
Cyclic Comonomers for the Synthesis of Carboxylic Acid and Amine Functionalized Poly(l-Lactic Acid)

Abstract
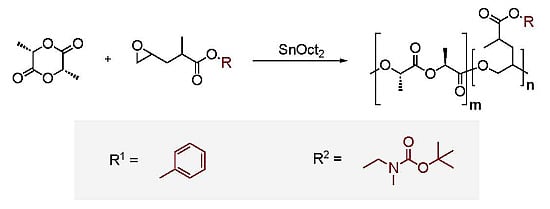
:
1. Introduction
2. Results and Discussion
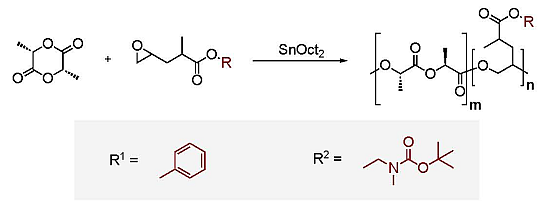
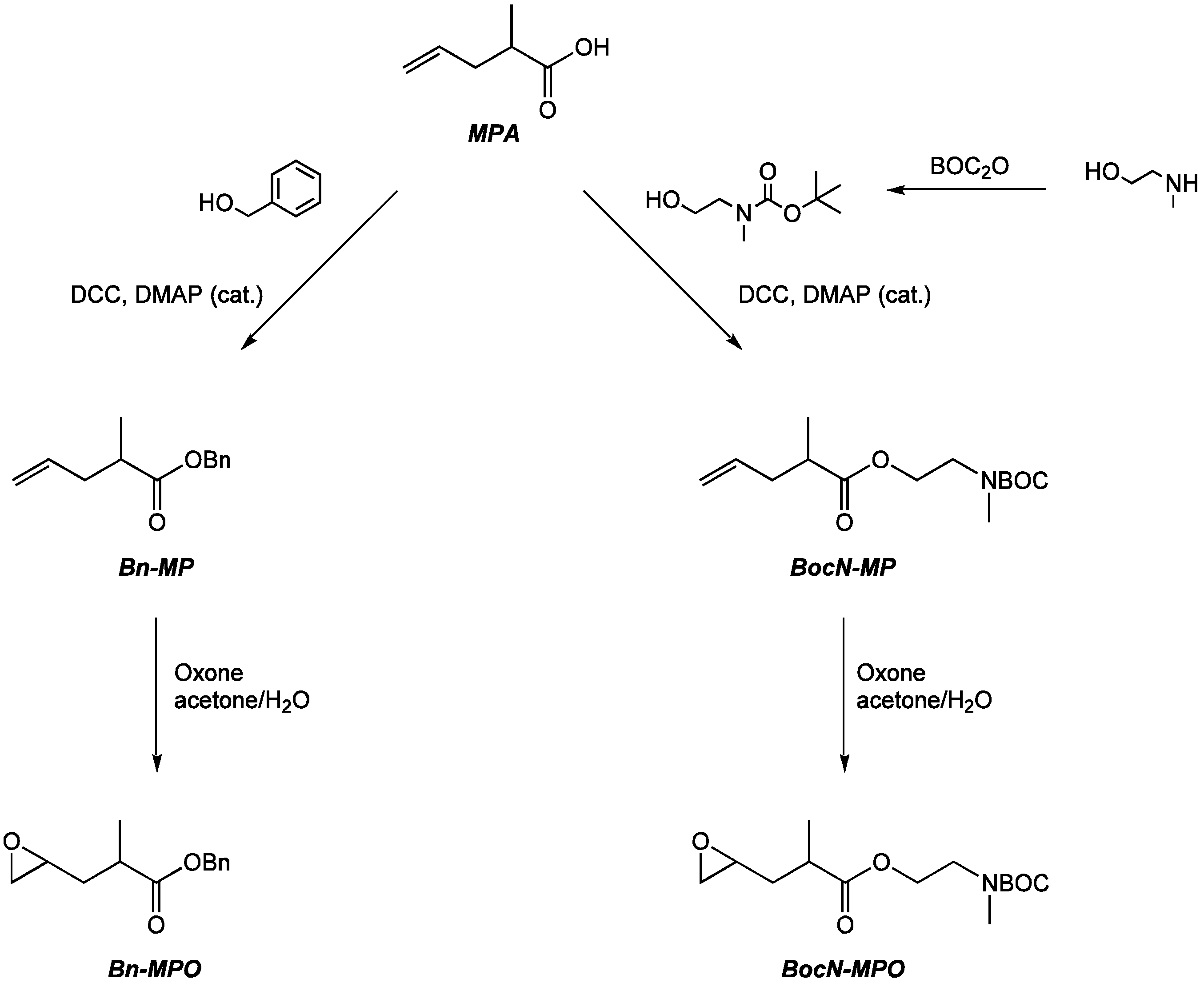
2.1. Monomer Synthesis


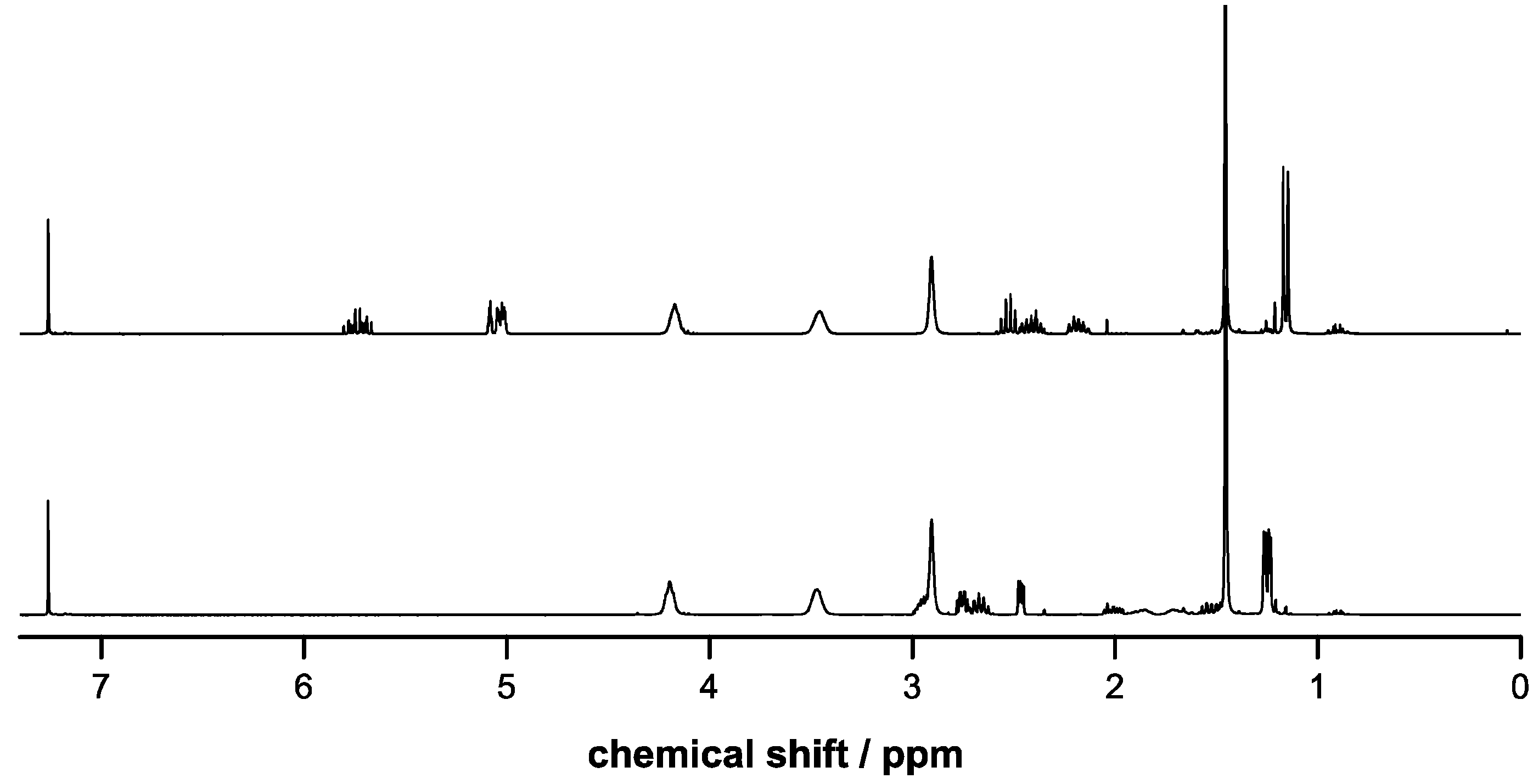
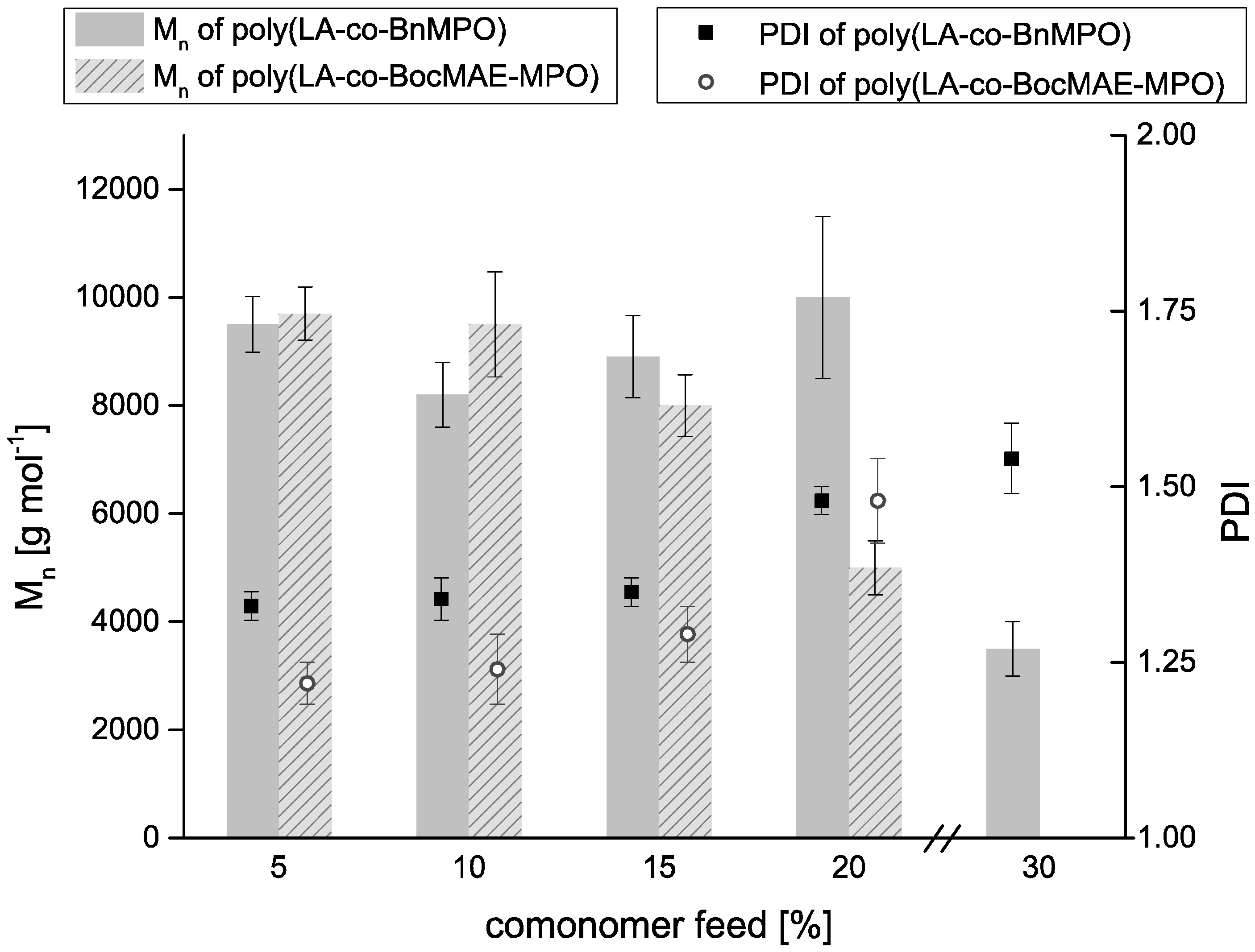
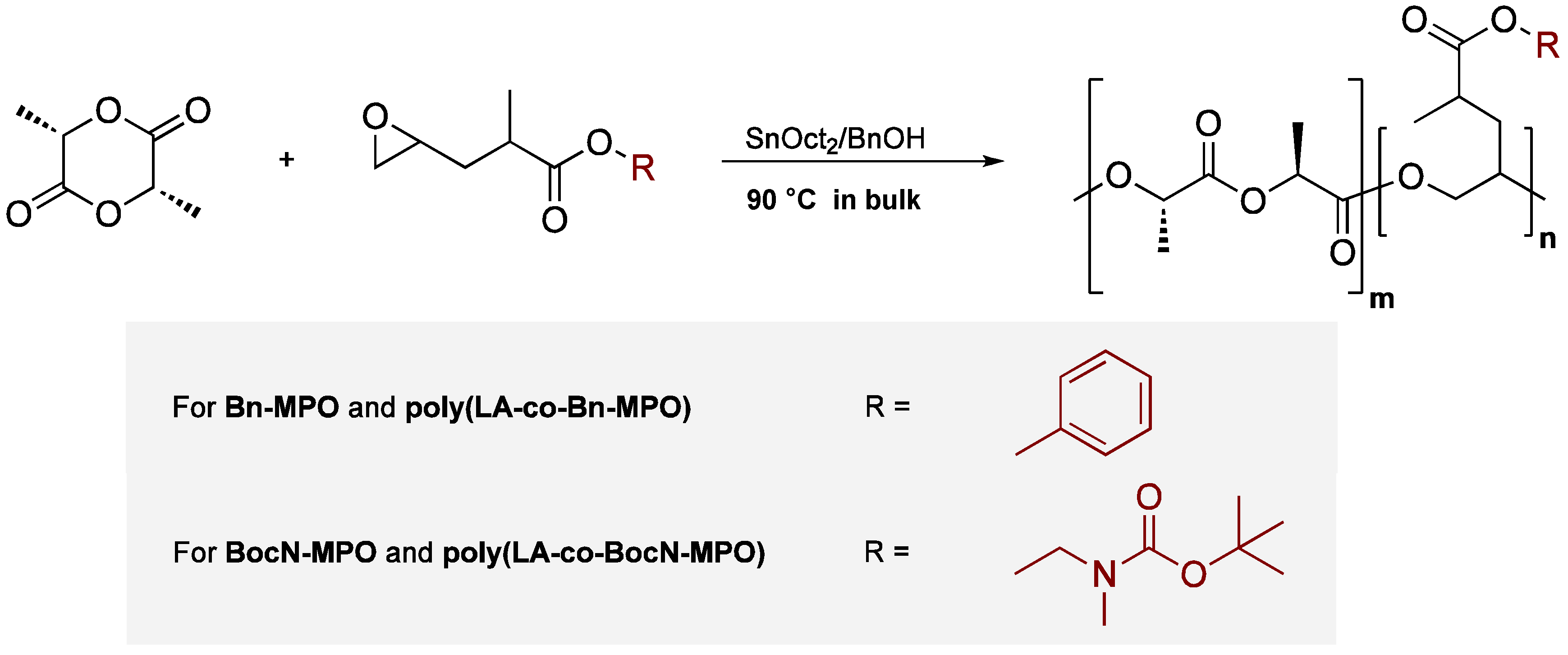
2.2. Copolymerizations of Bn-MPO and BocN-MPO with LA


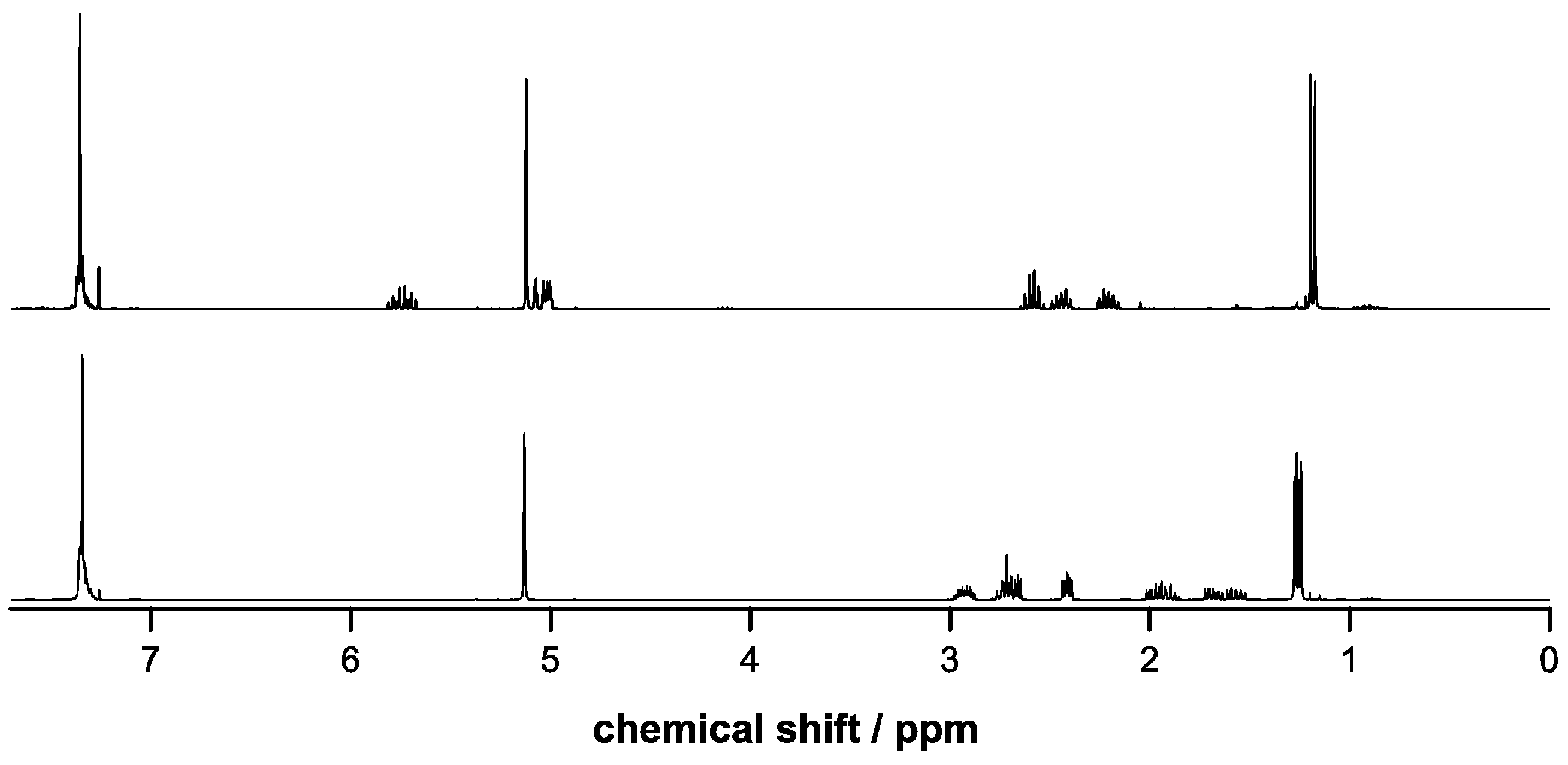
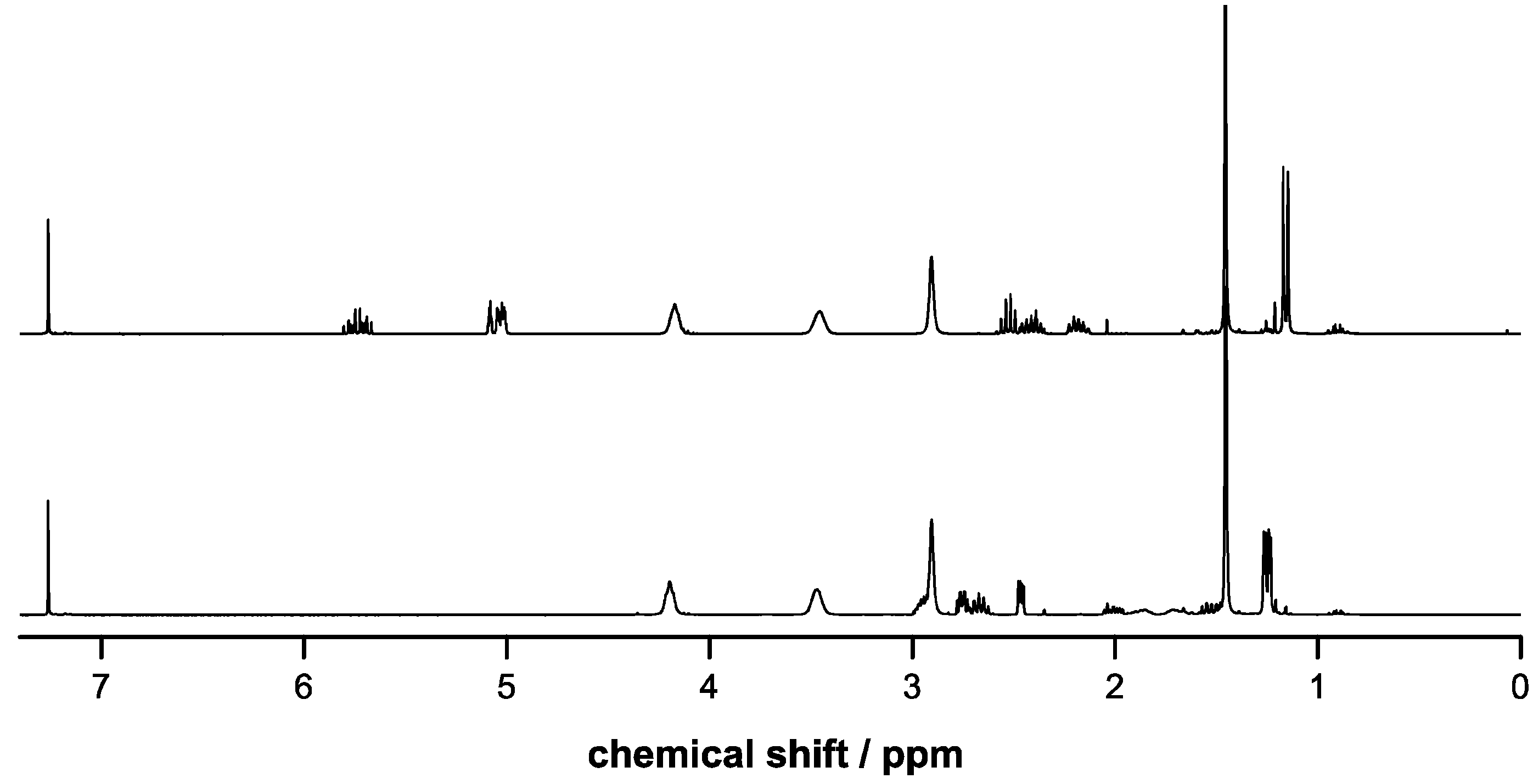
2.3. Incorporation of Comonomers

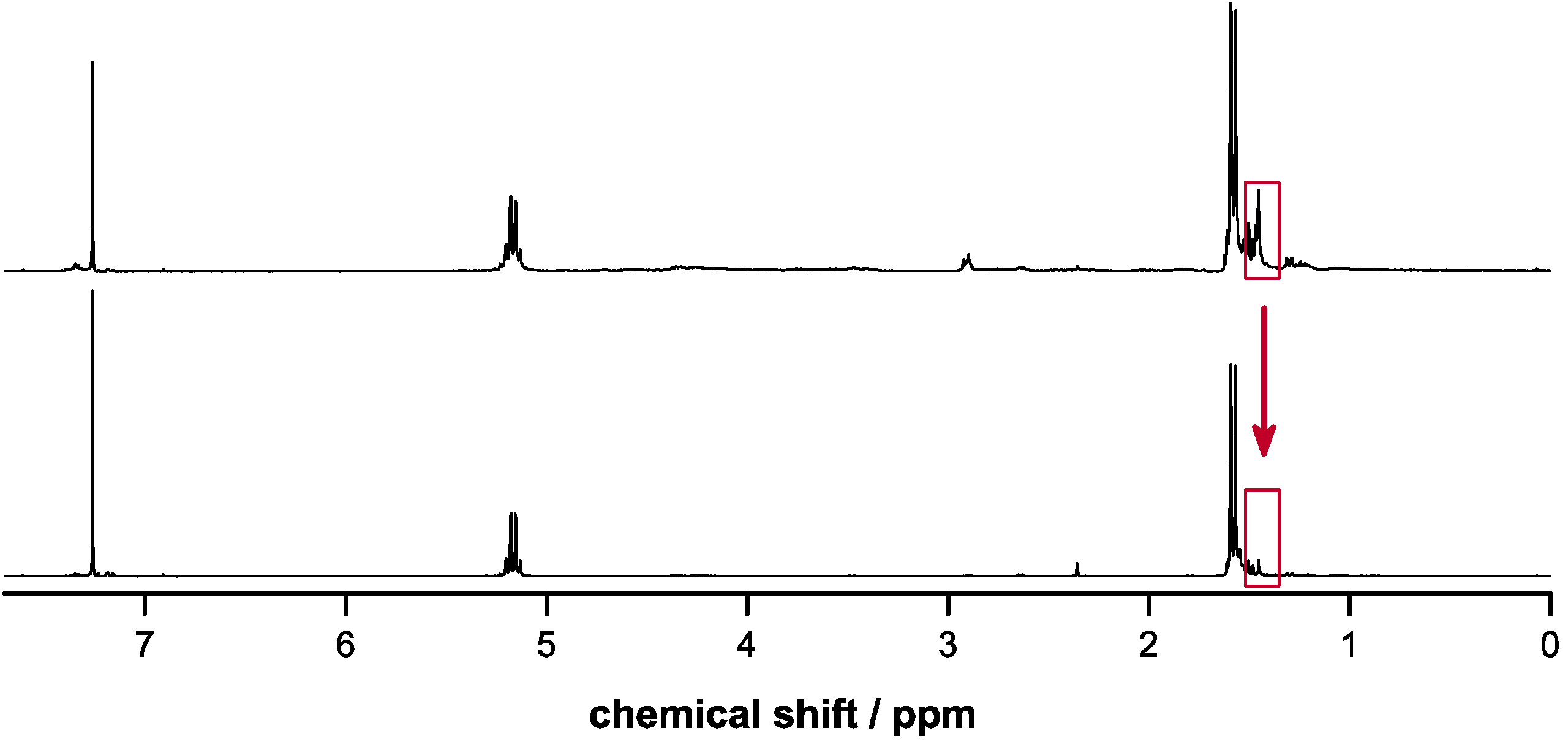

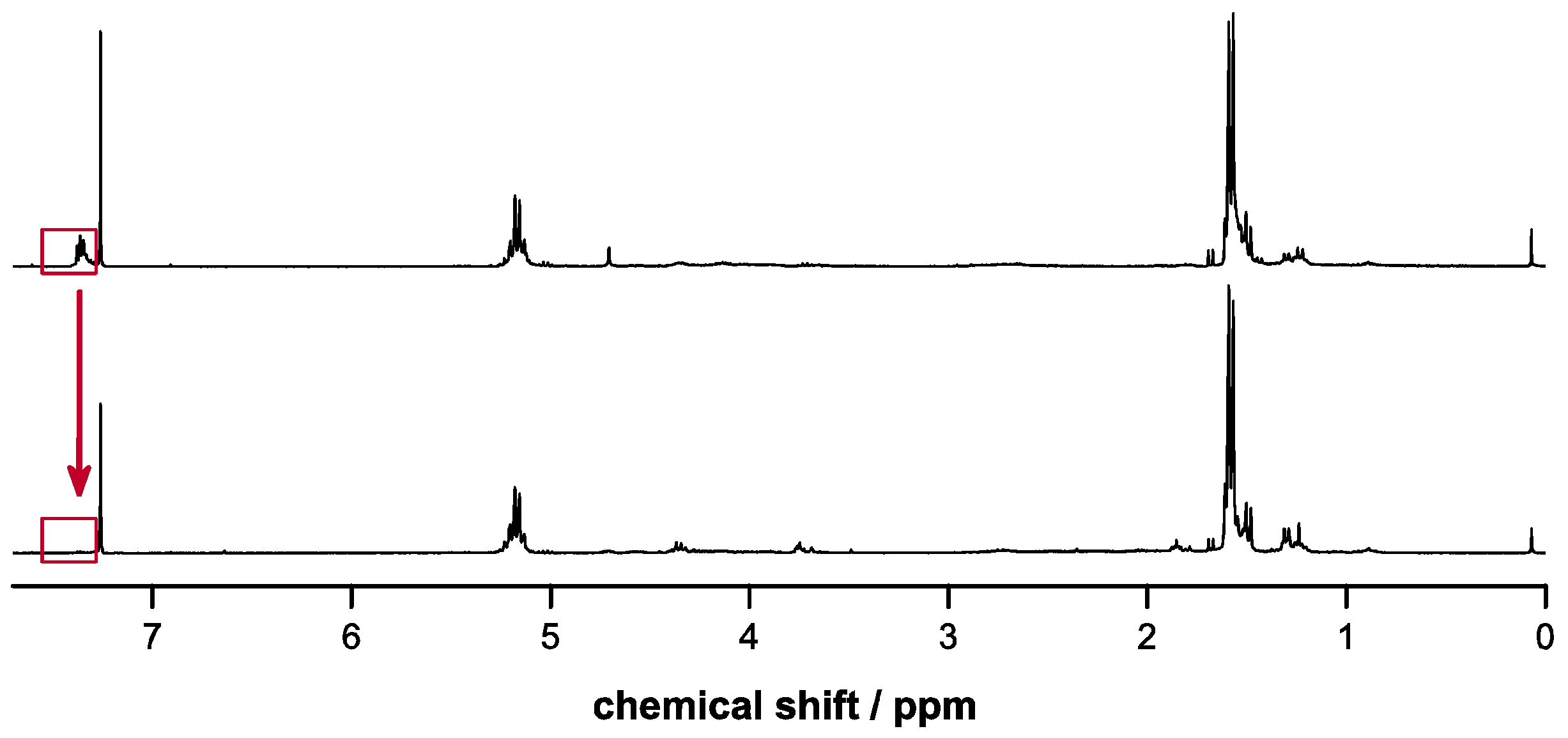
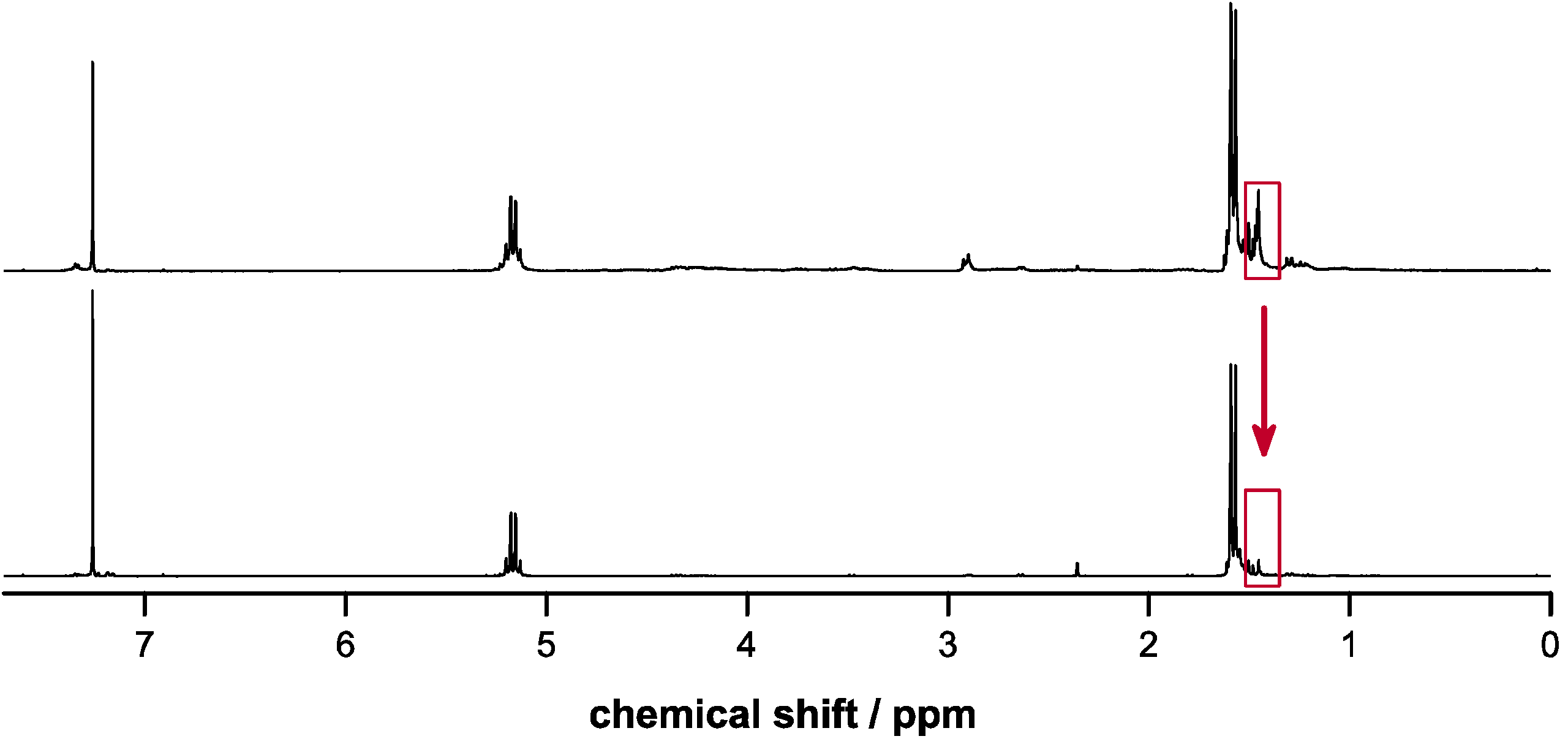
2.4. Deprotection of Copolymers
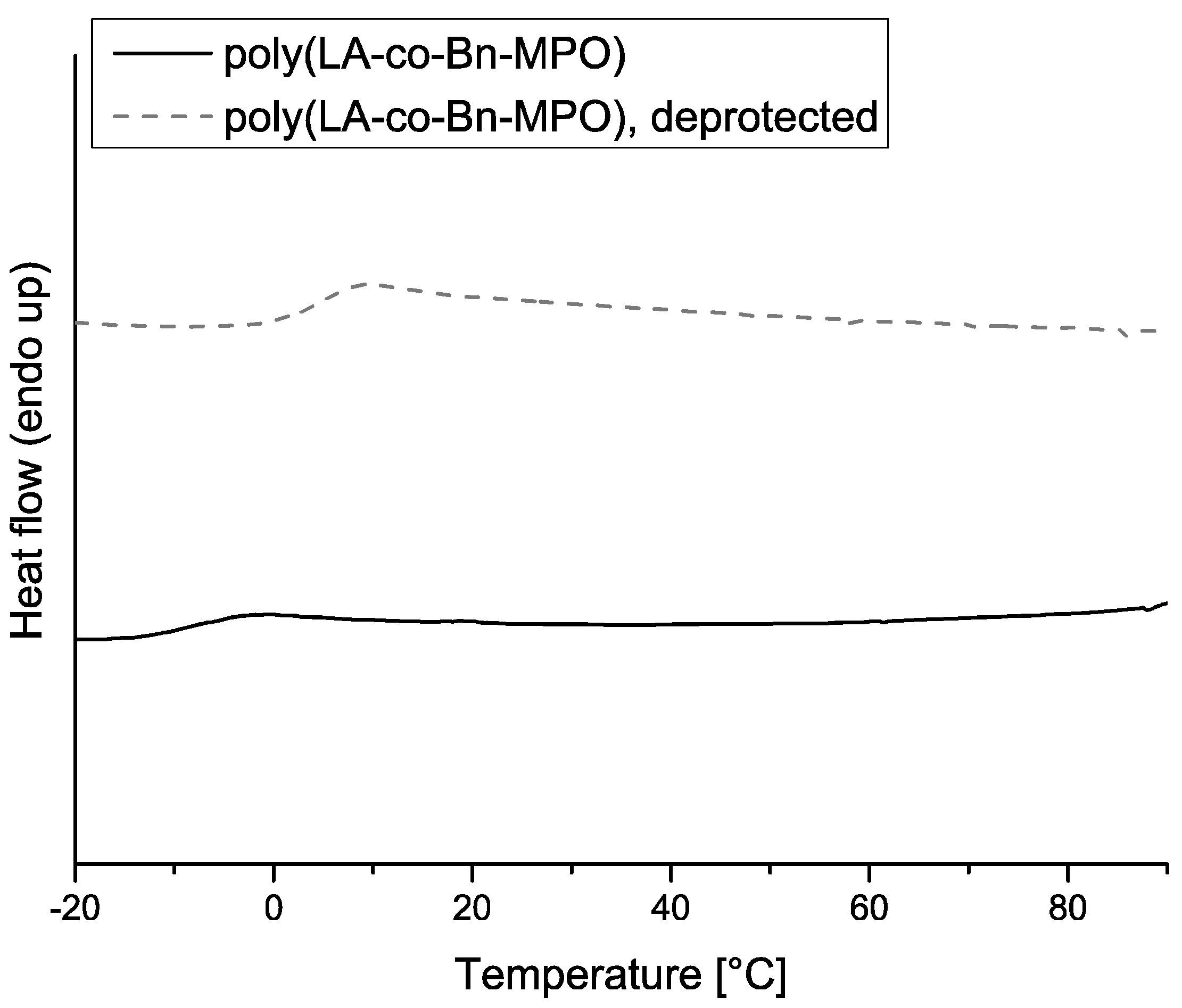
2.5. Thermal Properties of Copolymers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mn g·mol−1 | Comonomer Incorp. [%] | Td [°C] | Tg [°C] | Tg, after deprotection [°C] | |
|---|---|---|---|---|---|
| PLA | 9500 | - | 252 | 54 | - |
| poly(LA-co-Bn-MPO) | 8500 | 7.5 | 251 | 7 | 14 |
| 9000 | 8.6 | 252 | 4 | 13 | |
| 10,000 | 14.1 | 241 | −9 | 5 | |
| 3500 | 25.9 | 240 | −11 | 0 | |
| poly(LA-co-BocN-MPO) | 9500 | 7.7 | 262 | 15 | 23 |
