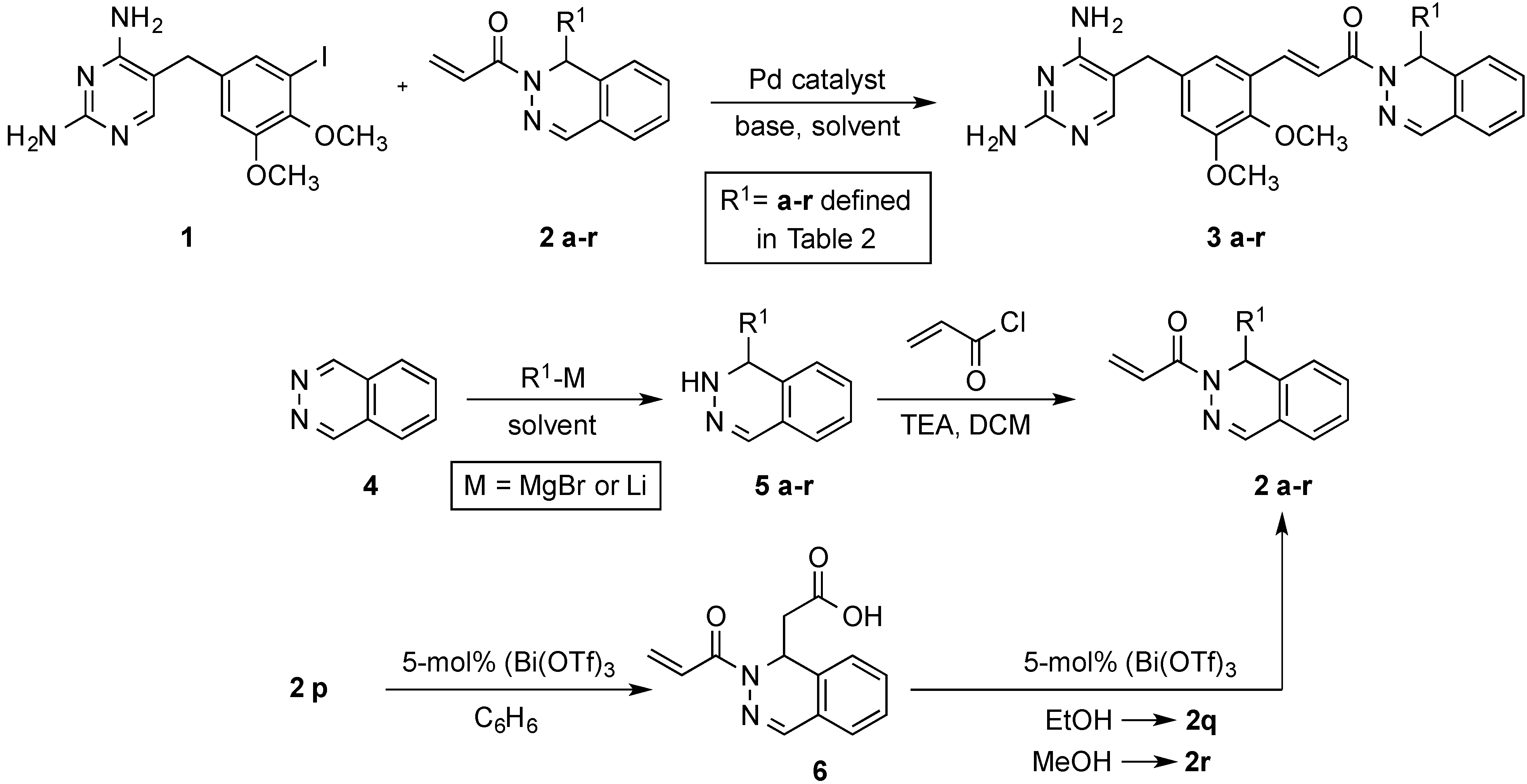
3.3. Preparation of Phthalazin-2(1H)-yl]-2-propen-1-ones 2a–r
(±)-1-[1-(2-Methyl-1-propen-1-yl)phthalazin-2(1H)-yl]-2-propen-1-one (
2a). This compound was prepared from
4 and 2-methyl-1-propenylmagnesium bromide in 62% yield on a 15.4-mmol scale according to the literature procedure [
15]. The spectral data matched those reported [
15].
(±)-1-(1-Allylphthalazin-2(1H)-yl)-2-propen-1-one (2b). A stirred solution of 4 (2.50 g, 19.2 mmol) in dry THF (60 mL) was treated dropwise with a solution of allylmagnesium bromide (12.4 mL, 24.9 mmol, 2 M in THF) for 30 min at 0 °C. The reaction mixture was warmed to room temperature and stirred for 1 h. After completion, the reaction mixture was treated with saturated NH4Cl (50 mL) and extracted with EtOAc (3 × 100 mL). The combined organic extracts were washed with saturated NaCl (50 mL), dried (MgSO4), filtered, and concentrated under vacuum to give 3b as a dark brown, viscous liquid. The crude 5b was dissolved in DCM (70 mL) and TEA (5.87 mL, 41.7 mmol) was added, followed by dropwise addition of acryloyl chloride (1.35 mL, 16.7 mmol) at 0 °C. The reaction mixture was stirred at this temperature for 2 h. The mixture was then treated with saturated NaCl (100 mL), the organic layer was separated, and the aqueous layer was extracted with DCM (2 × 50 mL). The combined extracts were washed with saturated NaCl (50 mL), dried (MgSO4), filtered, and concentrated to afford the crude product. This material was purified on a silica gel column eluted with hexanes–EtOAc (7:3) to afford 2b (2.47 g, 57%) as a viscous, yellow liquid. IR: 1662 cm−1; 1H-NMR (CDCl3): δ 7.60 (s, 1H), 7.43 (td, J = 7.4, 1.4 Hz, 1H), 7.35 (td, J = 7.6, 1.2 Hz, 1H), 7.30 (dd, J = 17.1, 10.5 Hz, 1H), 7.28 (d, J = 7.6, 0.6 Hz, 1H), 7.15 (dd, J = 7.6, 0.6 Hz, 1H), 6.42 (dd, J = 17.1, 2.1 Hz, 1H), 5.91 (t, J = 6.4 Hz, 1H), 5.78 (dd, J = 10.5, 2.1 Hz, 1H), 5.68 (ddt, J = 17.1, 10.1, 7.3 Hz, 1H), 4.98 (dm, J = 10.2 Hz, 1H), 4.90 (dm, J = 17.0 Hz, 1H), 2.41 (m, 2H); 13C-NMR (CDCl3): δ 166.3, 142.2, 133.2, 132.7, 131.4, 128.4, 128.1, 127.1, 126.6, 125.6, 123.9, 118.7, 51.1, 39.8.
