
Discovery of Novel Small Molecule Anti-HCV Agents via the CypA Inhibitory Mechanism Using O-Acylation-Directed Lead Optimization

Abstract
:1. Introduction
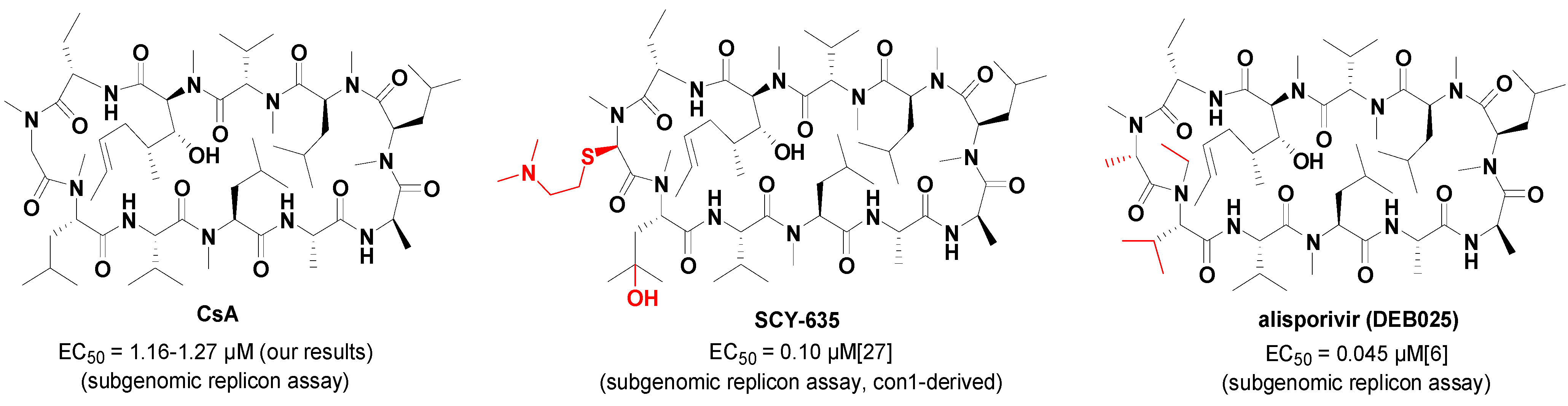
2. Results and Discussion
2.1. Screening of Compounds for Anti-HCV Activity

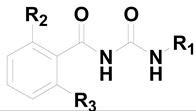
| Compound | R1 | R2 | R3 | Anti-HCV JFH1-2a Replicon Assay EC50 (μM) ± SD b |
|---|---|---|---|---|
| 1 |  | OH | OH | 0.92 ± 0.12 |
| 2 |  | OH | OH | >10 |
| 3 | 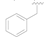 | OH | OH | >50 |
| 4 | 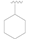 | OH | OH | >50 |
| 5 |  | Cl | H | >50 |
| 6 |  | NO2 | H | >50 |
| 7 | 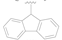 | CN | H | >50 |
| 8 | 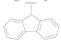 | F | H | 1.65 ± 0.32 |
| 9 |  | CF3 | H | >10 |
| 10 |  | MeS | H | >50 |
| 11 | 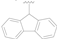 | F | F | >50 |
| 12 |  | F | MeS | >50 |
| 13 | 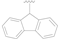 | Cl | MeO | >50 |
| 14 |  | 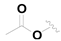 | OH | 0.55 ± 0.03 |
| 15 |  |  |  | 0.46 ± 0.05 |
| 16 |  |  | OH | >20 |
| 17 | 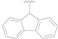 |  |  | >20 |
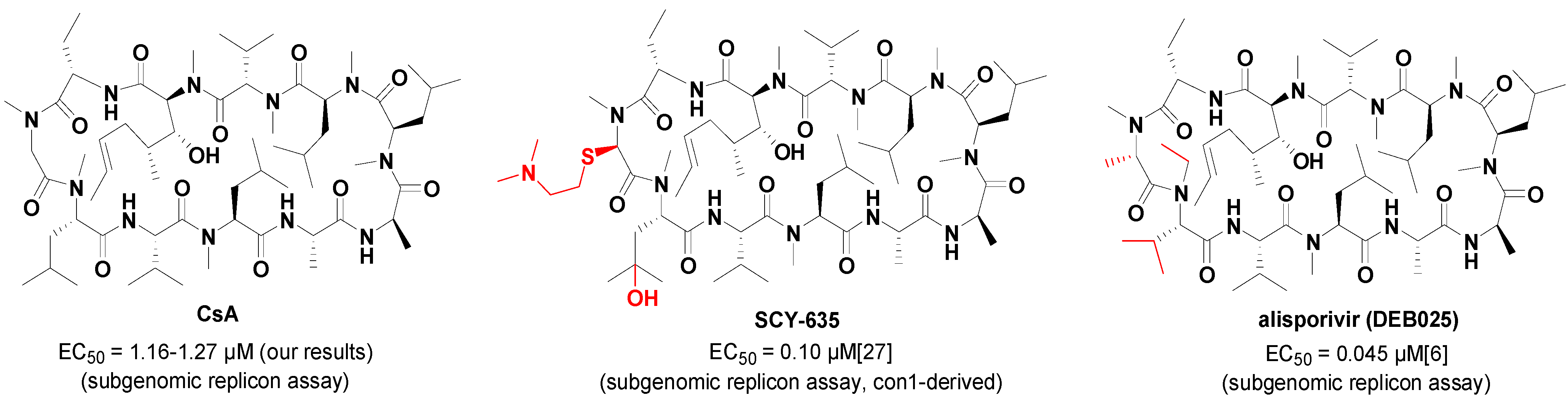
| CsA | 1.16 ± 0.14 |
2.2. Spleen Cells Proliferation Inhibition Assay
| Cmpd | T Spleen Cells IC50 (μM) ± SD a | B Spleen Cells IC50 (μM) ± SD a |
|---|---|---|
| 1 | 33.03 ± 5.72 | 51.93 ± 8.68 |
| CsA | 0.01 ± 0.005 | 0.04 ± 0.008 |
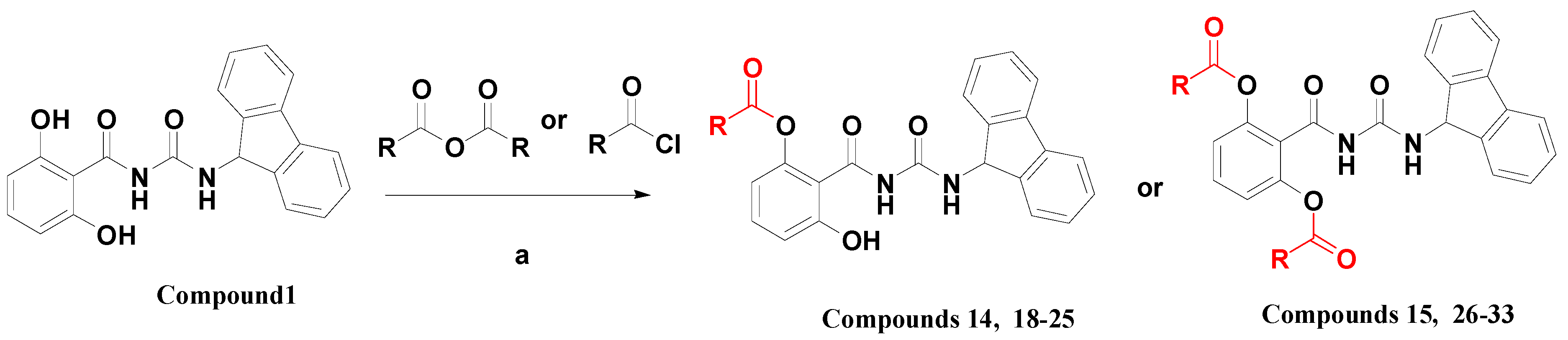