| 8000 | 9.9 | 257 | 22 | 31 | |
| 5000 | 15.7 | 254 | 23 | 31 |


3. Experimental Section
3.1. Materials
3.2. Synthetic Procedures
3.2.1. Synthesis of Benzyl 2-methylpent-4-enoate (Bn-MP)
3.2.2. Synthesis of Benzyl 2-methyl-3-(oxiran-2-yl)propanoate) (Bn-MPO)
3.2.3. Synthesis of tert-Butyl (2-hydroxyethyl)(methyl)carbamate (BocN)
3.2.4. Synthesis of 2-((tert-Butoxycarbonyl)(methyl)amino)ethyl 2-methylpent-4-enoate (BocN-MP)
3.2.5. Synthesis of 2-((tert-Butoxycarbonyl)(methyl)amino)ethyl 2-methyl-3-(oxiran-2-yl)propanoate (BocN-MPO)
3.2.6. Copolymerization of L-Lactide with Bn-MPO and BocN-MPO
3.2.7. Deprotection of Poly(LA-co-Bn-MPO)
3.2.8. Deprotection of Poly(LA-co-BocN-MPO)
3.3. Methods
3.3.1. Proton Nuclear Magnetic Resonance Spectroscopy (NMR)
3.3.2. Gel Permeation Chromatography (GPC)
3.3.3. Differential Scanning Calorimetry (DSC)
3.3.4. Elemental Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anderson, J.M.; Shive, M.S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 2012, 64, 72–82. [Google Scholar] [CrossRef]
- Nicolas, J.; Mura, S.; Brambilla, D.; Mackiewicz, N.; Couvreur, P. Design, functionalization strategies and biomedical applications of targeted biodegradable/biocompatible polymer-based nanocarriers for drug delivery. Chem. Soc. Rev. 2013, 42, 1147–1235. [Google Scholar] [CrossRef] [PubMed]