(±)-1-(1-Vinylphthalazin-2(1H)-yl)-2-propen-1-one (2c). This compound was prepared via the procedure described for 2b using 4 (2.50 g, 19.2 mmol) and vinylmagnesium bromide (11.5 mL, 23.0 mmol, 2.0 M in THF) in dry THF (60 mL) to afford 3c, followed by acylation using TEA (5.3 mL, 37.8 mmol) and acryloyl chloride (1.20 mL, 15.1 mmol) in DCM (70 mL) to give 2c (2.36 g, 58%) as a colorless, viscous liquid. IR: 1665 cm−1; 1H-NMR (CDCl3): δ 7.59 (s, 1H), 7.45 (td, J = 7.4, 0.8 Hz, 1H), 7.40–7.28 (m, 3H), 7.21 (dd, J = 7.6, 0.4 Hz, 1H), 6.51 (dd, J = 17.4, 2.1 Hz, 1H), 6.37 (d, J = 4.9 Hz, 1H), 5.81 (m, 2H), 5.10 (dd, J = 10.3, 0.4 Hz, 1H), 4.86 (d, J = 17.4 Hz, 1H); 13C-NMR (CDCl3): δ 166.2, 141.7, 134.7, 131.9, 131.7, 128.8, 128.4, 126.9, 126.8, 125.9, 123.8, 116.2, 53.0.
(±)-1-(1-Propylphthalazin-2(1H)-yl)-2-propen-1-one (
2d). This compound was prepared in 70% yield from
4 (2.00 g, 15.4 mmol) and propylmagnesium chloride (8.45 mL, 16.9 mmol, 2.0 M in ether) in dry THF (50 mL) to afford
5d, followed by acylation using TEA (1.82 g, 2.50 mL, 18.0 mmol) and acryloyl chloride (1.40 g, 1.26 mL, 15.5 mmol) in DCM (70 mL) to afford
2d according to the literature procedure [
15]. The spectral data matched those reported [
15].
(±)-1-(1-Pentylphthalazin-2(1H)-yl)-2-propen-1-one (2e). This compound was prepared via the procedure described for 2b from 4 (2.50 g, 19.2 mmol) and n-pentylmagnesium bromide (18.4 mL, 18.4 mmol, 1 M in THF) in dry THF (60 mL) to afford 5e, followed by acylation using TEA (3.27 g, 4.51 mL, 32.4 mmol) and acryloyl chloride (1.05 mL, 12.9 mmol) in DCM (70 mL) to give 2e (3.05 g, 62%) as an off-white solid, mp 52–54 °C. IR: 1664 cm−1; 1H-NMR (CDCl3): δ 7.61 (s, 1H), 7.42 (td, J = 7.4, 1.1 Hz, 1H), 7.33 (m, 2H), 7.26 (d, J = 7.0 Hz, 1H), 7.15 (dd, J = 7.4, 0.7 Hz, 1H), 6.47 (dd, J = 17.2, 2.4 Hz, 1H), 5.83 (t, J = 6.7 Hz, 1H), 5.77 (dd, J = 10.5, 2.4 Hz, 1H), 1.63 (m, 2H), 1.21 (m, 6H), 0.81 (t, J = 6.6 Hz, 3H); 13C-NMR (CDCl3): δ 166.1, 142.3, 134.0, 131.3, 128.2, 127.9, 127.1, 126.4, 125.6, 123.8, 51.2, 34.9, 31.4, 24.4, 22.3, 13.8.
(±)-1-(1-Cyclopropylphthalazin-2(1H)-yl)-2-propen-1-one (
2f). This compound was prepared in 87% yield from
4 (2.00 g, 15.4 mmol) and cyclopropylmagnesium chloride (33.8 mL, 16.9 mmol, 0.5 M in THF) in dry THF (50 mL) to give
5f, followed by acylation using TEA (1.86 g, 2.56 mL, 18.4 mmol) and acryloyl chloride (1.39 g, 1.25 mL, 15.4 mmol) in DCM (70 mL) to afford
2f according to the literature procedure [
13]. The spectral data matched those reported [
13].
(±)-1-(1-Cyclobutylphthalazin-2(1H)-yl)-2-propen-1-one (2g). This compound was prepared via the procedure described for 2b using 4 (2.50 g, 19.2 mmol) and cyclobutylmagnesium bromide (24.9 mL, 24.9 mmol, 1 M in THF) in dry THF (60 mL) to give 5g, followed by acylation using TEA (3.25 g, 4.48 mL, 32.2 mmol) and acryloyl chloride (1.16 g, 1.04 mL, 12.8 mmol) in DCM (70 mL) to afford 2g (3.04 g, 66%) as a yellow liquid. IR: 1663 cm−1; 1H-NMR (CDCl3): δ 7.61 (s, 1H), 7.44 (td, J = 7.4, 1.5 Hz, 1H), 7.35 (td, J = 7.4, 1.5 Hz, 1H), 7.31 (dd, J = 17.2, 10.5 Hz, 1H), 7.29 (m, 1H), 7.16 (dd, J = 7.5, 0.7 Hz, 1H), 6.48 (dd, J = 17.2, 2.0 Hz, 1H), 5.83 (d, J = 8.4 Hz, 1H), 5.77 (dd, J = 10.5, 2.0 Hz, 1H), 2.60 (sextet, J = 8.4 Hz, 1H), 1.98 (m, 1H), 1.90–1.73 (m, 2H), 1.67 (m, 3H); 13C-NMR (CDCl3): δ 166.5, 143.8, 132.6, 131.3, 128.2, 128.0, 127.1, 126.5, 125.6, 124.0, 54.5, 40.2, 25.6, 24.9, 17.6.
(±)-1-(1-Cyclopentylphthalazin-2(1H)-yl)-2-propen-1-one (2h). This compound was prepared via the procedure described for 2b from 4 (2.50 g, 19.2 mmol) and cyclopentylmagnesium bromide (24.9 mL, 24.9 mmol, 1 M in THF) in dry THF (60 mL) to give 5h, followed by acylation using TEA (3.78 g, 5.20 mL, 37.4 mmol) and acryloyl chloride (1.35 g, 1.21 mL, 14.9 mmol) in DCM (70 mL) to afford 2h (2.73 g, 56%) as a viscous, colorless liquid. IR: 1663 cm−1; 1H-NMR (CDCl3): δ 7.69 (s, 1H), 7.43 (td, J = 7.4, 1.4 Hz, 1H), 7.35 (td, J = 7.4, 1.4 Hz, 1H), 7.32 (dd, J = 17.2, 10.5 Hz, 1H), 7.28 (dd, J = 7.4, 1.4 Hz, 1H), 7.18 (dd, J = 7.4, 0.5 Hz, 1H), 6.46 (dd, J = 17.2, 2.1 Hz, 1H), 5.80 (d, J = 8.6 Hz, 1H), 5.77 (dd, J = 10.5, 2.1 Hz, 1H), 2.12 (m, 1H), 1.61 (m, 3H), 1.42 (m, 4H), 1.23 (m, 1H); 13C-NMR (CDCl3): δ 166.3, 143.6, 133.5, 131.2, 128.3, 128.0, 127.1, 126.8, 125.4, 124.2, 54.1, 45.2, 29.3, 28.7, 24.3, 24.2.
(±)-1-(1-(tert-Butyl)phthalazin-2(1H)-yl)-2-propen-1-one (2i). A stirred solution of 4 (2.00 g, 17.6 mmol) in 50 mL of dry THF was treated dropwise with a solution of t-BuLi (16.9 mL, 16.9 mmol, 1.0 M in heptanes) over a period of 15 min at −78 °C. The reaction was stirred at this temperature for 1 h and then slowly warmed to room temperature and stirred for an additional 30 min. The reaction mixture was added to cold saturated NH4Cl (50 mL) and extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with saturated NaCl (50 mL), dried (MgSO4), filtered, and concentrated under vacuum to give 5i as a dark brown liquid. The crude product was dissolved in DCM (100 mL), and TEA (4.00 g, 5.50 mL, 39.6 mmol) was added, followed by dropwise addition of acryloyl chloride (1.44 g, 1.29 mL, 15.9 mmol) at 0 °C. The reaction mixture was stirred at this temperature for 2 h. The mixture was then added to saturated NaCl (100 mL), the organic layer was separated, and the aqueous layer was extracted with DCM (2 × 50 mL). The combined extracts were washed with saturated NaCl (50 mL), dried (MgSO4), filtered, and concentrated to afford the crude product. The crude product was purified on a silica gel column eluted with hexanes–EtOAc (7:3) to afford 2i as a viscous, yellow liquid (2.79 g, 75%). IR: 1666 cm−1; 1H-NMR (CDCl3): δ 7.65 (s, 1H), 7.46 (td, J = 7.4, 1.4 Hz, 1H), 7.37 (td, J = 7.4, 1.4 Hz, 1H), 7.34 (dd, J = 17.1, 10.1 Hz, 1H), 7.29 (dd, J = 7.4, 0.8 Hz, 1H), 7.19 (d, J = 7.8 Hz, 1H), 6.43 (dd, J = 17.1, 2.3 Hz, 1H), 5.78 (s, 1H), 5.75 (dd, J = 10.1, 2.3 Hz, 1H), 0.86 (s, 9H); 13C-NMR (CDCl3): δ 167.0, 144.6, 133.4, 131.0, 130.6, 128.6, 128.2, 128.0, 127.4, 125.4, 125.1, 58.1, 39.4, 26.6, 25.5.
(±)-1-(1-(Furan-2-yl)phthalazin-2(1H)-yl)-2-propen-1-one (
2j). This compound was prepared in 72% yield from
4 (2.00 g, 15.4 mmol), furan-2-yllithium [from furan (1.20 g, 17.6 mmol) and
n-BuLi (7.30 mL, 18.3 mmol, 2.5 M in hexanes)] in dry THF (50 mL) to give
5j, followed by acylation using TEA (2.37 g, 3.26 mL, 23.5 mmol) and acryloyl chloride (1.59 g, 1.43 mL, 17.6 mmol) in DCM (70 mL) to afford
2j according to the literature procedure [
13]. The spectral data matched those reported [
13].
(±)-1-[1-(Thiophen-2-yl)phthalazin-2(1H)-yl]-2-propen-1-one (
2k). This compound was prepared in 60% yield from
4 (2.00 g, 15.4 mmol), thiophen-2-ylmagnesium bromide [from 2-bromothiophene (1.77 g, 1.69 mL, 21.0 mmol) and magnesium (0.69 g, 28.4 mmol)] in dry THF (50 mL) to give
5k. The crude
5k was acylated using TEA (2.80 g, 3.86 mL, 27.7 mmol) and acryloyl chloride (1.90 g, 1.71 mL, 21.0 mmol) in DCM (70 mL) to afford
2k according to the literature procedure [
13]. The spectral data matched those reported [
13].
(±)-1-[1-(1-Methyl-1H-indol-2-yl)phthalazin-2(1H)-yl]-2-propen-1-one (2l). To a stirred solution of 1-methylindole (1.50 g, 11.4 mmol) in dry THF (25 mL) was added dropwise n-BuLi (6.86 mL, 17.2 mmol, 2.5 M in hexanes) over a period of 30 min at −78 °C. The solution was warmed to −25 °C, and stirring was continued at this temperature for 1 h. The reaction mixture was cooled to −78 °C, and a solution of 4 (1.48 g, 11.4 mmol) in dry THF (20 mL) was added dropwise over 30 min. The reaction mixture was stirred at this temperature for 2 h. The mixture was poured into saturated NH4Cl (100 mL) and extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with saturated NaCl (50 mL), dried (MgSO4), filtered, and concentrated under vacuum to give 5l as a light yellow liquid. The crude product 5l was dissolved in DCM (100 mL), and TEA (2.08 g, 2.87 mL, 20.6 mmol) was added, followed by dropwise addition of acryloyl chloride (0.81 g, 0.73 mL, 8.95 mmol) at 0 °C. The reaction mixture was stirred at this temperature for an additional 2 h. The aqueous layer was added to saturated NaCl (50 mL), and the organic layer was separated. The aqueous layer was extracted with DCM (2 × 30 mL), and the combined organic extracts were washed with saturated NaCl (50 mL), dried (MgSO4), filtered, and concentrated to afford the crude product. The product was purified on a silica gel column eluted with hexanes–EtOAc (7:3) to afford 2l (3.01 g, 62%) as a light yellow solid, mp 69–71 °C. IR: 1657 cm−1; 1H-NMR (CDCl3): δ 7.79 (s, 1H), 7.42–7.34 (complex, 5H), 7.22 (dd, J = 17.2, 10.5 Hz, 1H), 7.18 (s, 1H), 7.16 (m, 2H), 7.00 (t, J = 7.6 Hz, 1H), 6.45 (dd, J = 7.2, 2.0 Hz, 1H), 5.89 (s, 1H), 5.75 (dd, J = 10.5, 2.0 Hz, 1H), 3.99 (s, 3H); 13C-NMR (CDCl3): δ 166.0, 143.3, 138.9, 137.4, 132.3, 132.1, 129.0, 128.6, 127.2, 127.0, 126.7, 126.0, 123.7, 122.0, 120.5, 119.6, 109.6, 103.3, 46.6, 30.6.
(±)-1-[1-(Benzofuran-2-yl)phthalazin-2(1H)-yl]-2-propen-1-one (2m). The compound was prepared using benzofuran-2-yllithium [from benzofuran (2.00 g, 16.9 mmol) and n-BuLi (6.8 mL, 17.0 mmol, 2.5 M in hexanes)] and 4 (2.20 g, 16.9 mmol) in dry THF (75 mL) to give 5m. The crude 5m was acylated using TEA (3.05 g, 4.20 mL, 30.2 mmol) and acryloyl chloride (1.09 g, 0.97 mL, 12.0 mmol) in DCM (120 mL) to afford 2m (2.37 g, 51%) as a light yellow solid, mp 55–57 °C. IR: 1657 cm−1; 1H-NMR (CDCl3): δ 7.84 (d, J = 7.8 Hz, 1H), 7.63 (dd, J = 7.8, 0.6 Hz, 1H), 7.56 (d, J = 8.2 Hz, 1H), 7.48–7.40 (complex, 3H), 7.36 (td, J = 7.5, 1.2 Hz, 2H), 7.27 (t, J = 7.5 Hz, 1H), 7.23 (s, 1H), 7.23 (d, J = 7.5 Hz, 1H), 6.54 (dd, J = 17.1, 2.1 Hz, 1H), 5.84 (dd, J = 10.4, 2.0 Hz, 1H), 5.02 (s, 1H); 13C-NMR (CDCl3): δ 166.4, 154.9, 151.1, 141.8, 131.7, 131.0, 139.1, 138.1, 137.7, 126.5, 126.3, 126.0, 125.4, 123.6, 123.3, 121.5, 111.5, 108.0, 42.0.