2.3. Derivatives Design and Synthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | R1 | R2 | Cmpd | R1 | R2 |
|---|---|---|---|---|---|
| 18 |  | H | 26 |  |  |
| 19 | 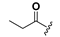 | H | 27 |  |  |
| 20 | 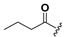 | H | 28 | 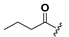 |  |
| 21 |  | H | 29 |  | 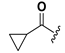 |
| 22 |  | H | 30 |  | 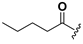 |
| 23 | 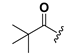 | H | 31 |  | 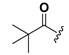 |
| 24 |  | H | 32 | 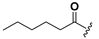 |  |
| 25 |  | H | 33 |  |  |
2.4. Virus Assays
| Cmpd | Anti-HCV Activity | ||||
|---|---|---|---|---|---|
| EC50 (μM) ± SD a | CC50 (μM) | ||||
| Replicon Assay | Virus Assay | ||||
| H77-1a | JFH1-2a | Con1-1b | JFH1-2a | ||
| 1 | 1.11 ± 0.33 | 0.92 ± 0.12 | 0.78 ± 0.19 | 0.67 ± 0.23 | >16 |
| 14 | 0.77 ± 0.37 | 0.55 ± 0.03 | 0.80 ± 0.23 | 0.71 ± 0.34 | >16 |
| 15 | 0.80 ± 0.13 | 0.46 ± 0.05 | 0.36 ± 0.04 | 0.74 ± 0.36 | >16 |
| 18 | 0.60 ± 0.35 | 1.05 ± 0.01 | 1.03 ± 0.02 | 0.70 ± 0.23 | >16 |
| 19 | 0.38 ± 0.22 | 0.94 ± 0.07 | 0.72 ± 0.20 | 0.47 ± 0.04 | >16 |
| 20 | 0.98 ± 0.03 | 0.70 ± 0.02 | 0.49 ± 0.02 | 0.28 ± 0.02 | >16 |
| 21 | 0.57 ± 0.20 | 0.43 ± 0.08 | 0.85 ± 0.24 | 0.78 ± 0.35 | >16 |
| 22 | 1.00 ± 0.06 | 0.47 ± 0.02 | 0.44 ± 0.10 | 0.44 ± 0.03 | >16 |
| 23 | 0.67 ± 0.05 | 0.48 ± 0.02 | 0.83 ± 0.28 | 0.53 ± 0.05 | >16 |