- Armentano, I.; Dottori, M.; Fortunati, E.; Mattioli, S.; Kenny, J.M. Biodegradable polymer matrix nanocomposites for tissue engineering: A review. Polym. Degrad. Stab. 2010, 95, 2126–2146. [Google Scholar] [CrossRef]
- Agrawal, C.M.; Ray, R.B. Biodegradable polymeric scaffolds for musculoskeletal tissue engineering. J. Biomed. Mater. Res. 2001, 55, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, M.S. Polymer scaffolds for biomaterials applications. Macromolecules 2010, 43, 581–591. [Google Scholar] [CrossRef]
- Gulati, K.; Ramakrishnan, S.; Aw, M.S.; Atkins, G.J.; Findlay, D.M.; Losic, D. Biocompatible polymer coating of titania nanotube arrays for improved drug elution and osteoblast adhesion. Acta Biomater. 2012, 8, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yuan, L.; Song, W.; Wu, Z.; Li, D. Biocompatible polymer materials: Role of protein-surface interactions. Prog. Polym. Sci. 2008, 33, 1059–1087. [Google Scholar] [CrossRef]
- Ikada, Y.; Tsuji, H. Biodegradable polyesters for medical and ecological applications. Macromol. Rapid Commun. 2000, 21, 117–132. [Google Scholar] [CrossRef]
