3.1. General Methods
All reactions were carried out under dry nitrogen or argon unless otherwise indicated. Commercially available reagents were used without further purification. Solvents and gases were dried according to standard procedures. Organic solvents were evaporated with reduced pressure using a rotary evaporator. Analytical thin layer chromatography (TLC) was performed using glass plates precoated with silica gel (0.25 mm). TLC plates were visualized by exposure to UV light (UV), and then were visualized with a KMnO4 stain followed by brief heating on hot plate. Flash column chromatography was performed using silica gel 60 (230–400 mesh, Merck, Kenilworth, NJ, USA) with the indicated solvents. The melting points of solid products was measured by using capillary tubes for the determination of melting points (Marienfeld-Superior, Lauda-Königshofen, Germany) and an automated melting point system (OptiMelt, Stanford Research Systems, Inc. Sunnyvale, CA, USA). The tubes containing solid compounds were inserted in OptiMelt, and the temperature was slowly raised from 100 °C to 400 °C. 1H- and 13C-NMR spectra were recorded on Bruker 300 or 400 spectrometers (Bruker Co. Billerica, MA, USA). 1H-NMR spectra are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet, td = triplet of doublet, brs = broad singlet, brt = broad triplet), integration, and coupling constant (J) in Hertz (Hz). 1H-NMR chemical shifts are reported relative to CDCl3 (7.26 ppm). 13C-NMR was recorded relative to the central line of CDCl3 (77.0 ppm). HRMS analyses were performed on Bruker compact ESI+ positive mode. HPLC purifications were performed on an Alliance Waters 2489 UV/Visible detector, e2695 Separations module using a Waters Xterra prep RP-18 10 μM, 10 × 250 mm column (Waters Inc. Milford, MA, USA) and a 30%–100% acetonitrile in water solvent gradient.
