Abstract
A novel molecular scaffold, dihydropyridothienopyrimidin-4,9-dione, was synthesized from benzylamine or p-methoxybenzylamine in six steps involving successive ring closure to form a fused ring system composed of dihydropyridone, thiophene and pyrimidone. The pharmacological versatility of the dihydropyridothenopyrimidin-4,9-dione scaffold was demonstrated by inhibitory activity against metabotropic glutamate receptor subtype 1 (mGluR1), which shows that the title compounds can serve as an interesting scaffold for the discovery of potential bioactive molecules for the treatment of human diseases.
1. Introduction
Much attention has been paid to the thienopyrimidine and thienopyridine scaffolds due to their notable biological and pharmacological activities (Figure 1) [1,2,3,4,5,6]. Thienopyrimidines, like (R)-N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)-6-(pyrrolidin-2-ylethynyl)thieno[3,2-d]pyrimidin-4-amine and 6-(1-benzyl-1H-indol-3-yl)-2-(piperidin-1-ylmethyl)thieno[3,2-d]pyrimidin-4(3H)-one, show potent anticancer activity by inhibition of epidermal growth factor receptor (EGFR) [1] and vascular endothelial growth factor receptor-2 (VEGFR-2) [2], respectively. On the other hand, thienopyridine derivatives such as ticlopidine, clopidogrel, and prasugrel block P2Y12 receptors and thus inhibit platelet activation and aggregation [3,4,5,6].
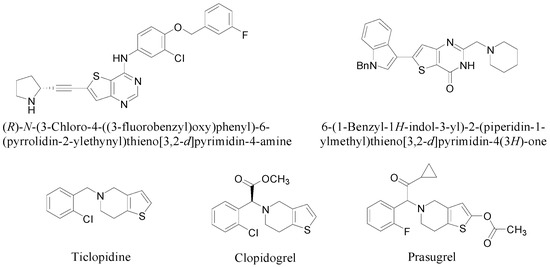
Figure 1.
Bioactive thienopyrimidine and thienopyridine derivatives.
Figure 1.
Bioactive thienopyrimidine and thienopyridine derivatives.
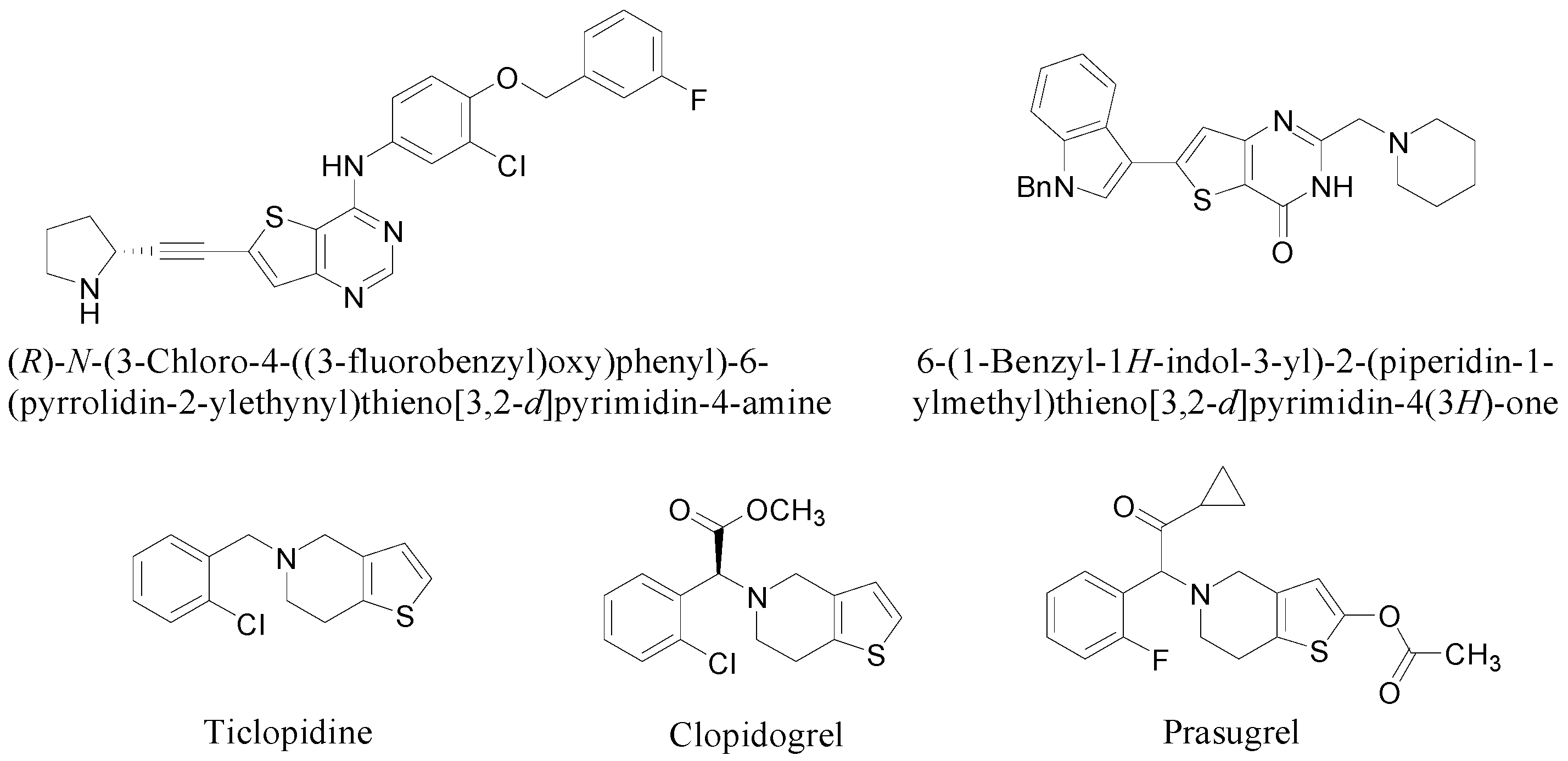
Accordingly, combining both the thienopyrimidine and thienopyridine moieties in the same molecular framework (i.e., pyridothienopyrimidine 1, Figure 2) has served as an attractive strategy for designing a novel scaffold with more favorable pharmacological properties [7]. In this study, as a part of our ongoing efforts to synthesize fused tricyclic heterocycles, dihydropyridinothienopyrimidin-4,9-dione derivatives 2 were prepared starting from benzylamine or p-methoxybenzylamine in six steps involving successive ring formations. Also, to demonstrate the biological and pharmacological versatility, the synthesized compounds were biologically evaluated against metabotropic glutamate receptor subtype 1 (mGluR1) which is one of molecular targets for treatment of neuropathic pain.