(±)-1-[1-(Benzo[b]thiophen-2-yl)phthalazin-2(1H)-yl]-2-propen-1-one (2n). The compound was prepared using benzothiophen-2-yllithium [from benzothiophene (2.00 g, 14.9 mmol) and n-BuLi (6.0 mL, 15.0 mmol, 2.5 M in hexanes)] and 4 (1.94 g, 14.9 mmol) in dry THF (75 mL) to give 5n. The crude 5n was acylated using TEA (2.86 g, 3.94 mL, 28.3 mmol) and acryloyl chloride (1.02 g, 0.92 mL, 11.3 mmol) in DCM (120 mL) to afford 2n (2.94 g, 62%) as a light yellow solid, mp 69–71 °C. IR: 1663 cm−1; 1H-NMR (CDCl3): δ 7.86 (m, 1H), 7.81 (m, 2H), 7.70 (s, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.46–7.36 (complex, 5H), 7.32 (d, J = 7.2 Hz, 1H), 6.54 (dd, J = 17.2, 1.8 Hz, 1H), 5.87 (dd, J = 10.3, 1.8 Hz, 1H), 5.05 (s, 1H); 13C-NMR (CDCl3): δ 166.4, 146.4, 139.8, 139.7, 139.0, 131.8, 131.7, 129.2, 128.2, 126.6, 126.1, 125.8 (2C), 125.7, 124.6, 124.4, 124.2, 122.2, 42.2.
(±)-1-[1-(Benzo[d]thiazol-2-yl)phthalazin-2(1H)-yl]-2-propen-1-one (2o). The compound was prepared using benzothiazol-2-yllithium [from (2.00 g, 14.8 mmol) and n-BuLi (6.5 mL, 16.3 mmol, 2.5 M in hexanes)] and 4 (1.92 g, 14.8 mmol) in dry THF (75 mL) to give 5o. The crude 5o was acylated using TEA (2.86 g, 3.94 mL, 28.3 mmol) and acryloyl chloride (1.02 g, 0.92 mL, 11.3 mmol) in DCM (120 mL) to afford 5o (3.20 g, 68%) as a light yellow solid, mp 68–70 °C. IR: 1663 cm−1; 1H-NMR (CDCl3): δ 7.93 (d, J = 8.2 Hz, 1H), 7.75 (dt, J = 8.0, 0.6 Hz, 1H), 7.70 (s, 1H), 7.56 (d, J = 7.6 Hz, 1H), 7.51 (td, J = 7.4, 1.4 Hz, 1H), 7.45–7.27 (complex, 6H), 6.56 (dd, J = 17.4, 2.0 Hz, 1H), 5.87 (dd, J = 10.5, 2.0 Hz, 1H); 13C-NMR (CDCl3): δ 169.5, 166.6, 153.0, 141.5, 135.2, 132.2, 130.3, 129.9, 129.3, 127.7, 126.5, 126.3, 125.9, 125.2, 123.6, 123.2, 121.5, 53.2.
(±)-t-Butyl 2-(2-acryloylphthalazin-2(1H)-yl)acetate (
2p). This compound was prepared in 87% yield from
tert-butyl acetate (2.67 g, 3.08 mL, 23.0 mmol),
n-BuLi (7.7 mL, 19.3 mmol, 2.5 M in hexanes),
4 (2.99 g, 23.0 mmol), TEA (1.86 g, 2.56 mL, 18.4 mmol) and acryloyl chloride (1.39 g, 1.25 mL, 15.4 mmol) according to the literature procedure [
13]. The spectral data matched those reported [
13].
(±)-2-(2-Acryloylpthalazin-2(1H)-yl)acetic acid (
6). This compound was prepared in 94% yield from
2p (1.50 g, 5.00 mmol), and Bi(OTf)
3 (0.164 g, 0.25 mmol, 5 mol %) in benzene (25 mL) according to the literature procedure [
13]. The spectral data matched those reported [
13].
(±)-Ethyl 2-(2-acryloylphthalazin-2(1H)-yl)acetate (
2q). This compound was prepared in 95% yield from
6 (1.00 g, 4.10 mmol), and Bi(OTf)
3 (0.134 g, 0.20 mmol, 5 mol %) in ethanol (25 mL) according to the literature procedure [
13]. The spectral data matched those reported [
13].
(±)-Methyl 2-(2-acryloylphthalazin-2(1H)-yl)acetate (
2r). This compound was prepared in 95% yield from
6 (1.00 g, 4.10 mmol), Bi(OTf)
3 (0.134 g, 0.20 mmol, 5 mol %) in CH
3OH (25 mL) according to the literature procedure [
13]. The spectral data matched those reported [
13].
