2.2. Differential Scanning Calorimetry
The impact of antibiotic peptides on the phase behavior of phospholipids generally reflects their activity and provides a good hint regarding their mode of action [
38,
39]. Liposomes mimicking the cytoplasmic membrane of bacteria were composed of POPE and POPG at the ratio 70:30. Despite the fact that POPE can exhibit a hexagonal phase for temperatures above 70 °C, the combination with POPG resulted in vesicles, which remained in a stable lamellar phase in the studied temperature range of 3 °C to 75 °C, as the results of DSC revealed (
Figure 2). At 19.9 °C the transition from gel into a liquid crystalline phase was observed. The almost symmetrical peak indicated the good mixing of the phospholipids, and cooperative chain melting. A distinct alteration of the lipid phase behavior was observed when NKCS was added at a lipid:peptide molar ratio of 100:1, which corresponded to a concentration of 65 µM, well above the MIC in the antibacterial activity test discussed above (
Figure 1). Now, the phase transition was shifted to a higher temperature, namely 21.7 °C and a distinct shoulder appeared suggesting the separation of phospholipids induced by a strong interaction of the positively charged NKCS. Presumably, the positively charged NKCS interacts stronger with the negatively charged POPG and thus leads to a spatial separation of POPG and POPE.
Figure 1.
Biological activity of NKCS. (a) Hemolytic activity of the nonselective membranolytic peptide melittin and of NKCS; (b) Growth inhibition of labeled NKCS on E. coli. The activity of the dye-labeled peptides was comparable to the unlabeled species.
Figure 1.
Biological activity of NKCS. (a) Hemolytic activity of the nonselective membranolytic peptide melittin and of NKCS; (b) Growth inhibition of labeled NKCS on E. coli. The activity of the dye-labeled peptides was comparable to the unlabeled species.
Figure 2.
Heat capacity function of liposomes composed of POPE and POPG at a molar ratio of 70:30 without and with NKCS (lipid:peptide molar ratio of 100:1). The curves are vertically shifted for the sake of clarity.
Figure 2.
Heat capacity function of liposomes composed of POPE and POPG at a molar ratio of 70:30 without and with NKCS (lipid:peptide molar ratio of 100:1). The curves are vertically shifted for the sake of clarity.
2.3. Diffusion of Single Lipid Tracer Molecules in GUV Membranes
DSC provided a static overview of the peptide-bilayer interactions. Next, we studied the dynamics of the NKCS-membrane system. We started out by quantifying the lipid dynamics within respective model bilayers to obtain a reference for the NKCS dynamics. Fluorescent lipid analogs were inserted into GUVs created by electro-formation at very low molar ratios. The high dilution of the fluorescent tracer molecules made it possible to observe single diffraction limited signals by sensitive fluorescence microscopy well separated from each other, and to follow their diffusion pathways within the membrane by high speed imaging [
40]. From the single molecule trajectories the diffusion coefficients of the molecules could be determined. Unfortunately, it was not possible to generate GUVs from pure PE or pure PG by electroformation. Also PE:PG GUVs could not be formed due to their negative charge [
26]. Rather, we had to add a certain amount of PC to form stable GUVs. We determined the mobility of the fluorescent lipid tracer TR-DHPE in GUV membranes of three different compositions: GUVs comprising DOPC alone mimicking human erythrocyte membranes, GUVs comprising DOPC/POPE at a molar ratio of 60/40 and GUVs comprising DOPC/POPG at a molar ratio of 70/30 as approximations to bacterial membranes.
Figure 3.