Figure 2.
Pyridothienopyrimidine (1) and dihydropyridothienopyrimidin-4,9-diones 2.
Figure 2.
Pyridothienopyrimidine (1) and dihydropyridothienopyrimidin-4,9-diones 2.
2. Results and Discussion
To synthesize the target compounds 2, the reaction sequence shown in Scheme 1 was used. Benzylamines 3a and 3b were used as starting materials. Thus, benzylamine 3a or p-methoxybenzylamine 3b were treated with ethyl acrylate in the presence of catalytic Cu(OAc)2 to give the corresponding β-aminoester 4a or 4b (47% or 57% yield, respectively). The β-aminoesters 4a and 4b were coupled with cyanoacetic acid using N,Nʹ-dicyclohexylcarbodimide (DCC) and 1-hydroxybenzotriazole (HOBt) in methylene chloride to afford tertiary cyanoacetamides 5a and 5b in 46%~63% yields.
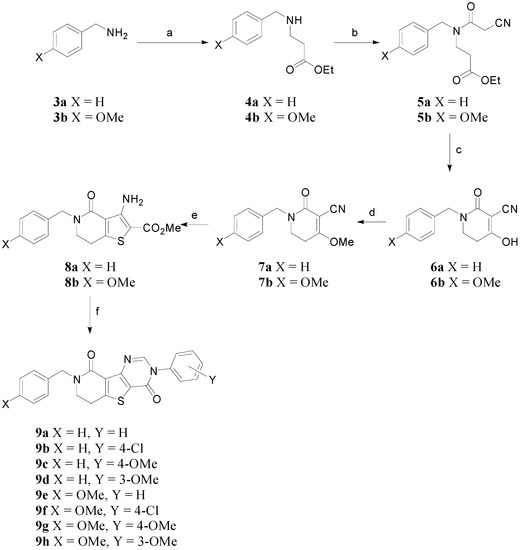
Scheme 1.
Synthesis of N3-aryl-N8-benzyldihydropyridothienopyrimidin-4,9-diones 9a–9h.
Scheme 1.
Synthesis of N3-aryl-N8-benzyldihydropyridothienopyrimidin-4,9-diones 9a–9h.
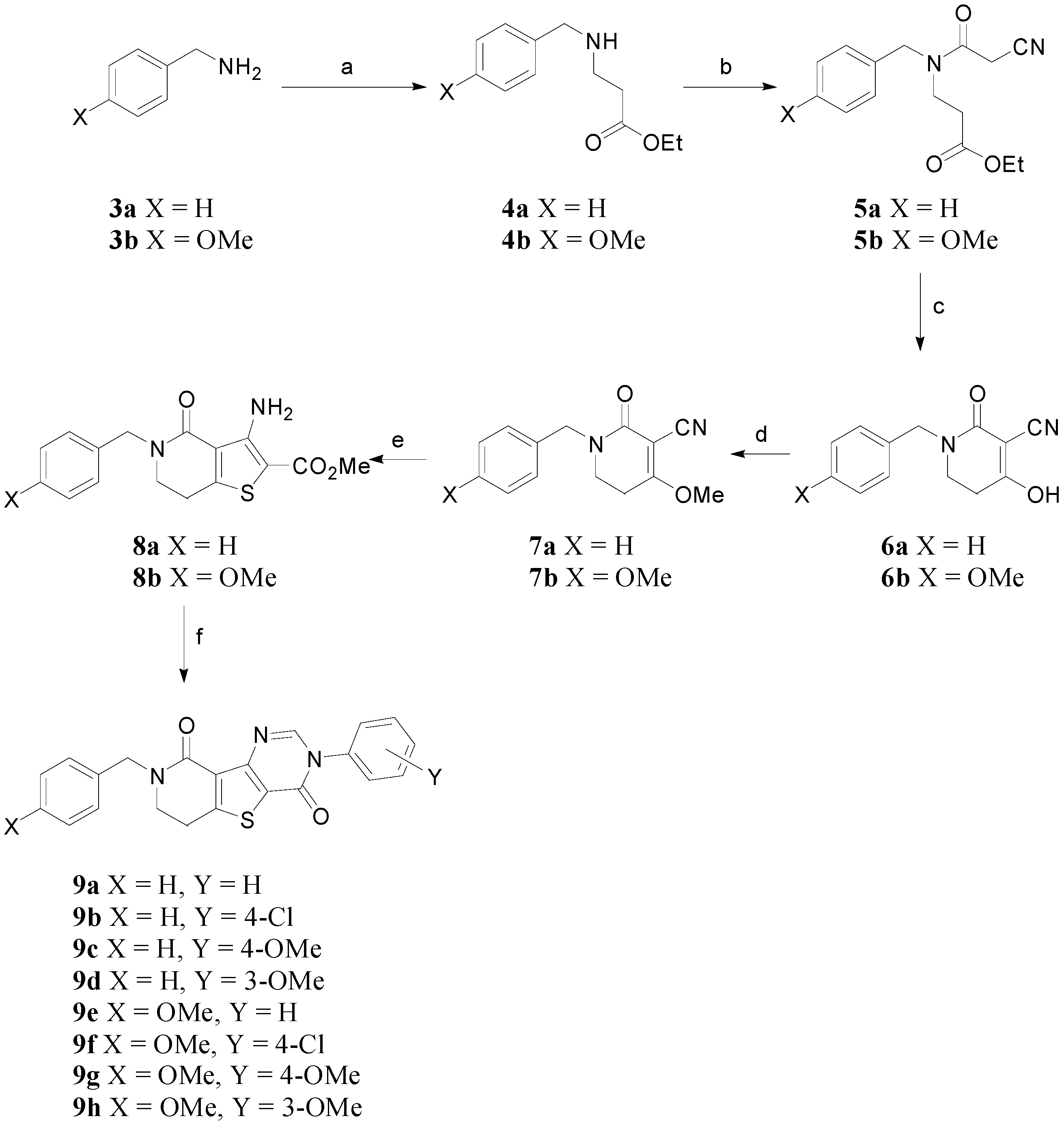
Reagents and conditions: (a) ethyl acrylate, Cu(OAc)2, H2O, rt; (b) cyanoacetic acid, DCC, HOBt, DCM, 0 °C to rt; (c) Amberlyst A-26 resin, MeOH, rt; (d) Me2SO4, NaH, THF, rt to 40 °C; (e) HSCH2CO2Me, NaOMe, MeOH, 65 °C; (f) anilines, CH(OEt)3, AcOH, 160 °C, sealed tube.
To form the dihydropyridone ring, the cyanoacetamides 5a and 5b underwent modified Dieckmann condensation under acidic conditions using Amberlyst A-26 resin as catalyst to give 6a and 6b in 55%~64% yields [8]. The hydroxy groups of the compounds 6a and 6b were methylated by dimethyl sulfate and NaH (37%~63% yields). The resulting methylated compounds 7a and 7b were then subjected to a series of heterocyclization reactions [9,10] to give the desired dihydropyridothienopyrimidin-4,9-diones 9a~9h in 20%~67% combined yields.
The synthesized compounds 9a~9h were biologically evaluated against Chem-3 cells with stably expressing mGluR1 which has been considered as a potential target for treating neuropathic pain [11], and the results are summarized in Table 1. Percent inhibitions (%-inhibitions) for all the synthesized compounds were measured at 1 μM. Overall, the dihydropyridothienopyrimidin-4,9-dione derivatives showed low to moderate inhibitory activity compared with the reference compound (93.37% inhibition) [9]: the compounds 9a, 9d, and 9f showed moderate inhibitory activity against mGluR1 with 23.04%, 16.54%, and 18.47% inhibition, respectively, while others showed negligible inhibitory activities.

Table 1.
Inhibitory activities of the synthesized dihydropyridothienopyrimidin-4,9-diones 9 against mGluR1. 
| Entry | Compound | X | Y | %Inhibition at 1 μM a |
|---|---|---|---|---|
| 1 | 9a | H | H | 23.04% |
| 2 | 9b | H | 4-Cl | −1.59% |
| 3 | 9c | H | 4-OMe | 3.15% |
| 4 | 9d | H | 3-OMe | 16.54% |
| 5 | 9e | OMe | H | 8.29% |
| 6 | 9f | OMe | 4-Cl | 18.47% |
| 7 | 9g | OMe | 4-OMe | 10.35% |
| 8 | 9h | OMe | 3-OMe | 11.86% |
| 9 | Reference compound b | 93.37% | ||
a: %-Inhibition of compounds at 1 μM against mGluR1; b: the reference compound is shown below. 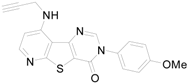
3. Experimental Section
3.1. General Methods
All reactions were carried out under dry nitrogen or argon unless otherwise indicated. Commercially available reagents were used without further purification. Solvents and gases were dried according to standard procedures. Organic solvents were evaporated with reduced pressure using a rotary evaporator. Analytical thin layer chromatography (TLC) was performed using glass plates precoated with silica gel (0.25 mm). TLC plates were visualized by exposure to UV light (UV), and then were visualized with a KMnO4 stain followed by brief heating on hot plate. Flash column chromatography was performed using silica gel 60 (230–400 mesh, Merck, Kenilworth, NJ, USA) with the indicated solvents. The melting points of solid products was measured by using capillary tubes for the determination of melting points (Marienfeld-Superior, Lauda-Königshofen, Germany) and an automated melting point system (OptiMelt, Stanford Research Systems, Inc. Sunnyvale, CA, USA). The tubes containing solid compounds were inserted in OptiMelt, and the temperature was slowly raised from 100 °C to 400 °C. 1H- and 13C-NMR spectra were recorded on Bruker 300 or 400 spectrometers (Bruker Co. Billerica, MA, USA). 1H-NMR spectra are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = doublet of doublet, td = triplet of doublet, brs = broad singlet, brt = broad triplet), integration, and coupling constant (J) in Hertz (Hz). 1H-NMR chemical shifts are reported relative to CDCl3 (7.26 ppm). 13C-NMR was recorded relative to the central line of CDCl3 (77.0 ppm). HRMS analyses were performed on Bruker compact ESI+ positive mode. HPLC purifications were performed on an Alliance Waters 2489 UV/Visible detector, e2695 Separations module using a Waters Xterra prep RP-18 10 μM, 10 × 250 mm column (Waters Inc. Milford, MA, USA) and a 30%–100% acetonitrile in water solvent gradient.
Ethyl 3-(benzylamino)propanoate (4a). To a solution of benzylamine 3a (2.6 g, 24.0 mmol) in H2O (100 mL) were added ethyl acrylate (2 g, 20.0 mmol), and Cu(OAc)2 (182 mg, 1.0 mmol). The resulting mixture was stirred at room temperature for 6 h. After termination of the reaction, the solution was extracted with EtOAc, dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (n-hexane-EtOAc = 1:1) to afford the desired product 4a (2.34 g, 11.3 mmol, 47% yield) as a yellow oil: 1H-NMR (300 MHz, CDCl3) δ 7.36–7.23 (m, 5H), 4.14 (q, J = 7.2 Hz, 2H), 3.81 (s, 2H), 2.90 (t, J = 6.3 Hz, 2H), 2.53 (t, J = 6.3 Hz, 2H), 1.66 (s, 1H), 1.25 (t, J = 7.2 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ 172.79, 140.17, 128.42, 128.09, 126.96, 60.41, 53.77, 44.51, 34.79, 14.23; LC/MS (ESI+): m/z: calcd for C12H17NO2: 207.27, [M+H]+; found: 208.00.
Ethyl 3-((4-methoxybenzyl)amino)propanoate (4b). Following the same procedure used for the synthesis of 4a, the reaction of (4-methoxyphenyl)methanamine 3b (7.8 mL, 60.0 mmol), ethyl acrylate (5 g, 50.0 mmol), and Cu(OAc)2 (454 mg, 2.5 mmol) gave the title compound 4b (6.72 g, 28.3 mmol, 57% yield) as a brown oil: 1H-NMR (400 MHz, CDCl3) δ 7.24–7.22 (m, 2H), 6.87–6.85 (m, 2H), 4.13 (q, J = 7.1 Hz, 2H), 3.79 (s, 3H), 3.74 (s, 2H), 2.88 (t, J = 6.4 Hz, 2H), 2.52 (t, J = 6.4 Hz, 2H), 1.63 (s, 1H), 1.25 (t, J = 7.2 Hz, 3H); 13C-NMR (100 MHz, CDCl3) δ 172.80, 158.66, 132.30, 129.26, 113.81, 60.40, 55.26, 53.16, 44.41, 34.78, 14.22; LC/MS (ESI+): m/z: calcd for C13H19NO3: 237.30, [M+H]+; found: 238.30.
Ethyl 3-(N-benzyl-2-cyanoacetamido)propanoate (5a). DCC (1.39 g, 6.75 mmol) were added to the a solution of ethyl 3-(benzylamino)propanoate 4a (1 g, 4.82 mmol), cyanoacetic acid (492 mg, 5.79 mmol), and HOBt (782 mg, 5.79 mmol) in MC (30 mL) at 0 °C. The solution was stirred at room temperature for 12 h. After termination of the reaction, the solution was filtered. The filtrate was washed with NaHCO3, and brine, dried over MgSO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (n-hexane- EtOAc = 1:2) to afford the desired product 5a (858 mg, 3.13 mmol, 65% yield) as a yellow oil: 1H NMR (300 MHz, CDCl3) δ 7.42–7.15 (m, 5H), 4.62 (d, J = 2.7 Hz, 2H), 4.12 (q, J = 7.2 Hz, 2H), 3.87 (s, 1H), 3.70 (t, J = 6.6 Hz, 1H), 3.54 (t, J = 6.3 Hz, 1H), 3.47 (s, 1H), 2.66 (t, J = 6.6 Hz, 1H), 2.56 (t, J = 6.6 Hz, 1H), 1.28–1.22 (m, 3H); 13C-NMR (100MHz, CDCl3) (isomers) δ 171.71, 171.07, 162.63, 162.57, 136.24, 135.39, 129.34, 128.86, 128.23, 127.95, 127.85, 126.08, 114.42, 113.92, 61.27, 60.81, 52.94, 48.35, 44.11, 43.00, 32.55, 32.51, 25.35, 25.23, 14.16, 14.10; LC/MS (ESI+): m/z: calcd for C15H18N2O3: 274.32, [M+H]+; found: 275.15.
Ethyl 3-(2-cyano-N-(4-methoxybenzyl)acetamido)propanoate (5b). Following the same procedure used for the synthesis of 5a, the reaction of ethyl 3-((4-methoxybenzyl)amino)propanoate 4b (6.72 g, 28.3 mmol), cyanoacetic acid (2.89 g, 34.0 mmol), DCC (8.17 g, 39.6 mmol) and HOBt (4.59 g, 34.0 mmol) in MC (125 mL) gave the title compound 5b (3.99 g, 13.1 mmol, 46% yield) as a yellow oil: 1H-NMR (300 MHz, CDCl3) δ 7.13 (dd, J = 8.6, 26.1 Hz, 2H), 6.88 (dd, J = 8.6, 16.6 Hz, 2H), 4.55 (s, 2H), 4.12 (q, J = 7.1 Hz, 2H), 3.83–3.80 (m, 4H), 3.67 (t, J = 6.6 Hz, 1H), 3.51 (t, J = 6.4 Hz, 1H), 3.47 (s, 1H), 2.64 (t, J = 6.7 Hz, 1H), 2.56 (t, J = 6.4 Hz, 1H), 1.28–1.22 (m, 3H); 13C-NMR (100 MHz, CDCl3) (isomers) δ 171.70, 171.07, 162.48, 162.41, 159.53, 159.29, 129.46, 128.26, 127.52, 127.13, 114.70, 114.43, 114.22, 113.99, 61.25, 60.79, 55.37, 55.29, 52.44, 47.70, 43.81, 42.69, 32.51, 25.36, 25.24, 14.15, 14.09; LC/MS (ESI+): m/z: calcd for C16H20N2O4: 304.35, [M+H]+; found: 305.15.
Benzyl-4-hydroxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (6a). To a solution of ethyl 3-(N-benzyl-2-cyanoacetamido)propanoate 5a (850 mg, 3.10 mmol) in MeOH (20 mL) were added Amberlyst A-26 resin (1.5 g). The resulting mixture was stirred at room temperature for 16 h. After termination of the reaction, the resin was filtered and washed with MeOH. Then, the resin was added to the solution of TFA (2 mL) in MeOH (20 mL), and stirred at room temperature for 30 min. The mixture solution was filtered, and concentrated in vacuo to afford the desired product 6a (390 mg, 1.71 mmol, 55% yield) as a white solid: mp 225–227 °C; 1H-NMR (300 MHz, DMSO-d6) δ 7.37–7.24 (m, 5H), 4.51 (s, 2H), 3.30 (t, J = 7.2 Hz, 2H), 2.68 (t, J = 6.9 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 181.05, 163.16, 137.98, 128.97, 128.05 127.60, 115.57, 83.49, 49.35, 42.82, 29.01; LC/MS (ESI+): m/z: calcd for C13H12N2O2: 228.25, [M+H]+; found: 229.15.
4-Hydroxy-1-(4-methoxybenzyl)-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (6b). Following the same procedure used for the synthesis of 6a, the reaction of ethyl 3-(2-cyano-N-(4-methoxybenzyl)acetamido)propanoate 5b (200 mg, 0.66 mmol), Amberlyst A-26 resin (354 mg), and TFA (2 mL) in MeOH (10 × 2 mL) gave the title compound 6b (108.7 mg, 0.42 mmol, 64% yield) as a white solid: mp 170–173 °C; 1H-NMR (400 MHz, DMSO-d6) δ 7.30–7.26 (m, 2H), 7.01–6.98 (m, 2H), 4.53 (s, 2H), 3.83 (s, 3H), 3.36 (t, J = 7.1 Hz, 2H), 2.74 (t, J = 7.0 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 180.87, 163.02, 158.95, 129.82, 129.56, 115.54, 114.37, 83.59, 55.52, 48.70, 42.53, 28.93; LC/MS (ESI+): m/z: calcd for C14H14N2O3: 258.28, [M+H]+; found: 259.15.
1-Benzyl-4-methoxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (7a). To a solution of benzyl-4-hydroxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile 6a (100 mg, 0.44 mmol) and THF (3 mL), NaH (21 mg, 0.88 mmol) was added and the reaction mixture was stirred at room temperature for 2 h. Then dimethylsulfate (0.071 mL, 0.75 mmol) was added to the mixture and the resulting solution was stirred at 40 °C for 12 h. The mixture was quenched with H2O and evaporated. Then it was diluted with EtOAc, extracted with EtOAc, and dried over MgSO4 and concentrated, the crude product was purified by silica gel column chromatography (n-hexane-EtOAc = 1:1) to give product 7a (67 mg, 0.28 mmol, 63% yield) as a white solid: mp 153–157 °C; 1H-NMR (300 MHz, CDCl3) δ 7.37–7.26 (m, 5H), 4.62 (s, 2H), 4.14 (s, 3H), 3.33 (t, J = 6.9 Hz, 2H), 2.61 (t, J = 6.9 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 177.55, 162.08, 136.66, 128.77, 128.19, 127.76, 114.15, 86.77, 58.13, 49.85, 42.07, 26.91; LC/MS (ESI+): m/z: calcd for C14H14N2O2: 242.28, [M+H]+; found: 243.15.
4-Methoxy-1-(4-methoxybenzyl)-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (7b). Following the same procedure used for the synthesis of 7a, the reaction of 4-hydroxy-1-(4-methoxybenzyl)-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile 6b (1.74 g, 6.70 mmol), NaH (321 mg, 13.4 mmol), and dimethylsulfate (1.1 ml, 11.4 mmol) gave the title compound 7b (677 mg, 2.45 mmol, 37% yield) as a white solid: mp 145–148 °C; 1H-NMR (300 MHz, CDCl3) δ 7.23 (d, J = 8.1 Hz, 2H), 6.88 (d, J = 8.4 Hz, 2H), 4.57 (s, 2H), 4.14 (s, 3H), 3.82 (s, 3H), 3.35 (t, J = 6.7 Hz, 2H), 2.65 (t, J = 6.7 Hz, 2H); 13C-NMR (75 MHz, CDCl3) δ 177.20, 161.95, 159.28, 129.62, 128.73, 114.16, 86.85, 58.14, 55.31, 49.29, 41.87, 27.10; LC/MS (ESI+): m/z: calcd for C15H16N2O3: 272.30, [M+H]+; found: 273.15.
Methyl 3-amino-5-benzyl-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate (8a). NaOMe (5M in MeOH, 36.0 mL, 17.9 mmol) in MeOH followed by methyl thioglycolate (1.8 mL, 20.5 mmol) were added to 1-benzyl-4-methoxy-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile 7a (3.1 g, 12.8 mmol). The reaction mixture was stirred and refluxed at 65 °C for 12 h. After the reaction, the mixture was filtered using Celite and washed with MC. The crude compound was then purified by by silica gel column chromatography (n-hexane-EtOAc = 4:1) to give white solid product 8a (2.44 g, 7.38 mmol, 58% yield) as a yellowish solid: mp 110–112 °C; 1H-NMR (300 MHz, CDCl3) δ 7.37–7.27 (m, 5H), 6.91 (brs, 2H), 4.70 (s, 2H), 3.82 (s, 3H), 3.53 (t, J = 6.9 Hz, 2H), 2.93 (t, J = 6.9 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 164.55, 162.73, 153.78, 151.00, 137.12, 128.77, 127.99, 127.63, 119.74, 96.83, 51.16, 49.03, 45.82, 25.00; LC/MS (ESI+): m/z: calcd for C16H16N2O3S: 316.38, [M+H]+; found: 317.05.
Methyl 3-amino-5-(4-methoxybenzyl)-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate (8b). Following the same procedure used for the synthesis of 8a, the reaction of 4-methoxy-1-(4-methoxybenzyl)-2-oxo-1,2,5,6-tetrahydropyridine-3-carbonitrile 7b (354 mg, 1.30 mmol), NaOMe (5M in MeOH, 0.40 mL, 2.08 mmol), and methyl thioglycolate (0.21 mL, 2.34 mmol) gave the title compound 8b (281 mg, 0.81 mmol, 62% yield) as a yellow oil: 1H-NMR (400 MHz, CDCl3) δ 7.24–7.22 (m, 2H), 6.88–6.86 (m, 2H), 4.62 (s, 2H), 3.82 (s, 3H), 3.80 (s, 3H), 3.50 (t, J = 6.9 Hz, 2H), 2.91 (t, J = 6.9 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 164.52, 162.63, 159.13, 153.74, 150.99, 129.37, 129.13, 119.77, 114.11, 96.80, 55.28, 51.12, 48.38, 45.59, 24.95; LC/MS (ESI+): m/z: calcd for C17H18N2O4S: 346.40, [M+H]+; found: 347.05.
8-Benzyl-3-phenyl-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9a). To a pressure bottle containing methyl 3-amino-5-benzyl-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8a (1g, 3.03 mmol), CH(OEt)3 (10 mL) was added, followed by aniline (0.524 mL, 5.75 mmol) and AcOH (1 mL). The reaction mixture was stirred and refluxed at 160 °C for 18 h. After the reaction, the mixture was evaporated then solidified with EtOAc and Et2O. The produced solid was filtered and dried in vacuo to give the title product 9a (743 mg, 1.92 mmol, 63% yield) as a reddish solid: mp 172–173 °C; 1H-NMR (300 MHz, DMSO-d6) δ 8.44–7.51 (m, 5H), 7.39–7.26 (m, 5H), 4.71 (s, 2H), 3.62 (t, J = 6.6 Hz, 2H), 3.23 (t, J = 6.6 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6) δ 159.61, 156.61, 156.48, 154.82, 149.69, 138.26, 137.32, 129.71, 129.49, 129.20, 128.08, 128.02, 127.63, 126.01, 122.13, 49.19, 46.26, 25.79; HRMS (ESI TOF-mass) calcd for C22H17N3O2S [M+Na]+ 410.0934, found 410.0936; purity (HPLC) 83.91%, tR 18.00 min.
8-Benzyl-3-(4-chlorophenyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9b). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-benzyl-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8a (100 mg, 0.32 mmol), CH(OEt)3 (1 mL), 4-chloroaniline (72.7 mg, 0.57 mmol) and AcOH (0.1 mL) gave the title compound 9b (89 mg, 0.21 mmol, 66% yield) as a yellowish solid: mp 220–223 °C; 1H-NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 7.54–7.27 (m, 9H), 4.80 (s, 2H), 3.63 (t, J = 6.7 Hz, 2H), 3.13 (t, J = 6.8 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.06, 156.36, 155.24, 154.74, 148.31, 137.33, 135.55, 135.10, 129.93, 128.72, 128.46, 128.39, 127.65, 126.17, 122.65, 49.53, 45.64, 25.89; HRMS (ESI TOF-mass) calcd for C22H16ClN3O2S [M+Na]+ 444.0544, found 444.0545; purity (HPLC) 100.00%, tR 19.21 min.
8-Benzyl-3-(4-methoxyphenyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9c). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-benzyl-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8a (100 mg, 0.32 mmol), CH(OEt)3 (1 mL), p-anisidine (72.7 mg, 0.57 mmol) and AcOH (0.1 mL) gave the title compound 9c (44 mg, 0.11 mmol, 33% yield) as a reddish solid: mp 223–225 °C; 1H-NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 7.40–7.04 (m, 9H), 4.81 (s, 2H), 3.87 (s, 3H), 3.63 (t, J = 6.7 Hz, 2H), 3.14 (t, J = 6.7 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.19, 160.17, 156.84, 154.92, 154.81, 149.05, 137.41, 129.35, 128.71, 128.39, 128.23, 127.61, 126.13, 122.79, 114.88, 55.64, 49.48, 45.64, 25.89; HRMS (ESI TOF-mass) calcd for C23H19N3O3S [M+Na]+ 440.1039, found 440.1039; purity (HPLC) 100.00%, tR 18.09 min.
8-Benzyl-3-(3-methoxyphenyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9d). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-benzyl-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8a (200 mg, 0.63 mmol), CH(OEt)3 (2 mL), m-anisidine (135 μL, 1.21 mmol) and AcOH (0.2 mL) gave the title compound 9d (53 mg, 0.13 mmol, 20% yield) as a white solid: mp 223–226 °C; 1H-NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 7.47–6.95 (m, 9H), 4.81 (s, 2H), 3.85 (s, 3H), 3.63 (t, J = 6.8 Hz, 2H), 3.14 (t, J = 6.7 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.43, 160.17, 156.50, 154.98, 154.78, 148.72, 137.72, 137.38, 130.45, 128.72, 128.39, 127.63, 126.18, 122.86, 119.15, 115.39, 112.93, 55.60, 49.49, 45.63, 25.91; HRMS (ESI TOF-mass) calcd for C23H19N3O3S [M+Na]+ 440.1039, found 440.1038; purity (HPLC) 99.64%, tR 18.31 min.
8-(4-Methoxybenzyl)-3-phenyl-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9e). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-(4-methoxybenzyl)-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8b (118 mg, 0.34 mmol), CH(OEt)3 (1.2 mL), aniline (62 μL, 0.68 mmol) and AcOH (0.12 mL) gave the title compound 9e (95 mg, 0.23 mmol, 67% yield) as a yellowish solid: mp 227–230 °C; 1H-NMR (400 MHz, DMSO-d6) δ 8.27 (s, 1H), 7.58–6.85 (m, 9H), 4.73 (s, 2H), 3.79 (s, 3H), 3.61 (t, J = 6.7 Hz, 2H), 3.12 (t, J = 6.8 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.16, 159.18, 156.56, 154.96, 154.78, 148.80, 136.70, 134.10, 129.76, 129.70, 129.43, 127.06, 126.22, 122.87, 114.09, 55.31, 48.87, 45.43, 25.91; HRMS (ESI TOF-mass) calcd for C23H19N3O3S [M+Na]+ 440.1039, found 440.1039; purity (HPLC) 98.75%, tR 17.88 min.
3-(4-Chlorophenyl)-8-(4-methoxybenzyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9f). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-(4-methoxybenzyl)-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8b (100 mg, 0.29 mmol), CH(OEt)3 (1 mL), 4-chloroaniline (74 mg, 0.58 mmol) and AcOH (0.1 mL) gave the title compound 9f (68 mg, 0.15 mmol, 52% yield) as a yellowish solid: mp 269–272 °C; 1H-NMR (400 MHz, CDCl3) δ 8.24 (s, 1H), 7.55–6.86 (m, 8H), 4.74 (s, 2H), 3.80 (s, 3H), 3.62 (t, J = 6.7 Hz, 2H), 3.13 (t, J = 6.7 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 159.98, 159.17, 156.36, 155.19, 154.72, 148.27, 135.53, 135.11, 129.91, 129.77, 129.42, 128.47, 126.24, 122.61, 114.08, 55.31, 48.92, 45.46, 25.88; HRMS (ESI TOF-mass) calcd for C23H18ClN3O3S [M+Na]+ 474.0650, found 474.0652; purity (HPLC) 100.00%, tR 19.03 min.
8-(4-Methoxybenzyl)-3-(4-methoxyphenyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9g). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-(4-methoxybenzyl)-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8b (100 mg, 0.29 mmol), CH(OEt)3 (1 mL), p-anisidine (73 mg, 0.58 mmol) and AcOH (0.1 mL) gave the title compound 9g (67 mg, 0.15 mmol, 51% yield) as a reddish solid: mp 225–228 °C; 1H-NMR (400 MHz, CDCl3) δ 8.25 (s, 1H), 7.33–7.30 (m, 4H), 7.06–6.84 (m, 4H), 4.73 (s, 2H), 3.86 (s, 3H), 3.78 (s, 3H), 3.60 (t, J = 6.7 Hz, 2H), 3.12 (t, J = 6.7 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.18, 160.16, 159.17, 156.85, 154.83, 154.82, 149.04, 129.76, 129.47, 129.35, 128.20, 126.23, 122.82, 114.89, 114.08, 55.63, 55.31, 48.86, 45.44, 25.91; HRMS (ESI TOF-mass) calcd for C24H21N3O4S [M+Na]+ 470.1145, found 470.1146; purity (HPLC) 100.00%, tR 17.95 min.
8-(4-Methoxybenzyl)-3-(3-methoxyphenyl)-7,8-dihydropyrido[3',4':4,5]thieno[3,2-d]pyrimidine-4,9(3H,6H)-dione (9h). Following the same procedure used for the synthesis of 9a, the reaction of methyl 3-amino-5-(4-methoxybenzyl)-4-oxo-4,5,6,7-tetrahydrothieno[3,2-c]pyridine-2-carboxylate 8b (100 mg, 0.29 mmol), CH(OEt)3 (1 mL), m-anisidine (65 μL, 0.58 mmol) and AcOH (0.1 mL) gave the title compound 9h (66 mg, 0.15 mmol, 51% yield) as a yellowish solid: mp 171–175 °C; 1H-NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 7.46–6.84 (m, 8H), 4.74 (s, 2H), 3.85 (s, 3H), 3.79 (s, 3H), 3.61 (t, J = 6.7 Hz, 2H), 3.12 (t, J = 6.8 Hz, 2H); 13C-NMR (100 MHz, CDCl3) δ 160.42, 160.12, 159.16, 156.51, 154.98, 154.72, 148.77, 137.71, 130.44, 129.76, 129.44, 126.20, 122.81, 119.15, 115.41, 114.08, 112.92, 55.60, 55.31, 48.87, 45.44, 25.89; HRMS (ESI TOF-mass) calcd for C24H21N3O4S [M+Na]+ 470.1145, found 470.1146; purity (HPLC) 98.23%, tR 18.13 min.
3.2. mGluR1 Assay using FDSS6000
Chem-3 cells which stably express mGluR1 were purchased from Milipore Co. (Billerica, MA, USA). Cells were grown in DMEM medium supplemented with 10% (v/v) fetal bovine serum, penicillin (100 U/mL), streptomycin (100 μg/mL), and puromycin (10 μg/mL) at 37 °C in a humid atmosphere of 5% CO2 and 95% air. For calcium assay, cells were harvested and dispensed into 96-well black wall clear bottom plates at a density of 40,000 cells per a well. After 18 h of incubation, cells were treated with Calcium-5 assay reagent, which is prepared by manufacture’s instruction (Molecular Devices Co. Sunnyvale, CA, USA). During fluorescence-based FDSS6000 assay, mGluR1 was activated using a high concentration of l-glutamate (10 μM) in HBSS, and proper concentrations of synthesized compounds were treated to cells 75 s before mGluR activation. All data were collected and analyzed using FDSS6000 and related software (Hamamatsu Photonics, Hamamatsu, Japan).
4. Conclusions
In this proof-of-concept study, a synthetic protocol was devised to allow access to a fused tricyclic heterocycle composed of dihydropyridone, thiophene and pyrimidinone. Also, even though only a small set of the dihydropyridothienopyrimidin-4,9-dione derivatives were synthesized, promising biological activity was identified in the title compounds. Thus, by using variously substituted benzylamines and anilines, an extensive structure-activity relationship study is warranted to investigate the pharmacological versatility of the dihydropyridothienopyrimidin-4,9-dione derivatives.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/20/03/5074/s1.
Acknowledgments
This research was supported by the Basic Science Research Program (NRF-2013-R1A1A2A10009907) funded by the National Research Foundation of Korea (NRF). Additional funding was provided by the Korea Institute of Science and Technology (KIST) Institutional Program (2E24510, 2E25473, and 2E25240).
Author Contributions
YK, JT, D-JB, and HC designed research; YK and MK performed research; YK, MK, MP, and KDP analyzed the data; YK and HC wrote the paper. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wood, E.R.; Shewchuk, L.M.; Ellis, B.; Brignola, P.; Brashear, R.L.; Caferro, T.R.; Dickerson, S.H.; Dickson, H.D.; Donaldson, K.H.; Gaul, M.; et al. 6-Ethynylthieno[3,2-d]- and 6-ethynylthieno[2,3-e]pyrimidin-4-anilines as tunable covalent modifiers of ErbB kinases. Proc. Natl. Acad. Sci. USA 2008, 105, 2773–2778. [Google Scholar] [CrossRef] [PubMed]
- Perspicace, E.; Jouan-Hureaux, V.; Ragno, R.; Ballante, F.; Sartini, S.; la Motta, C.; da Settimo, F.; Chen, B.; Kersch, G.; Schneider, S.; et al. Design, synthesis and biological evaluation of new classes of thieno[3,2-d]pyrimidinone and thieno[1,2,3]triazine as inhibitor of vascular endothelial growth factor receptor-s (VEGFR-2). Eur. J. Med. Chem. 2013, 63, 765–781. [Google Scholar] [CrossRef] [PubMed]
- CAPRIE Streering Committee. A randomized, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPREI). Lancet 1996, 348, 1329–1339. [Google Scholar]
- Sabatine, M.S. Clopidogrel in ST-elevation myocardial infarction. Eur. Heart J. 2006, 8, G31–G34. [Google Scholar] [CrossRef]
- Aalla, S.; Gilla, G.; Metil, D.S.; Anumula, R.R.; Vummenthala, P.R.; Padi, P.R. Process improvements of prasugrel hydrochloride: An adenosine diphosphate receptor antagonist. Org. Process Res. Dev. 2012, 16, 240–243. [Google Scholar] [CrossRef]
- Aradi, D.; Rideg, O.; Vorobcsuk, A.; Magyarlaki, T.; Magyari, B.; Konyi, A.; Pinter, T.; Horvath, I.G.; Komocsi, A. Justification of 150 mg clopidogrel in patients with high on-clopidogrel platelet reactivity. Eur. J. Clin. Investig. 2012, 42, 384–392. [Google Scholar] [CrossRef]
- Abdel-Wadood, F.K.; Abdel-Monem, M.I.; Fahmy, A.M.; Geies, A.A. Synthesis, reactions, and biological activities of some new thieno[3,2-c]quinoline and pyrrolo[3,2-c]quinoline derivatives. Arch. Pharm. Chem. Life Sci. 2014, 347, 142–152. [Google Scholar] [CrossRef]
- Kulkarni, B.A.; Ganesan, A. Ion-exchange resins for combinatorial synthesis: 2,4-pyrrolidinediones by Dieckmann condensation. Angew. Chem. Int. Ed. Engl. 1997, 36, 2454–2455. [Google Scholar] [CrossRef]
- Sasikumar, T.K.; Qiang, L.; Burnett, D.A.; Greenlee, W.J.; Li, C.; Heimark, L.; Pramanik, B.; Grilli, M.; Bertorelli, R.; Lozza, G.; Reggiani, A. Tricyclic thienopyridine-pyrimidones/thienopyrimidine-pyrimidines as orally efficacious mGluR1 antagonists for neuropathic pain. Bioorg. Med. Chem. Lett. 2009, 19, 3199–3203. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, J.; Kim, S.; Ki, Y.; Seo, S.H.; Tae, J.; Ko, M.K.; Jang, H.S.; Lim, E.J.; Song, C.; et al. Novel thienopyrimidinones as mGluR1 antagonists. Eur. J. Med. Chem. 2014, 85, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Fundytus, M.E.; Yashpal, K.; Chabot, J.G.; Osborne, M.G.; Lefebvre, C.D.; Dray, A.; Henry, J.L.; Coderre, T.J. Knockdown of spinal metabotropic glutamate receptor 1 (mGluR1) alleviates pain and restores opioid efficacy after nerve injury in rats. Br. J. Pharmacol. 2001, 132, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).