3.4. Preparation of Drug Candidates 3a–r
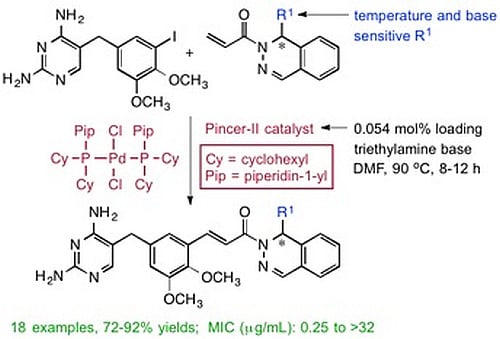
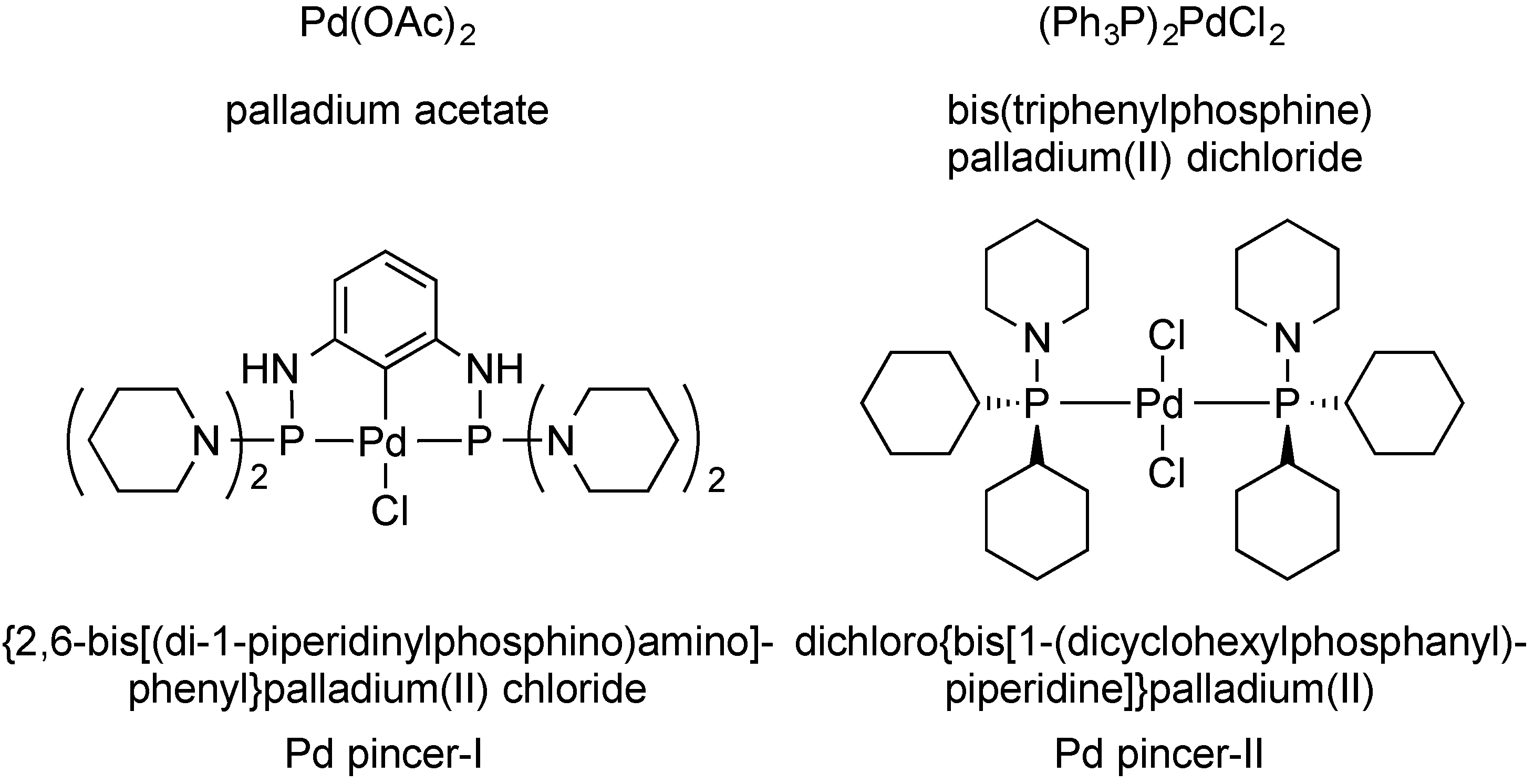
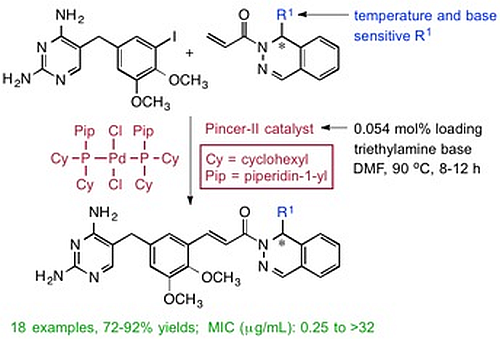
(±)-(E)-3-{5-[(2,4-Diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-1-[1-(2-methyl-1-propen-1-yl)phthalazin-2(1H)yl]-2-propen-1-one (
3a). To a stirred solution of
1 (1.00 g, 2.59 mmol) in dry DMF (10 mL) was added a solution of
2a (0.684 g, 2.85 mmol) in DMF (2 mL), followed by TEA (0.313 g, 0.431 mL, 3.10 mmol), and the Pd pincer-II catalyst (1 mg, 0.0014 mmol). The reaction was heated at 90 °C for 8 h and then cooled using an ice bath. The product was purified by directly pouring the crude reaction mixture onto a 50 cm × 2.5 cm silica gel chromatography column slurry packed with DCM. Impurities were eluted using DCM, and the final product was collected using DCM/MeOH/TEA (95:4:1). Evaporation of the solvent gave a yellow solid, which was dried under high vacuum for 2 h. MeOH (5 mL) was added to dissolve the crude product, followed by ether (10 mL), and the mixture was cooled for 4 h to crystallize the product. The product was filtered and dried under vacuum to afford
3a as a yellow solid (1.18, 88%). The melting point and spectral data matched those in the literature [
15].
(±)-(E)-1-(1-Allylphthalazin-2(1H)-yl)-3-{5-[(2,4-diaminopyrimidin-5-yl)methyl]-2,3-dimethoxy-phenyl}-2-propen-1-one (3b). This compound was prepared as above using 1 (2.00 g, 5.18 mmol), 2b (1.55 g, 6.73 mmol), TEA (0.680 g, 0.937 mL, 6.73 mmol), and the Pd pincer-II catalyst (2 mg, 0.0028) in dry DMF (15 mL) to give 3b (2.03 g, 81%) as a white solid, mp 215–217 °C. IR: 3418, 3123, 3122, 1657, 1641, 1598, 1564 cm−1; 1H-NMR (DMSO-d6): δ 7.91 (s, 1H), 7.86 (d, J = 16.2 Hz, 1H), 7.62 (d, J = 16.2 Hz, 1H), 7.60 (s, 1H), 7.56–7.37 (complex, 4H), 7.24 (s, 1H), 6.99 (s, 1H), 6.19 (br s, 2H), 5.95 (t, J = 6.3 Hz, 1H), 5.74 (br s, 2H), 5.66 (ddt, J = 17.4, 10.5, 6.3 Hz, 1H), 4.94 (dm, J = 10.5 Hz, 1H), 4.86 (dm, J = 17.4 Hz, 1H), 3.79 (s, 3H), 3.74 (s, 3H), 3.60 (s, 2H), 2.36 (m, 2H); 13C-NMR (DMSO-d6): δ 165.6, 162.3, 162.2, 155.8, 152.5, 146.0, 142.5, 136.6, 133.2, 132.9, 131.7, 128.3, 127.8, 126.6, 126.0, 123.7, 118.4, 118.3, 117.9, 114.8, 105.7, 60.8, 55.7, 54.9, 50.4, 32.4 (1 aromatic/alkene C unresolved); Anal. Calcd for C27H28N6O3·1.3 H2O: C, 63.84; H, 6.07; N, 16.54. Found: C, 63.84; H, 5.69; N, 16.28.
(±)-(E)-3-{5-[(2,4-Diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-1-(1-vinylphthalazin-2(1H)-yl)-2-propen-1-one (3c). This compound was prepared as above using 1 (1.60 g, 4.15 mmol), 2c (1.14 g, 5.38 mmol), TEA (0.543 g, 0.750 mL, 5.38 mmol) and the Pd pincer-II catalyst (1.5 mg, 0.0021 mmol) in dry DMF (15 mL) to give 3c (1.65 g, 85%) as a white solid, mp 210–212 °C. IR: 3354, 3169, 1638, 1593, 1567 cm−1; 1H-NMR (DMSO-d6): δ 7.91 (s, 1H), 7.89 (d, J = 16.0 Hz, 1H), 7.68 (d, J = 16.0 Hz, 1H), 7.60–7.44 (complex, 5H), 7.32 (d, J = 1.3 Hz, 1H), 7.25 (br s, 2H), 7.04 (d, J = 1.4, 1H), 6.73 (br s, 2H), 6.40 (d, J = 4.5 Hz, 1H), 5.80 (ddd, J = 1.5, 10.2, 4.5 Hz, 1H), 5.08 (d, J = 10.2 Hz, 1H), 4.78 (d, J = 17.0 Hz, 1H), 3.81 (s, 3H), 3.76 (s, 3H), 3.65 (s, 2H); 13C-NMR (DMSO-d6): δ 165.6, 163.2, 157.7, 152.6, 146.8, 146.3, 142.1, 136.9, 135.4, 135.0, 132.0, 131.7, 128.6, 127.9, 126.9, 126.3, 123.5, 118.8, 117.9, 115.3, 115.0, 107.6, 60.8, 55.9, 52.5, 31.9; Anal. Calcd for C26H26N6O3·4.6 H2O·0.1 C2H5OH: C, 56.21; H, 5.32; N, 15.01. Found: C, 56.25; H, 4.99; N, 15.04.
(±)-(E)-3-{5-[(2,4-Diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-1-(1-propylphthalazin-2(1H)-yl)-2-propen-1-one (
3d). This compound was prepared as above using
1 (1.00 g, 2.59 mmol),
2d (0.701 g, 3.10 mmol), TEA (0.313 g, 0.432 mL, 3.10 mmol), and the Pd pincer-II catalyst (1 mg, 0.0014 mmol) in dry DMF (15 mL) to give
3d (1.08 g, 86%) as a white solid. The melting point and spectral data matched those in the literature [
15].
(±)-(E)-3-{5-[(2,4-Diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-1-(1-pentylphthalazin-2(1H)-yl)-2-propen-1-one (3e). This compound was prepared as above using 1 (1.60 g, 4.15 mmol), 2e (1.39 g, 5.35 mmol), TEA (0.500 g, 0.689 mL, 4.95 mmol), and the Pd pincer-II catalyst (1.5 mg, 0.0021 mmol) in dry DMF (15 mL) to give 3e (1.91 g, 90%) as a white solid, mp 212–213 °C. IR: 3363, 3173, 1638, 1590, 1560 cm−1; 1H-NMR (DMSO-d6): δ 7.92 (s, 1H), 7.86 (d, J = 16.4 Hz, 1H), 7.64 (d, J = 16.4 Hz, 1H), 7.54 (s, 1H), 7.52 (m, 2H), 7.43 (td, J = 7.4, 0.8 Hz, 1H), 7.38 (d, J = 1.6 Hz, 1H), 7.28 (d, J = 1.6 Hz, 1H), 7.08 (br s, 2H), 7.02 (d, J = 1.5 Hz, 1H), 6.58 (br s, 2H), 5.82 (t, J = 6.7 Hz, 1H), 3.79 (s, 3H), 3.74 (s, 3H), 3.63 (s, 2H), 1.53 (m, 2H), 1.16 (m, 6H) 0.77 (t, J = 6.6 Hz, 3H); 13C-NMR (DMSO-d6): δ 165.6, 163.1, 158.3, 152.6, 148.2, 146.2, 142.8, 136.5, 135.2, 133.6, 131.7, 128.3, 127.9, 126.5, 126.1, 123.6, 118.7, 118.0, 114.9, 107.3, 60.8, 55.8, 50.5, 34.5, 32.0, 30.9, 24.0, 21.9, 13.8; Anal. Calcd for C29H34N6O3·3.9 H2O·0.3 C2H5OH: C, 59.94; H, 6.42; N, 14.21. Found: C, 59.61; H, 6.42; N, 14.21.
(±)-(E)-1-(1-Cyclopropylphthalazin-2(1H)-yl)-3-{5-[(2,4-diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-2-propen-1-one (
3f). This compound was prepared as above using
1 (1.00 g, 2.59 mmol),
2f (0.645 g, 2.80 mmol), TEA (0.313 g, 0.432 mL, 3.10 mmol), and the Pd pincer-II catalyst (1 mg, 0.0014 mmol) in dry DMF (15 mL) to give
3f (1.05 g, 84%) as a white solid. The melting point and spectral data matched those in the literature [
13].
(±)-(E)-1-(1-Cyclobutylphthalazin-2(1H)-yl)-3-{5-[(2,4-diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-2-propen-1-one (3g). This compound was prepared as above using 1 (1.50 g, 3.89 mmol), 2g (1.21 g, 5.04 mmol), TEA (0.510 g, 0.702 mL, 5.05 mmol), and the Pd pincer-II catalyst (1.5 mg, 0.0021 mmol) in dry DMF (15 mL) to give 3g (1.68 g, 87%) as a white solid, mp 125–127 °C. IR: 3397, 3272, 1642, 1605, 1562 cm−1; 1H-NMR (DMSO-d6): δ 7.93 (s, 1H), 7.85 (d, J = 16.0 Hz, 1H), 7.64 (d, J = 16.0 Hz, 1H), 7.59 (s, 1H), 7.52 (m, 2H), 7.44 (m, 2H), 7.25 (s, 1H), 7.00 (s, 1H), 6.39 (br s, 2H), 5.92 (br s, 2H), 5.86 (d, J = 8.2 Hz, 1H), 3.79 (s, 3H), 3.74 (s, 3H), 3.60 (s, 2H), 2.54 (sextet, J = 8.2 Hz, 1H), 1.84 (m, 3H), 1.63 (m, 3H); 13C-NMR (DMSO-d6): δ 165.9, 162.4, 161.4, 154.0, 152.5, 146.0, 143.3, 136.6, 136.3, 132.1, 131.7, 128.3, 127.8, 126.6, 126.1, 123.8, 118.4, 117.9, 114.8, 106.1, 60.8, 55.8, 53.8, 32.3, 25.1, 24.2, 17.1; Anal. Calcd for C28H30N6O3·1.6 H2O: C, 60.27; H, 6.31; N, 14.84. Found: C, 60.08; H, 6.07; N, 15.01.
(±)-(E)-1-(1-Cyclopentylphthalazin-2(1H)-yl)-3-{5-[(2,4-diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-2-propen-1-one (3h). This compound was prepared as above using 1 (1.50 g, 3.89 mmol), 2h (1.28 g, 5.04 mmol), TEA (0.510 g, 0.702 mL, 5.05 mmol), and the Pd pincer-II catalyst (1.5 mg, 0.0021 mmol) in dry DMF (15 mL) to give 3h (1.83 g, 92%) as a white solid, mp 190–192 °C. IR: 3338, 3174, 1637, 1594, 1560 cm−1; 1H-NMR (DMSO-d6, 400 MHz): δ 7.96 (s, 1H), 7.81 (d, J = 16.1 Hz, 1H), 7.60 (d, J = 16.1 Hz, 1H), 7.52 (s, 1H), 7.49 (m, 1H), 7.42 (d, J = 7.4 Hz, 2H), 7.38 (d, J = 7.4, 1H), 7.23 (d, J = 1.6 Hz, 1H), 6.97 (d, J = 1.6 Hz, 1H), 6.86 (br s, 2H), 6.37 (br s, 2H), 5.74 (d, J = 8.4 Hz, 1H), 3.76 (s, 3H), 3.70 (s, 3H), 3.58 (s, 2H), 2.10 (sextet, J = 8.4 Hz, 1H), 1.50 (m, 3H), 1.36 (m, 3H), 1.22 (m, 2H); 13C-NMR (DMSO-d6): δ 165.4, 162.9, 159.2, 152.6, 149.7, 146.1, 144.1, 136.6, 135.5, 133.1, 131.6, 128.3, 127.9, 126.9, 125.9, 124.1, 118.6, 118.0, 114.9, 107.0, 60.8, 55.8, 53.4, 44.6, 32.1, 29.0, 28.2, 24.1, 24.0; Anal. Calcd for C29H32N6O3·3.5 H2O: C, 60.32; H, 5.84; N, 14.55. Found: C, 60.19; H, 5.79; N, 14.57.
(±)-(E)-1-(1-tert-Butylphthalazin-2(1H)-yl)-3-{5-[(2,4-diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-2-propen-1-one (3i). This compound was prepared as above using 1 (2.20 g, 5.70 mmol), 2i (1.77 g, 7.32 mmol), TEA (0.748 g, 1.03 mL, 7.41 mmol), and the Pd pincer-II catalyst (2 mg, 0.0028 mmol) in dry DMF (15 mL) to give 3i (2.36 g, 83%) as a white solid, mp 228–230 °C. IR: 3354, 3174, 1637, 1590, 1561 cm−1; 1H-NMR (DMSO-d6): δ 7.97 (s, 1H), 7.84 (d, J = 16.0 Hz, 1H), 7.71 (d, J = 16.0 Hz, 1H), 7.56 (td, J = 7.5, 1.6 Hz, 1H), 7.53 (m, 2H), 7.48 (td, J = 7.4, 1.2 Hz, 1H), 7.46 (br s, 2H), 7.39 (d, J = 7.8 Hz, 1H), 7.32 (d, J = 1.5 Hz, 1H), 7.02 (d, J = 2.0 Hz, 1H), 6.92 (br s, 2H), 5.79 (s, 1H), 3.80 (s, 3H), 3.73 (s, 3H), 3.64 (s, 2H), 0.80 (s, 9H); 13C-NMR (DMSO-d6): δ 166.7, 163.4, 156.8, 152.6, 146.2, 145.1, 144.7, 136.3, 134.6, 131.3, 129.8, 128.5, 128.4, 128.0, 125.5, 125.1, 118.9, 118.4, 114.9, 107.9, 60.8, 53.7, 55.9, 39.0, 31.8, 26.4 (3C); Anal. Calcd for C28H32N6O3·2.4 H2O·1.1 C2H5OH: C, 56.50; H, 5.96; N, 14.21. Found: C, 56.31; H, 5.74; N, 13.93.
(±)-(E)-3-{5-[(2,4-Diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-1-[1-(furan-2-yl)phthalazin-2(1H)-yl]-2-propen-1-one (
3j). This compound was prepared as above using
1 (1.00 g, 2.59 mmol),
2j (0.785 g, 3.12 mmol), TEA (0.313 g, 0.432 mL, 3.10 mmol), and the Pd pincer-II catalyst (1 mg, 0.0014 mmol) in dry DMF (15 mL) to give
3j (1.19 g, 90%) as a white solid. The melting point and spectral data matched those in the literature [
13].
(±)-(E)-3-{5-[(2,4-Diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-1-[1-(thiophen-2-yl)phthalazin-2(1H)-yl]-2-propen-1-one (
3k). This compound was prepared as above using
1 (1.00 g, 2.59 mmol),
2k (0.836 g, 3.12 mmol), TEA (0.313 g, 0.432 mL, 3.10 mmol), and the Pd pincer-II catalyst (1 mg, 0.0014 mmol) in dry DMF (15 mL) to give
3k (1.10 g, 81%) as a white solid. The melting point and spectral data matched those in the literature [
13].
(±)-(E)-3-{5-[(2,4-Diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-1-[1-(1-methyl-1H-indol-2-yl)phthalazin-2(1H)-yl]-2-propen-1-one (3l). This compound was prepared as above using 1 (1.30 g, 3.37 mmol), 2l (1.27 g, 4.04 mmol), TEA (0.440 g, 0.60 mL, 4.36 mmol), and the Pd pincer-II catalyst (1.3 mg, 0.0018 mmol) in dry DMF (15 mL) to give 3l (1.45 g, 75%) as a white solid, mp 178–180 °C. IR: 3345, 3150, 1634, 1602, 1562 cm−1; 1H-NMR (DMSO-d6): δ 8.12 (s, 1H), 7.86 (d, J = 16.0 Hz, 1H), 7.63 (d, J = 16.0 Hz, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.55–7.48 (complex, 4H), 7.44 (d, J = 7.2 Hz, 1H), 7.36 (d, J = 7.8 Hz, 1H), 7.27 (d, J = 1.5 Hz, 1H), 7.26 (s, 1H), 7.11 (td, J = 7.4, 0.8 Hz, 1H), 7.02 (br s, 2H), 7.01 (d, J = 1.5 Hz, 1H), 6.95 (td, J = 7.4, 0.8 Hz, 1H), 6.52 (br s, 2H), 5.92 (s, 1H), 4.05 (s, 3H), 3.78 (s, 3H), 3.71 (s, 3H), 3.61 (s, 2H); 13C-NMR (DMSO-d6): δ 165.2, 163.0, 158.5, 152.6, 148.5, 146.2, 142.8, 140.3, 137.2, 136.6, 135.3, 132.3, 128.7, 127.8, 126.8, 126.7, 123.1, 121.6, 120.0, 119.4, 118.7, 117.8, 115.1, 110.1, 107.2, 100.9, 60.8, 55.8, 46.3, 32.0, 30.5 (2 aromatic C unresolved); Anal. Calcd for C33H31N7O3·4.1 H2O: C, 61.70; H, 5.28; N, 14.92. Found: C, 61.75; H, 5.13; N, 15.25.
(±)-(E)-1-[1-(Benzofuran-2-yl)phthalazin-2(1H)-yl]-3-{5-[(2,4-diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-2-propen-1-one (3m). This compound was prepared as above using 1 (1.80 g, 4.66 mmol), 2m (1.83 g, 6.06 mmol), TEA (0.615 g, 0.85 mL, 6.06 mmol), and the Pd pincer-II catalyst (2 mg, 0.0028 mmol) in dry DMF (15 mL) to give 3m (2.09 1.51 g, 80%) as a light yellow solid, mp 222–224 °C. IR: 3345, 3150, 1634, 1602, 1562 cm−1; 1H-NMR (DMSO-d6): δ 7.83 (d, J = 16.0 Hz, 1H), 7.83 (m, 2H), 7.73 (d, J = 16.0 Hz, 1H), 7.73 (obscured, 1H), 7.61 (s, 1H), 7.58 (t, J = 7.4 Hz, 1H), 7.53 (s, 1H), 7.52–7.42 (complex, 4H), 7.33 (t, J = 7.4 Hz, 1H), 7.17 (d, J = 1.7 Hz, 1H), 7.05 (d, J = 1.7 Hz, 1H), 6.18 (br s, 2H), 5.68 (br s, 2H), 5.05 (s, 1H), 3.78 (s, 3H), 3.76 (s, 3H), 3.60 (s, 2H); 13C-NMR (DMSO-d6): δ 166.1, 162.3, 162.1, 155.5, 154.4, 152.5, 150.5, 146.1, 141.2, 137.6, 136.6, 132.1, 131.3, 128.5, 127.8, 127.7, 126.8, 126.0, 125.7, 123.6, 123.2, 122.1, 119.2, 117.8, 115.2, 111.6, 108.7, 105.8, 60.7, 55.8, 42.0, 32.4; Anal. Calcd for C32H28N6O4: C, 68.56; H, 5.03; N, 14.92. Found: C, 68.30; H, 4.95; N, 14.92.
(±)-(E)-1-[1-(Benzo[b]thiophen-2-yl)phthalazin-2(1H)-yl]-3-{5-[(2,4-diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-2-propen-1-one (3n). This compound was prepared as above using 1 (1.60 g, 4.15 mmol), 2n (1.71 g, 5.38 mmol), TEA (0.543 g, 0.75 mL, 5.38 mmol), and the Pd pincer-II catalyst (1.5 mg, 0.0021 mmol) in dry DMF (15 mL) to give 3n (1.86 g, 78%) as a yellow solid, mp 256–258 °C. IR: 3347, 3180, 1637, 1601, 1559 cm−1; 1H-NMR (DMSO-d6): δ 8.27 (d, J = 6.4 Hz, 1H), 8.10–7.30 (complex, 10H), 7.15 (s, 2H), 6.18 (br s, 2H), 5.76 (s, 1H), 5.65 (br s, 2H), 5.06 (s, 2H), 3.84 (s, 3H), 3.80 (s, 3H), 3.67 (s, 2H); 13C-NMR (DMSO-d6): δ 165.9, 162.9, 158.8, 152.6, 149.1, 146.3, 144.5, 142.5, 139.1, 138.5, 137.6, 135.4, 132.4, 131.5, 129.3, 127.7, 127.3, 126.5, 124.6, 124.5, 123.8, 123.5, 122.4 (2C), 118.8, 117.6, 115.1, 107.1, 60.8, 55.8, 49.6, 32.0; Anal. Calcd for C32H28N6O3S·2.1 H2O: C, 62.55; H, 5.18; N, 13.14. Found: C, 62.72; H, 4.91; N, 13.06.
(±)-(E)-1-[1-(Benzo[d]thiazol-2-yl)phthalazin-2(1H)-yl]-3-{5-[(2,4-diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}-2-propen-1-one (3o). This compound was prepared as above using 1 (1.50 g, 3.89 mmol), 2o (1.61 g, 5.04 mmol), TEA (0.510 g, 0.70 mL, 5.05 mmol), and the Pd pincer-II catalyst (1 mg, 0.0014 mmol) in dry DMF (15 mL) to give 3o (1.61 g, 72%) as a white solid, mp 145–147 °C. IR: 3339, 3202, 1660, 1614, 1564 cm−1; 1H-NMR (DMSO-d6): δ 8.04 (s, 1H), 8.03 (obscured dm, 1H), 7.95 (d, J = 16.0 Hz, 1H), 7.91 (dd, J = 7.4, 0.8 Hz, 1H), 7.74 (d, J = 16.0 Hz, 1H), 7.63 (td, J = 7.4, 1.2 Hz, 1H), 7.60 (m, 2H), 7.54 (td, J = 7.4, 0.8 Hz, 1H), 7.47 (td, J = 7.4, 0.8 Hz, 1H), 7.45 (s, 1H), 7.40 (td, J = 7.4, 1.2 Hz, 1H), 7.34 (d, J = 1.6 Hz, 1H), 7.04 (d, J = 1.6 Hz, 1H), 6.66 (br s, 2H), 6.17 (br s, 2H), 5.77 (s, 1H), 3.81 (s, 3H), 3.75 (s, 3H), 3.63 (s, 2H); 13C-NMR (DMSO-d6): δ 170.1, 168.1, 162.6, 160.3, 152.5, 152.2, 151.9, 146.3, 141.9, 137.8, 136.0, 134.7, 132.3, 130.1, 129.5, 127.8, 127.6, 126.7, 126.4, 125.5, 123.2, 122.8, 122.4, 118.6, 117.3, 115.2, 106.5, 60.8, 55.8, 52.5, 32.2; Anal. Calcd for C31H27N7O3S·3.0 H2O·0.5 C2H5OH: C, 58.70; H, 5.12; N, 14.08. Found: C, 58.49; H, 4.81; N, 14.15.
(±)-tert-Butyl (E)-2-[2-(3-{5-[(2,4-diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}acryloyl)-1,2-dihydrophthalazin-1-yl]acetate (
3p). This compound was prepared as above using
1 (1.00 g, 2.59 mmol),
2p (0.935 g, 3.12 mmol), TEA (0.313 g, 0.432 mL, 3.10 mmol), and the Pd pincer-II catalyst (1 mg, 0.0014 mmol) in dry DMF (15 mL) to give
3p (1.20 g, 83%) as a white solid. The melting point and spectral data matched those in the literature [
13].
(±)-Ethyl (E)-2-[2-(3-{5-[(2,4-diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}acryloyl)-1,2-dihydrophthalazin-1-yl]acetate (
3q). This compound was prepared as above using
1 (1.00 g, 2.59 mmol),
2q (0.849 g, 3.12 mmol), TEA (0.313 g, 0.432 mL, 3.10 mmol), and the Pd pincer-II catalyst (1 mg, 0.0014 mmol) in dry DMF (15 mL) to give
3q (1.10 g, 80%) as a white solid. The melting point and spectral data matched those in the literature [
13].
(±)-Methyl (E)-2-[2-(3-{5-[(2,4-diaminopyrimidin-5-yl)methyl]-2,3-dimethoxyphenyl}acryloyl)-1,2-dihydrophthalazin-1-yl-acetate (
3r). This compound was prepared as above using
1 (1.00 g, 2.59 mmol),
2r (0.805 g, 3.12 mmol), TEA (0.313 g, 0.432 mL, 3.10 mmol), and the Pd pincer-II catalyst (1 mg, 0.0014 mmol) in dry DMF (15 mL) to give
3r (1.10 g, 82%) as a white solid. The melting point and spectral data matched those in the literature [
13].







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