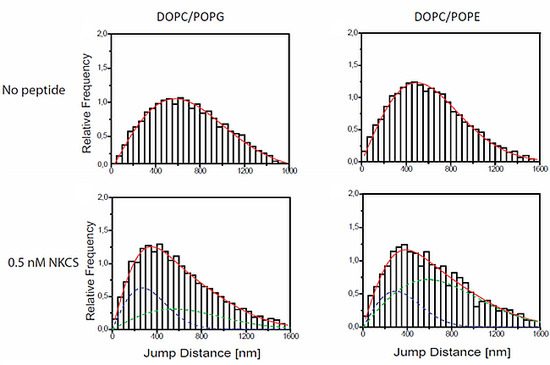
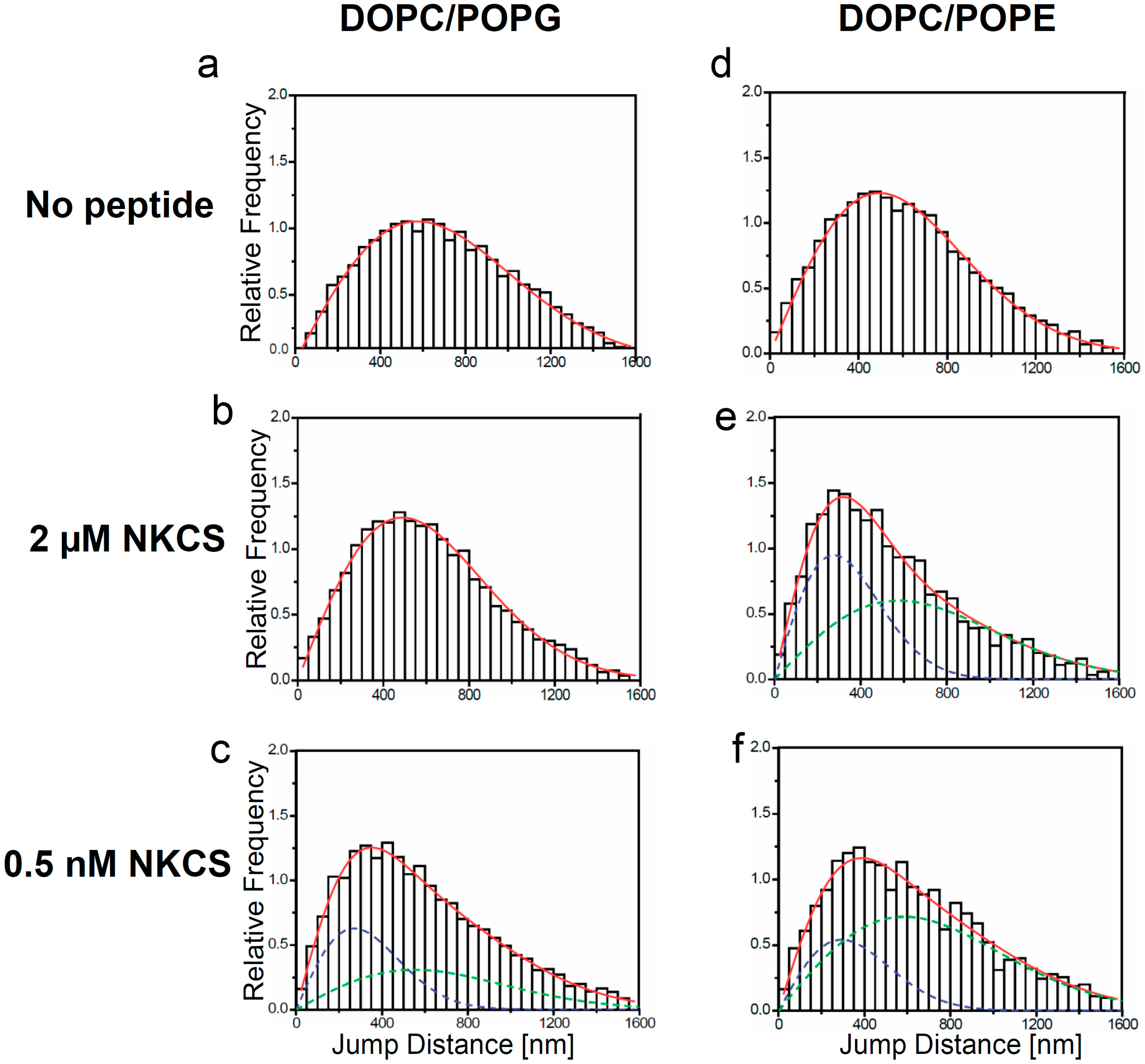
Jump distance distributions for TR-DHPE and NKCS molecules in GUV membranes as determined from single molecule trajectories. The jump distance analysis was performed for TR-DHPE in GUVs prepared from (a) DOPC/POPG and (d) DOPC/POPE, and for TR-DHPE in presence of 2 µM unlabeled NKCS in GUVs prepared from (b) DOPC/POPG and (e) DOPC/POPE. The lower panel shows the jump distance analysis for 0.5 nM Dy647-NKCS on GUVs formed by (c) DOPC/POPG and (f) DOPC/POPE, respectively, revealing the peptide mobility. All full red lines indicate the fitting results according to Equation (1) with one (a, b, d) or two (c, e, f) mobility terms. The green and blue dashed lines in (c, e, f) show separately the low (blue) and high (green) mobility fraction, when two diffusion terms were required to satisfactorily fit the data.