- Cutright, D.E.; Hunsuck, E.E. The repair of fractures of the orbital floor using biodegradable polylactic acid. Oral Surg. Oral Med. Oral Pathol. 1972, 33, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, W.; Ding, J.; Lu, S.; Wang, X.; Wang, C.; Chen, X. Cholesterol-enhanced polylactide-based stereocomplex micelle for effective delivery of doxorubicin. Materials 2015, 8, 216–230. [Google Scholar] [CrossRef]
- Farokhzad, O.C.; Jon, S.; Khademhosseini, A.; Tran, T.-N.T.; Lavan, D.A.; Langer, R. Nanoparticle-aptamer bioconjugates: A new approach for targeting prostate cancer cells. Cancer Res. 2004, 64, 7668–7672. [Google Scholar] [CrossRef] [PubMed]
- Athanasiou, K.A.; Agrawal, C.M.; Barber, F.A.; Burkhart, S.S. Orthopaedic applications for PLA-PGA biodegradable polymers. Arthroscopy 1998, 14, 726–737. [Google Scholar] [CrossRef] [PubMed]
- Thieme, M.; Agarwal, S.; Wendorff, J.H.; Greiner, A. Electrospinning and cutting of ultrafine bioerodible poly(lactide-co-ethylene oxide) tri- and multiblock copolymer fibers for inhalation applications. Polym. Adv. Technol. 2011, 22, 1335–1344. [Google Scholar]