| 24 | 1.05 ± 0.01 | 1.08 ± 0.05 | 1.00 ± 0.01 | 0.74 ± 0.37 | >16 |
| 25 | 0.85 ± 0.21 | 0.45 ± 0.08 | 0.81 ± 0.24 | 0.19 ± 0.08 | >16 |
| 26 | 0.53 ± 0.21 | 0.36 ± 0.03 | 0.39 ± 0.05 | 1.09 ± 0.03 | >16 |
| 27 | 0.52 ± 0.27 | 0.38 ± 0.03 | 0.38 ± 0.02 | 0.42 ± 0.08 | >16 |
| 28 | 0.79 ± 0.20 | 0.84 ± 0.32 | 0.62 ± 0.15 | 0.68 ± 0.47 | >16 |
| 29 | 0.52 ± 0.14 | 0.42 ± 0.14 | 0.60 ± 0.19 | 0.74 ± 0.35 | >16 |
| 30 | 0.73 ± 0.11 | 0.62 ± 0.05 | 0.83 ± 0.26 | 0.47 ± 0.01 | >16 |
| 31 | 0.87 ± 0.27 | 0.75 ± 0.29 | 0.56 ± 0.20 | 0.84 ± 0.21 | >16 |
| 32 | 0.63 ± 0.29 | 0.38 ± 0.05 | 0.73 ± 0.34 | 0.74 ± 0.31 | >16 |
| 33 | 0.54 ± 0.15 | 0.98 ± 0.01 | 0.73 ± 0.20 | 0.95 ± 0.02 | >16 |
| CsA | 1.26 ± 0.20 | 1.16 ± 0.14 | 1.27 ± 0.32 | 0.40 ± 0.11 | >8 |
| F680, F684 b | 0.20 c | ||||
| Cmpd | Anti-HCV Activity (Virus Assay) | ||
|---|---|---|---|
| EC50 (μM) ± SD a | CC50 (μM) | ||
| JFH1-2a (Wild Type) | JFH1-2a (S282T) | ||
| 1 | 0.52 ± 0.16 | 0.56 ± 0.13 | >20 |
| 25 | 0.27 ± 0.09 | 0.29 ± 0.07 | >20 |
| Sofosbuvir | 0.22 ± 0.07 | 1.14 ± 0.21 | >20 |
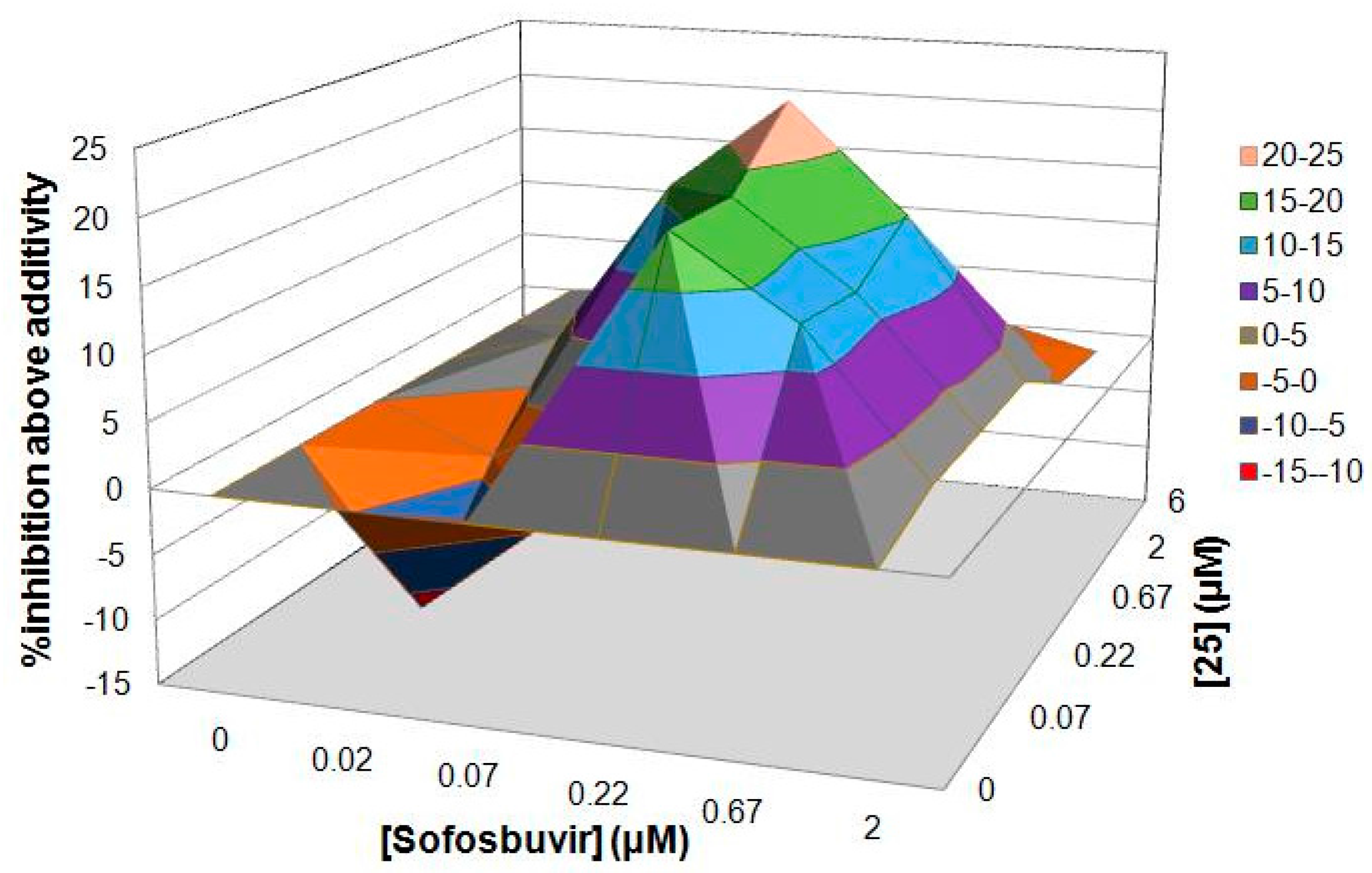
2.5. Drug Combination Assay

2.6. CypA Enzyme Inhibition Assay
| Cmpd | Enzyme Inhibition Assay IC50 (μM) ± SD a |
|---|---|
| 1 | 0.032 ± 0.002 |
| 14 | 2.43 ± 0.50 |
| 18 | 1.99 ± 0.47 |
| 15 and 19–33 | >5 |
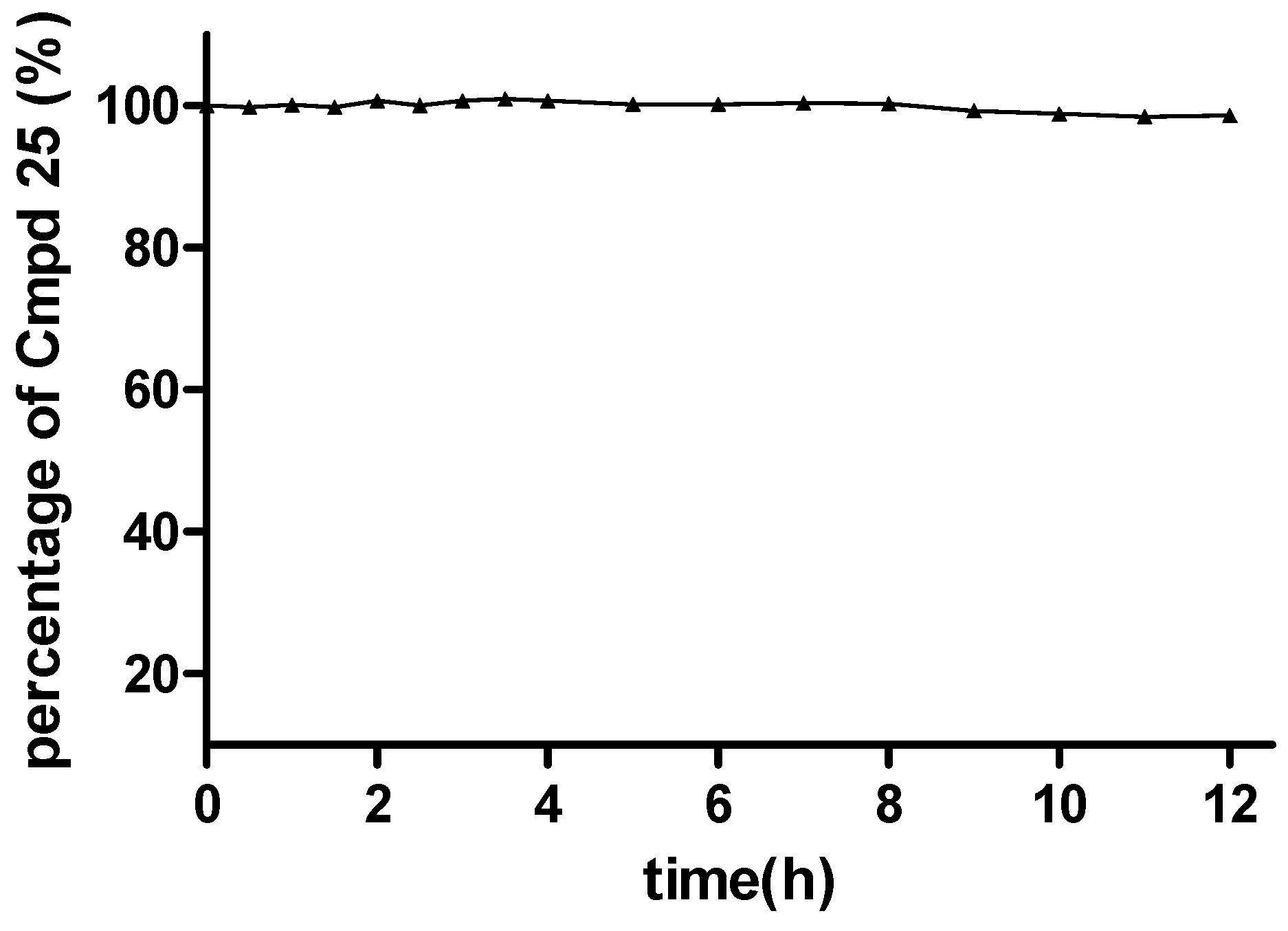
2.7. Preliminary Verification of the Hypothetical Prodrug Mechanism

3. Experimental Section
3.1. General Information
3.2. Biological Assays
3.2.1. Spleen Cells Proliferation Inhibition Assay
3.2.2. Virus Assay
3.2.3. Drug Combination Assay
3.2.4. CypA Enzyme Inhibition Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Te, H.S.; Jensen, D.M. Epidemiology of hepatitis B and C viruses: A global overview. Clin. Liver Dis. 2010, 14, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Shepard, C.W.; Finelli, L.; Alter, M.J. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 2005, 5, 558–567. [Google Scholar] [CrossRef]
- World Health Organization (WHO). WHO Report on the Hepatitis C, 2012; WHO: Geneva, Switzerland, 2012. [Google Scholar]
- Cui, H.; Qing, J.; Guo, Y.; Wang, Y.; Cui, L.; He, T.; Zhang, L.; Liu, L. Stapled peptide-based membrane fusion inhibitors of hepatitis C. Bioorg. Med. Chem. 2013, 21, 3547–3554. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Qing, J.; Wang, S.; Wang, H.; Zhang, L.; Tang, Y. Design and synthesis of imidazo[1,2-α][1,8]-naphthyridine derivatives as anti-HCV agents via direct C-H arylation. Org. Biomol. Chem. 2014, 12, 2344–2348. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Gallay, P. Curing a viral infection by targeting the host: The example of cyclophilin inhibitors. Antiviral Res. 2013, 99, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.; Wittmann-Liebold, B.; Lang, K.; Kiefhaber, T.; Schmid, F.X. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 1989, 337, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Peptidylprolyl cis/trans isomerases (immunophilins): Biological diversity-targets-functions. Curr. Top. Med. Chem. 2003, 3, 1315–1347. [Google Scholar] [CrossRef] [PubMed]