Figure 3.
Jump distance distributions for TR-DHPE and NKCS molecules in GUV membranes as determined from single molecule trajectories. The jump distance analysis was performed for TR-DHPE in GUVs prepared from (a) DOPC/POPG and (d) DOPC/POPE, and for TR-DHPE in presence of 2 µM unlabeled NKCS in GUVs prepared from (b) DOPC/POPG and (e) DOPC/POPE. The lower panel shows the jump distance analysis for 0.5 nM Dy647-NKCS on GUVs formed by (c) DOPC/POPG and (f) DOPC/POPE, respectively, revealing the peptide mobility. All full red lines indicate the fitting results according to Equation (1) with one (a, b, d) or two (c, e, f) mobility terms. The green and blue dashed lines in (c, e, f) show separately the low (blue) and high (green) mobility fraction, when two diffusion terms were required to satisfactorily fit the data.
Extensive single molecule tracking experiments showed that the lipid tracer diffused with a diffusion coefficient of about 5 ± 0.4 µm
2/s within membranes made from DOPC alone and within membranes made from DOPC/POPG. The mobility was reduced to 3.7 ± 0.2 µm
2/s in a membrane comprising DOPC/POPE (see
Figure 3a,d, and
Table 1). Accordingly, DOPC and DOPC/POPG membranes have a comparable viscosity, but that of DOPC/POPE-GUVs was higher by approximately 30%. This was not surprising since it is known that PE establishes more intermolecular hydrogen bonds yielding a higher viscosity [
41].
Table 1.
Mobility of lipid tracers (grey background) and NKCS (white background) within GUV membranes of different composition determined by single molecule tracking. Two rows of numbers in one field mean: Upper rows are the diffusion constants, lower rows are the fraction of this mobility component with respect to the whole system.
Table 1.
Mobility of lipid tracers (grey background) and NKCS (white background) within GUV membranes of different composition determined by single molecule tracking. Two rows of numbers in one field mean: Upper rows are the diffusion constants, lower rows are the fraction of this mobility component with respect to the whole system.
| Probe Molecule | DSPT [µm2/s] |
|---|
| DOPC | DOPC/POPG | DOPC/POPE |
|---|
| TR-DHPE | 5.3 ± 0.4 | 5.0 ± 0.2 | 3.7 ± 0.2 |
| TR-DHPE | 5.4 ± 0.2 | 3.6 ± 0.3 | 5.0 ± 0.6 | 1.2 ± 0.1 |
| +2 µM NKCS | 0.57 ± 0.02 | 0.43 ± 0.02 |
| NKCS | 5.6 ± 0.1 * | 4.9 ± 0.3 | 1.1 ± 0.1 | 5.2 ± 0.7 | 1.3 ± 0.3 |
| 0.59 ± 0.03 | 0.41 ± 0.03 | 0.7 ± 0.07 | 0.3 ± 0.08 |
2.5. Single NKCS Peptides Traced within GUV Membranes
To examine the behavior of NKCS peptides within the GUV membranes Dy647-labeled NKCS was used as probe in single molecule tracking experiments. GUVs were incubated for 30 min with 0.5 nM of the fluorescent peptides. During this time the peptides bound to the GUV membranes. As described above for the lipid tracer molecules the diffusion pathways of single Dy647-peptides in the GUV membrane could be recorded and analyzed. As in the previous experiments the mobility characteristics were quantified by jump distance histograms.
In GUVs formed from DOPC alone we detected only very rarely single peptide signals. This demonstrated again the weak interaction between the peptides and PC-lipids in DOPC membranes. In order to register enough events for jump distance histograms the NKCS concentration was increased to 1 nM. Then, the peptide displayed a mobility of 5.6 µm
2/s (
Table 1). This value corresponded well to the mobility of the lipid tracers, and suggested that there were no special interactions between the peptides and PC membranes. Probably, in rare cases the peptides loosely attached to and diffused together with single lipid molecules, but did not show distinct and long-lasting interactions, and certainly no incorporation into the bilayer. A similar outcome was previously obtained for TAT peptides, which was interpreted as a “floating” of the peptides on the membrane surface [
32].
The situation was strikingly different for GUVs made from DOPC/POPG and DOPC/POPE. Here, characteristic jump distance histograms exhibiting
two mobility components were obtained. However, an important difference was observed in the diffusion constants obtained for the different lipid mixtures. In the case of the PG-containing vesicles one diffusion constant was close to the lipid tracer mobility of the undisturbed system, namely 4.9 µm
2/s (as compared to 5.0 µm
2/s of the tracers in DOPC/POPG), but the second diffusion constant was strongly reduced to 1.1 µm
2/s (
Figure 3c). Thus it was even significantly smaller than the lipid tracer mobility of the system disturbed by the high NKCS concentration. For PE-containing vesicles the diffusion constant of the peptides was in the range of the two lipid tracer diffusion constants presented above (
Figure 3e,f).
2.6. CLSM Imaging of NKCS Peptide-GUV Interaction
The interaction of a fluorescently labeled peptide such as Dy647-NKCS with model lipid membranes can directly be visualized using confocal laser scanning microscopy [
32,
33,
35]. To this end, GUVs prepared from DOPC, DOPC/POPG and DOPC/POPE were incubated with 2 µM of Dy647-labeled NKCS. To monitor the integrity of the GUV membranes large FITC-labeled dextran molecules with a molecular mass of 40 kDa, which would diffuse into the GUVs in case of major membrane leakages, were added to the external solution.
In order to examine putative membrane pores, we prepared three batches of GUVs, and enclosed fluorescent tracer molecules of increasing molecular mass, namely Alexa Fluor 488 (0.64 kDa), 3 kDa AlexaFluor 546-dextran and 70 kDa AlexaFluor 546-dextran, within these batches, respectively. In case pores were formed upon peptide addition a release of the enclosed molecules into the buffer lead to a loss of the initial internal GUV fluorescence, and was monitored as a function of time. In this manner we obtained information about the possible existence and size of AMP-induced membrane pores.
Figure 4.
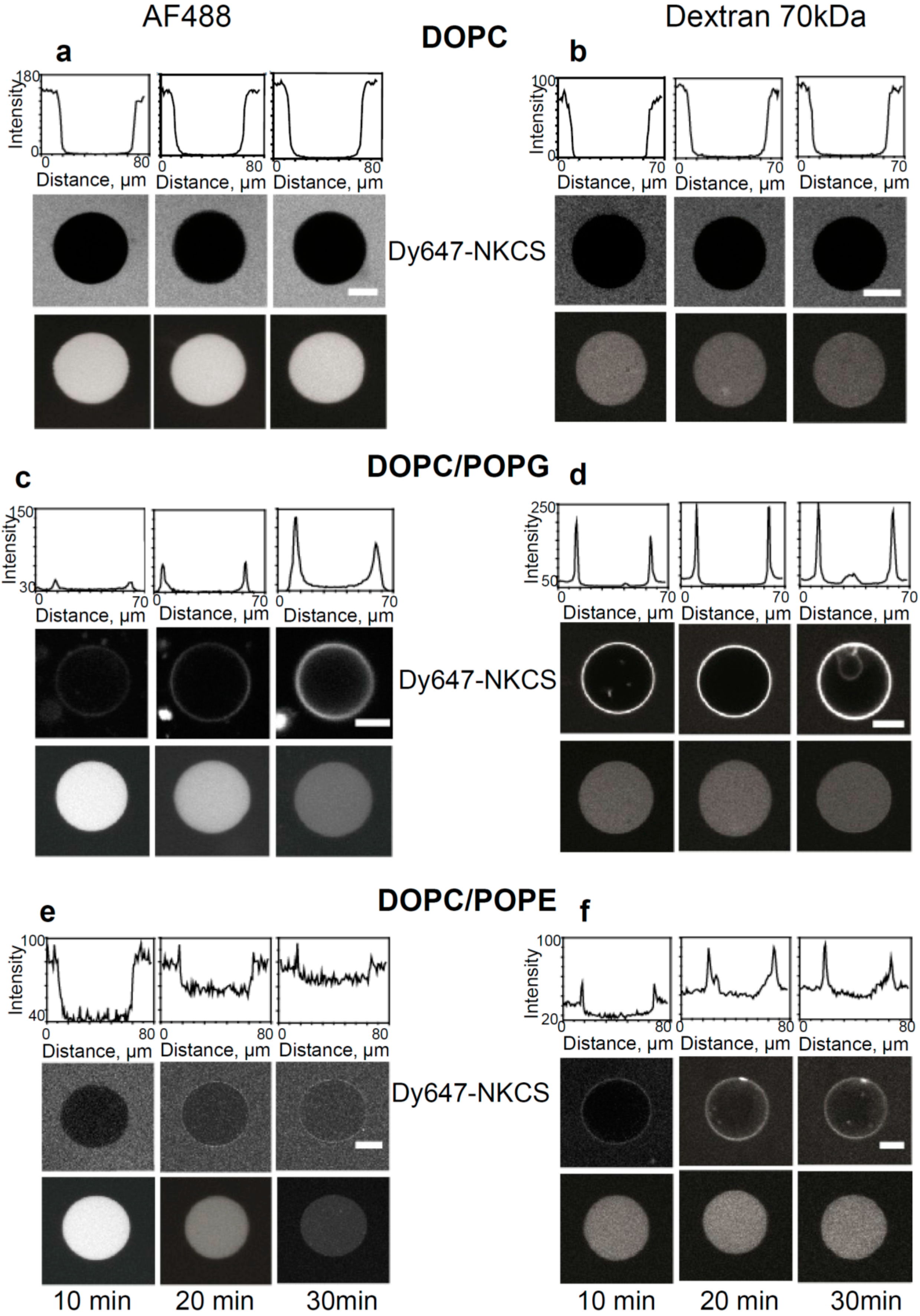
Internalization and tracer release induced by the NKCS peptide in GUVs. (a,b) Interaction of NKCS with GUVs prepared from DOPC. The middle panel shows neither accumulation nor internalization of the peptide on the GUV membrane. The top panel quantifies the fluorescence intensity along a horizontal line through the center of the GUV. The lower panel demonstrates that (a) AF488 and (b) 70 kDa dextran-Alexa Fluor 546 remained contained within the GUV. (c,d) Binding of Dy647-NKCS to the membrane formed by DOPC/POPG. Peptide internalization was indicated by the non-zero fluorescence in the GUV interior, as quantified in the upper panel showing the fluorescence intensity along a horizontal line through the center of the GUV. The lower panel demonstrates that there was leakage of (b) AF488 but not of (d) 70 kDa dextran-Alexa Fluor 546 out of the GUV. (c) Interaction of NKCS with GUVs prepared from DOPC/DOPE. The top panel quantifies the fluorescence intensity along a horizontal line through the center of the GUV. The middle panel shows the accumulation of the peptide in the membrane. But additionally over time a portion of the NKCS peptide is crossing the membrane and can be found inside the vesicle. The lower panel demonstrates that there was leakage of (e) AF488 but not of (f) 70 kDa dextran-Alexa Fluor 546 out of the GUV. All scale bars, 20 µm.
Figure 4.
Internalization and tracer release induced by the NKCS peptide in GUVs. (a,b) Interaction of NKCS with GUVs prepared from DOPC. The middle panel shows neither accumulation nor internalization of the peptide on the GUV membrane. The top panel quantifies the fluorescence intensity along a horizontal line through the center of the GUV. The lower panel demonstrates that (a) AF488 and (b) 70 kDa dextran-Alexa Fluor 546 remained contained within the GUV. (c,d) Binding of Dy647-NKCS to the membrane formed by DOPC/POPG. Peptide internalization was indicated by the non-zero fluorescence in the GUV interior, as quantified in the upper panel showing the fluorescence intensity along a horizontal line through the center of the GUV. The lower panel demonstrates that there was leakage of (b) AF488 but not of (d) 70 kDa dextran-Alexa Fluor 546 out of the GUV. (c) Interaction of NKCS with GUVs prepared from DOPC/DOPE. The top panel quantifies the fluorescence intensity along a horizontal line through the center of the GUV. The middle panel shows the accumulation of the peptide in the membrane. But additionally over time a portion of the NKCS peptide is crossing the membrane and can be found inside the vesicle. The lower panel demonstrates that there was leakage of (e) AF488 but not of (f) 70 kDa dextran-Alexa Fluor 546 out of the GUV. All scale bars, 20 µm.
![Molecules 20 06941 g004]()
As expected, for GUVs made solely from DOPC confocal microscopy did not show any binding of NKCS to the GUV surfaces. Also, we did not observe peptide internalization into the GUVs, nor efflux of any of the tracers (
Figure 4a,b).
The results were different for GUVs prepared from DOPC/POPE. Here, a reduced peptide binding to the GUV was seen, but the influx of the labeled peptides was more efficient and faster than for DOPC/POPG membranes (see
Figure 4e,f). The efflux experiments demonstrated the rapid and complete release of Alexa 488 and 3 kDa Alexa546-dextran molecules within 30 min in the presence of NKCS (see
Figure S11c for 3 kDa Alexa Fluor 546-dextran). Obviously, again the pores induced by peptides were small, since large dextran molecules with a molecular mass of 70 kDa were not released from the GUVs. The results of all confocal experiments were summarized in
Table 2.
Table 2.
Characteristics of NKCS peptide interaction with GUVs of different membrane composition loaded with tracer molecules of different size.
Table 2.
Characteristics of NKCS peptide interaction with GUVs of different membrane composition loaded with tracer molecules of different size.
| | DOPC | DOPC/POPG | DOPC/POPE |
|---|
| Peptide influx | − | + | + |
| AF488 efflux | − | + | + |
| AF546 Dextran 3 kDa efflux | − | − | + |
| AF546 Dextran 70 kDa efflux | − | − | − |
2.7. Discussion
In previous studies the peptide NKCS was shown to be very active against both Gram-negative and Gram-positive bacteria with insignificant toxicity towards human cells [
28,
30]. These biological characteristics make it an interesting and potent candidate as a novel antibacterial agent, and suggest it as a model for understanding AMP interaction with bacterial membranes. In this study we approached this question with a range of biophysical techniques.
NKCS showed low toxicity toward human erythrocytes. This observation was in good agreement with the result of the CLSM experiment, in which GUVs prepared from DOPC were used to mimic the membrane of human red blood cells, but upon incubation with NKCS there was no peptide accumulation on the membrane. Also, the peptides neither translocate into these GUVs nor did induce effect any efflux of tracer molecules. Finally, single molecule tracking of NKCS on DOPC membranes yielded diffusion coefficients, which were comparable to those of lipid tracer molecules. This indicated that only sporadically and for short periods, the peptides were non-specifically attached to the lipid head groups in the membrane. All this demonstrated that no significant interactions occurred with DOPC and implicitly with model erythrocyte membranes. This conclusion corroborated the results of Andrä
et al. [
28], who did not detect any interactions between NKCS and PC bilayers using circular dichroism and fluorescence resonance energy transfer.
Quite different observations were made, when model lipid membranes inspired by the bacterial system were studied. Measurements of the MIC of NKCS against the Gram-negative bacterium
E. coli showed that NKCS has a significant antibacterial activity. According to Monte Carlo simulations, upon the membrane interaction NKCS resembles two α-helices connected by a flexible region [
30].
Calorimetric studies of POPE and POPG at a molar ratio of 70:30 indicated that NKCS interacted strongly with model bacterial membranes. Without NKCS both lipid components were well mixed and did not form any domains. Upon NKCS addition, however, two different fractions appeared. This indicated that the cationic peptide induced the formation of lipid-peptide domains. The shoulder, which appeared in the thermogram upon addition of NKCS should be attributed to the binding of the peptide to the lipid head groups. Since the intercalation of the peptide between the head groups presumably leads to more space for the acyl chains it can be assumed that the acyl chain melting temperature (Tm) of lipids bound to NKCS is lower than that of the homogenous lipid mixture. Noteworthy, this experiment was performed at lipid:peptide molar ratio of 100:1, which corresponded to a concentration well above the MIC.
To get further insights into the effects of NKCS onto bacterial membranes we employed two bilayer model systems, namely GUVs formed from DOPC/POPE (molar ratio, 60/40), and DOPC/POPG (molar ratio of 70/30). A certain fraction of PC had to be included into the lipid mixture, because pure POPE or POPG did not form GUVs by electro-formation. As mentioned above, there were no special interactions between the peptides and DOPC membranes. In GUVs formed from DOPC alone we detected only very rarely single peptide signals and the value of diffusion coefficient for NKCS on DOPC membranes was the same as for lipids in membrane.
Both lipid and peptide dynamics were studied extensively in these systems. As a reference we determined the diffusion coefficients of lipid tracers in the model membranes. The dynamics in the DOPC/POPG membrane was comparable to that of pure DOPC, 5.0 µm
2/s, whereas the membrane comprising DOPC/POPE showed a lower mobility, 3.7 µm
2/s. The reason for this difference could be that the lipids DOPC and POPG, which are at room temperature both in the liquid crystalline phase, form very homogeneous and relatively stable mixed GUVs. Both lipids prefer structures with little or positive curvature. Because the low concentration of POPG in comparison to the neutral PC, stress induced by curvature does not play a role and the lipids can freely diffuse. In contrast to POPG, POPE has a small head group thus having the tendency to form inverse curvatures. Furthermore, POPE is slightly negatively charged [
26], it exhibits a gel phase at room temperature and the concentration summed up to almost half of the lipid population. Therefore a reduction of lipid mobility was likely, especially when considering the demonstrated existence of a trend to intermolecular hydrogen bonds [
41]. We could assume that we did not have very homogenous GUVs in a somewhat meta-stable situation.
Upon the addition of 2 µM NKCS to the solution in DOPC/POPG-vesicles the diffusion constant of the lipid tracer was reduced to 3.6–3.9 µm
2/s. This could be explained by a strong, probably charge-driven interaction of the peptides with the lipids. Probably we observed the motion of small peptide-linked lipid clusters, presumably of transient nature. However, looking at the mobility of the NKCS peptides—at a much lower concentration—we observed two separate NKCS diffusion constants in the same membrane. We assumed that one constant was due to peptides, which were loosely interacting with the lipid head groups and “floating” on the membrane. Such a behavior was previously observed for positively charged TAT peptides on GUV membranes [
32]. The second diffusion showed a striking, about 4-fold reduction in mobility. This suggested a strong interaction of the NKCS peptides with membrane components,
i.e., a strong binding to a larger number of lipid molecules. Probably distinct NKCS-POPG clusters were formed, in which the contained molecules diffused together.
Interestingly, for NKCS peptides diffusing on GUVs made from DOPC/POPE we observed a comparable mobility behavior as before for the lipid tracers, when the GUVs were bathed in 2 µM NKCS. Two mobility components were seen for NKCS exhibiting diffusion coefficients of 5.2 and 1.3 µm2/s, almost as before for the tracers (5.0 and 1.2 µm2/s).
Still, we did not expect that the extremely low concentration of NKCS molecules (0.5 nM) was capable of causing a significant phase separation, as it was obviously achieved at higher concentrations (65 µM, see
Figure 2). Rather, we concluded that the same effect as in the case of the DOPC/POPG-GUVs was responsible for the two mobility fractions: a certain fraction of peptides was “floating” on the bilayer, and another fraction was tightly bound to a major number of lipids, which drifted with a low mobility in the membrane.
Most interestingly, however, tracer efflux experiments clearly demonstrated that NKCS induced the efflux of small tracer molecules out of GUVs. Obviously, NKCS, which adopts an α-helical but bent conformation leading to a partial intercalation into the membrane, formed small lesions in the examined model bilayers. The effect was stronger for PE-containing vesicles than for PG-containing GUVs and could again be explained by the existence of POPE-rich microdomains in the membranes of the DOPC/POPE GUVs.
Altogether, both lipids PG and PE seem to be important for NKCS peptide activity. While in DOPC/POPG model membranes the peptide showed a stronger binding due to the electrostatic attraction, in DOPC/POPE membranes, we observed a reduced peptide binding but the influx of the labeled peptides was more efficient and faster than for DOPC/POPG GUVs. At concentrations well above the MIC, NKCS modifies also the biophysical properties of the model membrane. The tracer mobility in DOPC/POPG membranes was reduced by approximately 30% in presence of 2 µM unlabeled peptide. A different effect was observed when the peptides were added to DOPC/POPE model membranes. In this case, the lipid tracer mobility displayed a bimodal distribution with a diffusion coefficient close to the value of pure DOPC membranes and the other diffusion coefficient strongly slowed down. These observations indicated the occurrence of a peptide-induced lipid phase separation in DOPC/POPE membranes. Most likely, in the antibacterial activity of NKCS, the role of PG is to promote the accumulation of the peptide on the membrane and PE induces the pore formation or micellization and finally the destruction of bacterial membrane.
The comparison of the presented results with our previous data on TAT peptides suggests a quite different mode of action for NKCS than for TAT. For TAT, we observed only the mentioned floating of the peptides on the membrane surface. TAT peptides were loosely attached to the head group region of the lipid bilayer, but at higher concentrations they massively accumulated on the membrane, and destroyed it locally probably by creating strong local electric fields due to their positive charge. This process was proposed by molecular dynamics simulations of TAT peptide translocation [
42]. Probably the internalization at thinned and destabilized membrane patches led simultaneously to the formation of transient pores. NKCS, in contrast to TAT, showed a distinct attachment to PG/PE in GUV membranes. The peptides partly floated, but partly showed a strongly reduced mobility indicating the occurrence of distinct peptide-lipid clusters, which presumably also distorted the existing membrane structure. This effect is probably enhanced when lipids have a small head group, like PE. This will increase the trend to the formation of inverted micelles, in which the lipid head groups surround the peptides and form star-like structures. Such micelles then destroy the membrane integrity and can translocate into the membrane-enclosed space—a process that is accompanied by the creation of membrane lesions. In bacterial membranes this is certainly a lethal process.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