- Guimarães, P.P.G.; Oliveira, S.R.; de Castro Rodrigues, G.; Gontijo, S.M.L.; Lula, I.S.; Cortés, M.E.; Denadai, Â.M.L.; Sinisterra, R.D. Development of sulfadiazine-decorated PLGA nanoparticles loaded with 5-fluorouracil and cell viability. Molecules 2015, 20, 879–899. [Google Scholar] [CrossRef] [PubMed]
- Punet, X.; Mauchauffé, R.; Giannotti, M.I.; Rodríguez-Cabello, J.C.; Sanz, F.; Engel, E.; Mateos-Timoneda, M.A.; Planell, J.A. Enhanced cell-material interactions through the biofunctionalization of polymeric surfaces with engineered peptides. Biomacromolecules 2013, 14, 2690–2702. [Google Scholar] [CrossRef] [PubMed]
- Heiny, M.; Wurth, J.J.; Shastri, V.P. Progress in functionalized biodegradable polyesters. In Natural and Synthetic Biomedical Polymers; Kumbar, S., Laurencin, C., Deng, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 167–180. [Google Scholar]
- Makiguchi, K.; Kikuchi, S.; Satoh, T.; Kakuchi, T. Synthesis of block and end-functionalized polyesters by triflimide-catalyzed ring-opening polymerization of ε-caprolactone, 1,5-dioxepan-2-one, and rac-lactide. J. Polym. Sci. Part A: Polym. Chem. 2013, 51, 2455–2463. [Google Scholar] [CrossRef]
- Yang, J.; Wan, Y.; Tu, C.; Cai, Q.; Bei, J.; Wang, S. Enhancing the cell affinity of macroporous poly(l-lactide) cell scaffold by a convenient surface modification method. Polym. Int. 2003, 52, 1892–1899. [Google Scholar] [CrossRef]
- Kim, T.G.; Park, T.G. Biodegradable polymer nanocylinders fabricated by transverse fragmentation of electrospun nanofibers through aminolysis. Macromol. Rapid Commun. 2008, 29, 1231–1236. [Google Scholar] [CrossRef]
- Vert, M. Biomedical polymers from chiral lactides and functional lactones: Properties and applications. Makromol. Chem. Macromol. Symp. 1986, 6, 109–122. [Google Scholar] [CrossRef]
- Kimura, Y.; Shirotani, K.; Yamane, H.; Kitao, T. Ring-opening polymerization of 3(S)-[(benzyloxycarbonyl)methyl]-1,4-dioxane-2,5-dione: A new route to a poly(α-hydroxy acid) with pendant carboxyl groups. Macromolecules 1988, 21, 3338–3340. [Google Scholar] [CrossRef]
- Gerhardt, W.W.; Noga, D.E.; Hardcastle, K.I.; García, A.J.; Collard, D.M.; Weck, M. Functional lactide monomers: Methodology and polymerization. Biomacromolecules 2006, 7, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Lu, J.; Behl, M.; Lendlein, A. Progress in depsipeptide-based biomaterials. Macromol. Biosci. 2010, 10, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Shastri, V.P. Biodegradable Polymers from Derivatized Ring-Opened Epoxides. U.S. Patent 6730772, 4 May 2004. [Google Scholar]
- Sussman, E.M.; Clarke, M.B.; Shastri, V.P. Single-step process to produce surface-functionalized polymeric nanoparticles. Langmuir 2007, 23, 12275–12279. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Jallouk, A.P.; Vasquez, D.; Thomann, R.; Forget, A.; Pino, C.J.; Shastri, V.P. Surface functionality as a means to impact polymer nanoparticle size and structure. Langmuir 2013, 29, 4092–4095. [Google Scholar] [CrossRef] [PubMed]
- Wurth, J.J.; Shastri, V.P. Synthesis and characterization of functionalized poly(ɛ-caprolactone). J. Polym. Sci. Part A: Polym. Chem. 2013, 51, 3375–3382. [Google Scholar] [CrossRef]
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef] [PubMed]
- Schwach, G.; Coudane, J.; Engel, R.; Vert, M. Stannous octoate-versus zinc-initiated polymerization of racemic lactide. Polym. Bull. 1994, 32, 617–623. [Google Scholar] [CrossRef]
- Teske, N.S.; Voigt, J.; Shastri, V.P. Clickable degradable aliphatic polyesters via copolymerization with alkyne epoxy esters: Synthesis and post functionalization with organic dyes. J. Am. Chem. Soc. 2014, 136, 10527–10533. [Google Scholar] [CrossRef] [PubMed]
- Lipik, V.T.; Abadie, M.J.M. Synthesis of block copolymers of varying architecture through suppression of transesterification during coordinated anionic ring opening polymerization. Int. J. Biomater. 2012, 2012. [Google Scholar] [CrossRef]
- Yoo, J.-W.; Mitragotri, S. Polymer particles that switch shape in response to a stimulus. Proc. Natl. Acad. Sci. USA 2010, 107, 11205–11210. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heiny, M.; Shastri, V.P. Cyclic Comonomers for the Synthesis of Carboxylic Acid and Amine Functionalized Poly(l-Lactic Acid). Molecules 2015, 20, 4764-4779. https://doi.org/10.3390/molecules20034764
Heiny M, Shastri VP. Cyclic Comonomers for the Synthesis of Carboxylic Acid and Amine Functionalized Poly(l-Lactic Acid). Molecules. 2015; 20(3):4764-4779. https://doi.org/10.3390/molecules20034764
Chicago/Turabian StyleHeiny, Markus, and V. Prasad Shastri. 2015. "Cyclic Comonomers for the Synthesis of Carboxylic Acid and Amine Functionalized Poly(l-Lactic Acid)" Molecules 20, no. 3: 4764-4779. https://doi.org/10.3390/molecules20034764
APA StyleHeiny, M., & Shastri, V. P. (2015). Cyclic Comonomers for the Synthesis of Carboxylic Acid and Amine Functionalized Poly(l-Lactic Acid). Molecules, 20(3), 4764-4779. https://doi.org/10.3390/molecules20034764