- Dornan, J.; Taylor, P.; Walkinshaw, M.D. Structures of immu-nophilins and their ligand complexes. Curr. Top. Med. Chem. 2003, 3, 1392–1409. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.J.; Mei, Q.; Li, J.T.; He, H.Y. Cyclophilin A and viral infections. Biochem. Biophys. Res. Commun. 2012, 424, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Patel, H.D.; Sapp, M. Target cell cyclophilins facilitate human papillomavirus type 16 infection. PLoS Pathog. 2009. [Google Scholar] [CrossRef] [PubMed]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 1984, 226, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Sedrani, R.; Kallen, J.; Cabrejas, L.M.M.; Papageorgiou, C.D.; Senia, F.; Rohrbach, S.; Wagner, D.; Thai, B.; Eme, A.M.J.; France, J.; et al. Sanglifehrin-cyclophilin interaction: Degradation work, synthetic macrocyclic analogues, X-ray crystal structure, and binding data. J. Am. Chem. Soc. 2003, 125, 3849–3859. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, J.; Gui, C.; Zhang, L.; Qin, Y.; Xu, Q.; Zhang, J.; Liu, H.; Shen, X.; Jiang, H. Discovering novel chemical inhibitors of human cyclophilin A: Virtual screening, synthesis, and bioassay. Bioorg. Med. Chem. 2006, 14, 2209–2224. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, J.; Zhang, L.; Wang, F.; Gui, C.; Zhang, L.; Qin, Y.; Xu, Q.; Liu, H.; Nan, F.; et al. One novel quinoxaline derivative as a potent human cyclophilin A inhibitor shows highly inhibitory activity against mouse spleen cell proliferation. Bioorg. Med. Chem. 2006, 14, 5527–5534. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, J.; Chen, J.; Luo, X.; Zhu, W.; Shen, J.; Liu, H.; Shen, X.; Jiang, H. Strategy for discovering chemical inhibitors of human cyclophilin A: Focused library design, virtual screening, chemical synthesis and bioassay. J. Comb. Chem. 2006, 8, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Guichou, J.F.; Viaud, J.; Mettling, C.; Subra, G.; Lin, Y.L.; Chavanieu, A. Structure-based design, synthesis, and biological evaluation of novel inhibitors of human cyclophilin A. J. Med. Chem. 2006, 49, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Hijikata, M.; Hosaka, M.; Yamaji, M.; Shimotohno, K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 2003, 38, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Robotham, J.M.; Nelson, H.B.; Irsigler, A.; Kenworthy, R.; Tang, H.L. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 2008, 82, 5269–5278. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, U.; Bobardt, M.; Selvarajah, S.; Yang, F.; Tang, H.L.; Sakamoto, N.; Vuagniaux, G.; Parkinson, T.; Gallay, P. The isomerase active site of Cyclophilin A is critical for hepatitis C virus replication. J. Biol. Chem. 2009, 284, 16998–17005. [Google Scholar] [CrossRef] [PubMed]
- Gaither, L.A.; Borawski, J.; Anderson, L.J.; Balabanis, K.A.; Devay, P.; Joberty, G.; Rau, C.; Schirle, M.; Bouwmeester, T.; