased conversion and an increase in carbon formation. It follows that the smaller the Ni deposits, the larger the available surface area of Ni (per mass of Ni present) and thus the highest conversion. A similar effect has been found by others for Ni-SiO
2 samples for the DRM at 500 °C, proving that catalytic activity decreased exponentially with increasing particle size in the range of ~1.6–7.3 nm [
33]. Their results demonstrated the correlation between surface Ni atoms and sample activity with the turn over frequencies (per mole of surface Ni) being independent of Ni size [
33]. Additionally, larger NiO, with minimal metal-support interaction has a greater tendency to form carbon through methane decomposition (Equation (3)) [
6,
31]. Research has suggested the size limit for carbon filaments is <10 nm, and as in this work the NiO deposit sizes exceed this limit, the formation of carbon filaments is feasible [
33,
34]. For the smaller Ni deposits it is evident that at lower temperatures (<650 °C), the Boudouard reaction dominates carbon formation and results in deactivation. Additionally, Baudouin
et al., proved the extent to which the RWGS occurs is highly dependent on conversion (and thus particle size). Essentially they concluded that larger Ni results in a greater extent of the RWGS occurring due to the kinetics of the initial dissociation [
33].









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