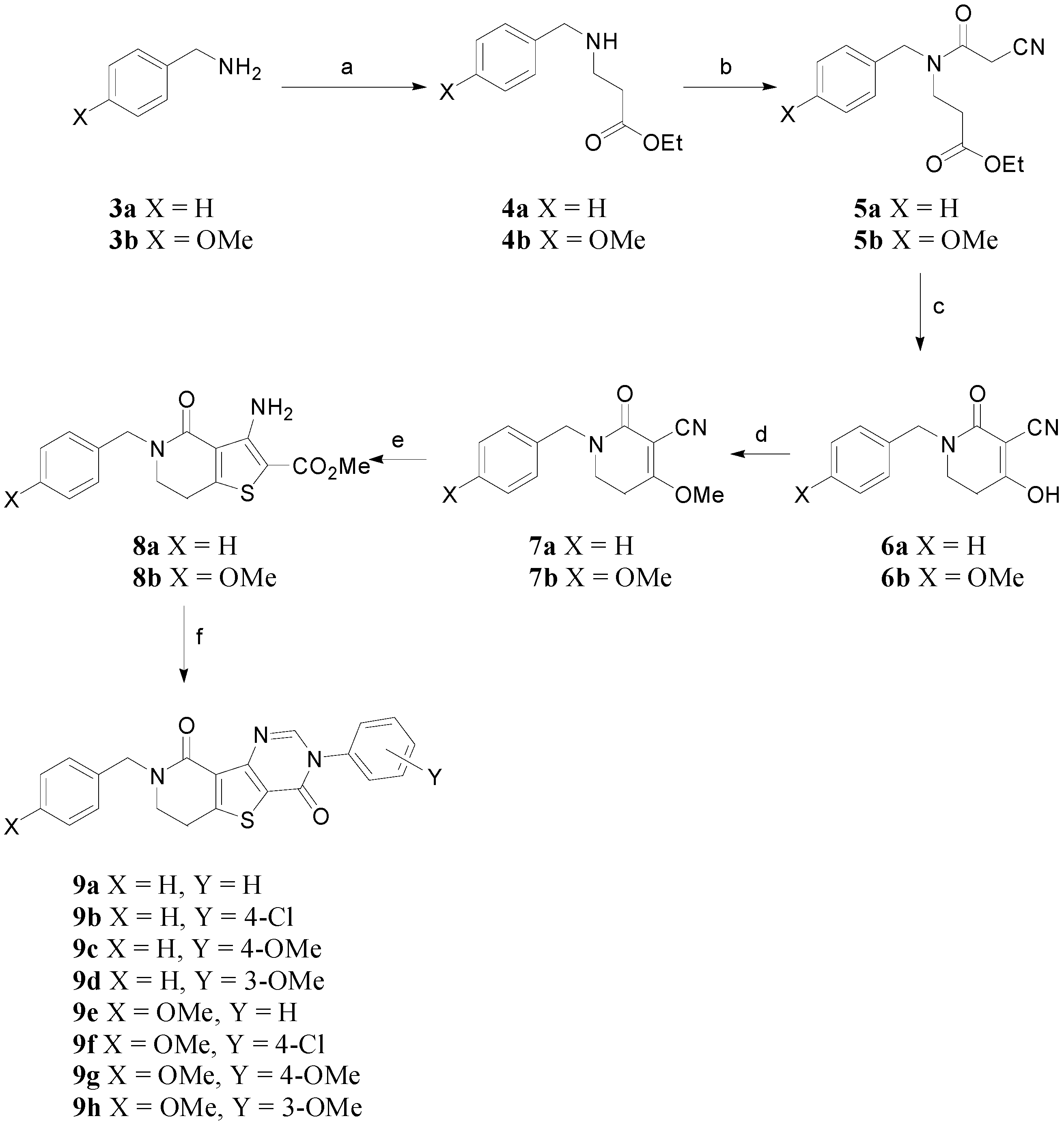
Ethyl 3-(benzylamino)propanoate (4a). To a solution of benzylamine 3a (2.6 g, 24.0 mmol) in H2O (100 mL) were added ethyl acrylate (2 g, 20.0 mmol), and Cu(OAc)2 (182 mg, 1.0 mmol). The resulting mixture was stirred at room temperature for 6 h. After termination of the reaction, the solution was extracted with EtOAc, dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (n-hexane-EtOAc = 1:1) to afford the desired product 4a (2.34 g, 11.3 mmol, 47% yield) as a yellow oil: 1H-NMR (300 MHz, CDCl3) δ 7.36–7.23 (m, 5H), 4.14 (q, J = 7.2 Hz, 2H), 3.81 (s, 2H), 2.90 (t, J = 6.3 Hz, 2H), 2.53 (t, J = 6.3 Hz, 2H), 1.66 (s, 1H), 1.25 (t, J = 7.2 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ 172.79, 140.17, 128.42, 128.09, 126.96, 60.41, 53.77, 44.51, 34.79, 14.23; LC/MS (ESI+): m/z: calcd for C12H17NO2: 207.27, [M+H]+; found: 208.00.
Ethyl 3-((4-methoxybenzyl)amino)propanoate (4b). Following the same procedure used for the synthesis of 4a, the reaction of (4-methoxyphenyl)methanamine 3b (7.8 mL, 60.0 mmol), ethyl acrylate (5 g, 50.0 mmol), and Cu(OAc)2 (454 mg, 2.5 mmol) gave the title compound 4b (6.72 g, 28.3 mmol, 57% yield) as a brown oil: 1H-NMR (400 MHz, CDCl3) δ 7.24–7.22 (m, 2H), 6.87–6.85 (m, 2H), 4.13 (q, J = 7.1 Hz, 2H), 3.79 (s, 3H), 3.74 (s, 2H), 2.88 (t, J = 6.4 Hz, 2H), 2.52 (t, J = 6.4 Hz, 2H), 1.63 (s, 1H), 1.25 (t, J = 7.2 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ 172.80, 158.66, 132.30, 129.26, 113.81, 60.40, 55.26, 53.16, 44.41, 34.78, 14.22; LC/MS (ESI+): m/z: calcd for C13H19NO3: 237.30, [M+H]+; found: 238.30.
Ethyl 3-(N-benzyl-2-cyanoacetamido)propanoate (5a). DCC (1.39 g, 6.75 mmol) were added to the a solution of ethyl 3-(benzylamino)propanoate 4a (1 g, 4.82 mmol), cyanoacetic acid (492 mg, 5.79 mmol), and HOBt (782 mg, 5.79 mmol) in MC (30 mL) at 0 °C. The solution was stirred at room temperature for 12 h. After termination of the reaction, the solution was filtered. The filtrate was washed with NaHCO3, and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (n-hexane- EtOAc = 1:2) to afford the desired product 5a (858 mg, 3.13 mmol, 65% yield) as a yellow oil: 1H NMR (300 MHz, CDCl3) δ 7.42–7.15 (m, 5H), 4.62 (d, J = 2.7 Hz, 2H), 4.12 (q, J = 7.2 Hz, 2H), 3.87 (s, 1H), 3.70 (t, J = 6.6 Hz, 1H), 3.54 (t, J = 6.3 Hz, 1H), 3.47 (s, 1H), 2.66 (t, J = 6.6 Hz, 1H), 2.56 (t, J = 6.6 Hz, 1H), 1.28–1.22 (m, 3H); 13C-NMR (100MHz, CDCl3) (isomers) δ 171.71, 171.07, 162.63, 162.57, 136.24, 135.39, 129.34, 128.86, 128.23, 127.95, 127.85, 126.08, 114.42, 113.92, 61.27, 60.81, 52.94, 48.35, 44.11, 43.00, 32.55, 32.51, 25.35, 25.23, 14.16, 14.10; LC/MS (ESI+): m/z: calcd for C15H18N2O3: 274.32, [M+H]+; found: 275.15.
Ethyl 3-(2-cyano-N-(4-methoxybenzyl)acetamido)propanoate (5b). Following the same procedure used for the synthesis of 5a, the reaction of ethyl 3-((4-methoxybenzyl)amino)propanoate 4b (6.72 g, 28.3 mmol), cyanoacetic acid (2.89 g, 34.0 mmol), DCC (8.17 g, 39.6 mmol) and HOBt (4.59 g, 34.0 mmol) in MC (125 mL) gave the title compound 5b (3.99 g, 13.1 mmol, 46% yield) as a yellow oil: 1H-NMR (300 MHz, CDCl3) δ 7.13 (dd, J = 8.6, 26.1 Hz, 2H), 6.88 (dd, J = 8.6, 16.6 Hz, 2H), 4.55 (s, 2H), 4.12 (q, J = 7.1 Hz, 2H), 3.83–3.80 (m, 4H), 3.67 (t, J = 6.6 Hz, 1H), 3.51 (t, J = 6.4 Hz, 1H), 3.47 (s, 1H), 2.64 (t, J = 6.7 Hz, 1H), 2.56 (t, J = 6.4 Hz, 1H), 1.28–1.22 (m, 3H); 13C-NMR (100 MHz, CDCl3) (isomers) δ 171.70, 171.07, 162.48, 162.41, 159.53, 159.29, 129.46, 128.26, 127.52, 127.13, 114.70, 114.43, 114.22, 113.99, 61.25, 60.79, 55.37, 55.29, 52.44, 47.70, 43.81, 42.69, 32.51, 25.36, 25.24, 14.15, 14.09; LC/MS (ESI+): m/z: calcd for C16H20N2O4: 304.35, [M+H]+; found: 305.15.
Benzyl-4-hydroxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (6a). To a solution of ethyl 3-(N-benzyl-2-cyanoacetamido)propanoate 5a (850 mg, 3.10 mmol) in MeOH (20 mL) were added Amberlyst A-26 resin (1.5 g). The resulting mixture was stirred at room temperature for 16 h. After termination of the reaction, the resin was filtered and washed with MeOH. Then, the resin was added to the solution of TFA (2 mL) in MeOH (20 mL), and stirred at room temperature for 30 min. The mixture solution was filtered, and concentrated in vacuo to afford the desired product 6a (390 mg, 1.71 mmol, 55% yield) as a white solid: mp 225–227 °C; 1H-NMR (300 MHz, DMSO-d6) δ 7.37–7.24 (m, 5H), 4.51 (s, 2H), 3.30 (t, J = 7.2 Hz, 2H), 2.68 (t, J = 6.9 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 181.05, 163.16, 137.98, 128.97, 128.05 127.60, 115.57, 83.49, 49.35, 42.82, 29.01; LC/MS (ESI+): m/z: calcd for C13H12N2O2: 228.25, [M+H]+; found: 229.15.
4-Hydroxy-1-(4-methoxybenzyl)-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (6b). Following the same procedure used for the synthesis of 6a, the reaction of ethyl 3-(2-cyano-N-(4-methoxybenzyl)acetamido)propanoate 5b (200 mg, 0.66 mmol), Amberlyst A-26 resin (354 mg), and TFA (2 mL) in MeOH (10 × 2 mL) gave the title compound 6b (108.7 mg, 0.42 mmol, 64% yield) as a white solid: mp 170–173 °C; 1H-NMR (400 MHz, DMSO-d6) δ 7.30–7.26 (m, 2H), 7.01–6.98 (m, 2H), 4.53 (s, 2H), 3.83 (s, 3H), 3.36 (t, J = 7.1 Hz, 2H), 2.74 (t, J = 7.0 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 180.87, 163.02, 158.95, 129.82, 129.56, 115.54, 114.37, 83.59, 55.52, 48.70, 42.53, 28.93; LC/MS (ESI+): m/z: calcd for C14H14N2O3: 258.28, [M+H]+; found: 259.15.
1-Benzyl-4-methoxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (7a). To a solution of benzyl-4-hydroxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile 6a (100 mg, 0.44 mmol) and THF (3 mL), NaH (21 mg, 0.88 mmol) was added and the reaction mixture was stirred at room temperature for 2 h. Then dimethylsulfate (0.071 mL, 0.75 mmol) was added to the mixture and the resulting solution was stirred at 40 °C for 12 h. The mixture was quenched with H2O and evaporated. Then it was diluted with EtOAc, extracted with EtOAc, and dried over MgSO4 and concentrated, the crude product was purified by silica gel column chromatography (n-hexane-EtOAc = 1:1) to give product 7a (67 mg, 0.28 mmol, 63% yield) as a white solid: mp 153–157 °C; 1H-NMR (300 MHz, CDCl3) δ 7.37–7.26 (m, 5H), 4.62 (s, 2H), 4.14 (s, 3H), 3.33 (t, J = 6.9 Hz, 2H), 2.61 (t, J = 6.9 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 177.55, 162.08, 136.66, 128.77, 128.19, 127.76, 114.15, 86.77, 58.13, 49.85, 42.07, 26.91; LC/MS (ESI+): m/z: calcd for C14H14N2O2: 242.28, [M+H]+; found: 243.15.
4-Methoxy-1-(4-methoxybenzyl)-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (7b). Following the same procedure used for the synthesis of 7a, the reaction of 4-hydroxy-1-(4-methoxybenzyl)-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile 6b (1.74 g, 6.70 mmol), NaH (321 mg, 13.4 mmol), and dimethylsulfate (1.1 ml, 11.4 mmol) gave the title compound 7b (677 mg, 2.45 mmol, 37% yield) as a white solid: mp 145–148 °C; 1H-NMR (300 MHz, CDCl3) δ 7.23 (d, J = 8.1 Hz, 2H), 6.88 (d, J = 8.4 Hz, 2H), 4.57 (s, 2H), 4.14 (s, 3H), 3.82 (s, 3H), 3.35 (t, J = 6.7 Hz, 2H), 2.65 (t, J = 6.7 Hz, 2H); 13C-NMR (75 MHz, CDCl3) δ 177.20, 161.95, 159.28, 129.62, 128.73, 114.16, 86.85, 58.14, 55.31, 49.29, 41.87, 27.10; LC/MS (ESI+): m/z: calcd for C15H16N2O3: 272.30, [M+H]+; found: 273.15.
Methyl 3-amino-5-benzyl-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate (8a). NaOMe (5M in MeOH, 36.0 mL, 17.9 mmol) in MeOH followed by methyl thioglycolate (1.8 mL, 20.5 mmol) were added to 1-benzyl-4-methoxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile 7a (3.1 g, 12.8 mmol). The reaction mixture was stirred and refluxed at 65 °C for 12 h. After the reaction, the mixture was filtered using Celite and washed with MC. The crude compound was then purified by by silica gel column chromatography (n-hexane-EtOAc = 4:1) to give white solid product 8a (2.44 g, 7.38 mmol, 58% yield) as a yellowish solid: mp 110–112 °C; 1H-NMR (300 MHz, CDCl3) δ 7.37–7.27 (m, 5H), 6.91 (brs, 2H), 4.70 (s, 2H), 3.82 (s, 3H), 3.53 (t, J = 6.9 Hz, 2H), 2.93 (t, J = 6.9 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 164.55, 162.73, 153.78, 151.00, 137.12, 128.77, 127.99, 127.63, 119.74, 96.83, 51.16, 49.03, 45.82, 25.00; LC/MS (ESI+): m/z: calcd for C16H16N2O3S: 316.38, [M+H]+; found: 317.05.
Methyl 3-amino-5-(4-methoxybenzyl)-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate (8b). Following the same procedure used for the synthesis of 8a, the reaction of 4-methoxy-1-(4-methoxybenzyl)-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile 7b (354 mg, 1.30 mmol), NaOMe (5M in MeOH, 0.40 mL, 2.08 mmol), and methyl thioglycolate (0.21 mL, 2.34 mmol) gave the title compound 8b (281 mg, 0.81 mmol, 62% yield) as a yellow oil: 1H-NMR (400 MHz, CDCl3) δ 7.24–7.22 (m, 2H), 6.88–6.86 (m, 2H), 4.62 (s, 2H), 3.82 (s, 3H), 3.80 (s, 3H), 3.50 (t, J = 6.9 Hz, 2H), 2.91 (t, J = 6.9 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 164.52, 162.63, 159.13, 153.74, 150.99, 129.37, 129.13, 119.77, 114.11, 96.80, 55.28, 51.12, 48.38, 45.59, 24.95; LC/MS (ESI+): m/z: calcd for C17H18N2O4S: 346.40, [M+H]+; found: 347.05.
8-Benzyl-3-phenyl-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9a). To a pressure bottle containing methyl 3-amino-5-benzyl-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8a (1g, 3.03 mmol), CH(OEt)3 (10 mL) was added, followed by aniline (0.524 mL, 5.75 mmol) and AcOH (1 mL). The reaction mixture was stirred and refluxed at 160 °C for 18 h. After the reaction, the mixture was evaporated then solidified with EtOAc and Et2O. The produced solid was filtered and dried in vacuo to give the title product 9a (743 mg, 1.92 mmol, 63% yield) as a reddish solid: mp 172–173 °C; 1H-NMR (300 MHz, DMSO-d6) δ 8.44–7.51 (m, 5H), 7.39–7.26 (m, 5H), 4.71 (s, 2H), 3.62 (t, J = 6.6 Hz, 2H), 3.23 (t, J = 6.6 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 159.61, 156.61, 156.48, 154.82, 149.69, 138.26, 137.32, 129.71, 129.49, 129.20, 128.08, 128.02, 127.63, 126.01, 122.13, 49.19, 46.26, 25.79; HRMS (ESI TOF-mass) calcd for C22H17N3O2S [M+Na]+ 410.0934, found 410.0936; purity (HPLC) 83.91%, tR 18.00 min.
8-Benzyl-3-(4-chlorophenyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9b). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-benzyl-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8a (100 mg, 0.32 mmol), CH(OEt)3 (1 mL), 4-chloroaniline (72.7 mg, 0.57 mmol) and AcOH (0.1 mL) gave the title compound 9b (89 mg, 0.21 mmol, 66% yield) as a yellowish solid: mp 220–223 °C; 1H-NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 7.54–7.27 (m, 9H), 4.80 (s, 2H), 3.63 (t, J = 6.7 Hz, 2H), 3.13 (t, J = 6.8 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.06, 156.36, 155.24, 154.74, 148.31, 137.33, 135.55, 135.10, 129.93, 128.72, 128.46, 128.39, 127.65, 126.17, 122.65, 49.53, 45.64, 25.89; HRMS (ESI TOF-mass) calcd for C22H16ClN3O2S [M+Na]+ 444.0544, found 444.0545; purity (HPLC) 100.00%, tR 19.21 min.
8-Benzyl-3-(4-methoxyphenyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9c). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-benzyl-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8a (100 mg, 0.32 mmol), CH(OEt)3 (1 mL), p-anisidine (72.7 mg, 0.57 mmol) and AcOH (0.1 mL) gave the title compound 9c (44 mg, 0.11 mmol, 33% yield) as a reddish solid: mp 223–225 °C; 1H-NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 7.40–7.04 (m, 9H), 4.81 (s, 2H), 3.87 (s, 3H), 3.63 (t, J = 6.7 Hz, 2H), 3.14 (t, J = 6.7 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.19, 160.17, 156.84, 154.92, 154.81, 149.05, 137.41, 129.35, 128.71, 128.39, 128.23, 127.61, 126.13, 122.79, 114.88, 55.64, 49.48, 45.64, 25.89; HRMS (ESI TOF-mass) calcd for C23H19N3O3S [M+Na]+ 440.1039, found 440.1039; purity (HPLC) 100.00%, tR 18.09 min.
8-Benzyl-3-(3-methoxyphenyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9d). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-benzyl-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8a (200 mg, 0.63 mmol), CH(OEt)3 (2 mL), m-anisidine (135 μL, 1.21 mmol) and AcOH (0.2 mL) gave the title compound 9d (53 mg, 0.13 mmol, 20% yield) as a white solid: mp 223–226 °C; 1H-NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 7.47–6.95 (m, 9H), 4.81 (s, 2H), 3.85 (s, 3H), 3.63 (t, J = 6.8 Hz, 2H), 3.14 (t, J = 6.7 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.43, 160.17, 156.50, 154.98, 154.78, 148.72, 137.72, 137.38, 130.45, 128.72, 128.39, 127.63, 126.18, 122.86, 119.15, 115.39, 112.93, 55.60, 49.49, 45.63, 25.91; HRMS (ESI TOF-mass) calcd for C23H19N3O3S [M+Na]+ 440.1039, found 440.1038; purity (HPLC) 99.64%, tR 18.31 min.
8-(4-Methoxybenzyl)-3-phenyl-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9e). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-(4-methoxybenzyl)-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8b (118 mg, 0.34 mmol), CH(OEt)3 (1.2 mL), aniline (62 μL, 0.68 mmol) and AcOH (0.12 mL) gave the title compound 9e (95 mg, 0.23 mmol, 67% yield) as a yellowish solid: mp 227–230 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.27 (s, 1H), 7.58–6.85 (m, 9H), 4.73 (s, 2H), 3.79 (s, 3H), 3.61 (t, J = 6.7 Hz, 2H), 3.12 (t, J = 6.8 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.16, 159.18, 156.56, 154.96, 154.78, 148.80, 136.70, 134.10, 129.76, 129.70, 129.43, 127.06, 126.22, 122.87, 114.09, 55.31, 48.87, 45.43, 25.91; HRMS (ESI TOF-mass) calcd for C23H19N3O3S [M+Na]+ 440.1039, found 440.1039; purity (HPLC) 98.75%, tR 17.88 min.
3-(4-Chlorophenyl)-8-(4-methoxybenzyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9f). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-(4-methoxybenzyl)-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8b (100 mg, 0.29 mmol), CH(OEt)3 (1 mL), 4-chloroaniline (74 mg, 0.58 mmol) and AcOH (0.1 mL) gave the title compound 9f (68 mg, 0.15 mmol, 52% yield) as a yellowish solid: mp 269–272 °C; 1H-NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.55–6.86 (m, 8H), 4.74 (s, 2H), 3.80 (s, 3H), 3.62 (t, J = 6.7 Hz, 2H), 3.13 (t, J = 6.7 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 159.98, 159.17, 156.36, 155.19, 154.72, 148.27, 135.53, 135.11, 129.91, 129.77, 129.42, 128.47, 126.24, 122.61, 114.08, 55.31, 48.92, 45.46, 25.88; HRMS (ESI TOF-mass) calcd for C23H18ClN3O3S [M+Na]+ 474.0650, found 474.0652; purity (HPLC) 100.00%, tR 19.03 min.
8-(4-Methoxybenzyl)-3-(4-methoxyphenyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9g). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-(4-methoxybenzyl)-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8b (100 mg, 0.29 mmol), CH(OEt)3 (1 mL), p-anisidine (73 mg, 0.58 mmol) and AcOH (0.1 mL) gave the title compound 9g (67 mg, 0.15 mmol, 51% yield) as a reddish solid: mp 225–228 °C; 1H-NMR (400 MHz, CDCl3) δ 8.25 (s, 1H), 7.33–7.30 (m, 4H), 7.06–6.84 (m, 4H), 4.73 (s, 2H), 3.86 (s, 3H), 3.78 (s, 3H), 3.60 (t, J = 6.7 Hz, 2H), 3.12 (t, J = 6.7 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.18, 160.16, 159.17, 156.85, 154.83, 154.82, 149.04, 129.76, 129.47, 129.35, 128.20, 126.23, 122.82, 114.89, 114.08, 55.63, 55.31, 48.86, 45.44, 25.91; HRMS (ESI TOF-mass) calcd for C24H21N3O4S [M+Na]+ 470.1145, found 470.1146; purity (HPLC) 100.00%, tR 17.95 min.
8-(4-Methoxybenzyl)-3-(3-methoxyphenyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9h). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-(4-methoxybenzyl)-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8b (100 mg, 0.29 mmol), CH(OEt)3 (1 mL), m-anisidine (65 μL, 0.58 mmol) and AcOH (0.1 mL) gave the title compound 9h (66 mg, 0.15 mmol, 51% yield) as a yellowish solid: mp 171–175 °C; 1H-NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 7.46–6.84 (m, 8H), 4.74 (s, 2H), 3.85 (s, 3H), 3.79 (s, 3H), 3.61 (t, J = 6.7 Hz, 2H), 3.12 (t, J = 6.8 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.42, 160.12, 159.16, 156.51, 154.98, 154.72, 148.77, 137.71, 130.44, 129.76, 129.44, 126.20, 122.81, 119.15, 115.41, 114.08, 112.92, 55.60, 55.31, 48.87, 45.44, 25.89; HRMS (ESI TOF-mass) calcd for C24H21N3O4S [M+Na]+ 470.1145, found 470.1146; purity (HPLC) 98.23%, tR 18.13 min.






{kind=link}
{kind=link}
{kind=link}

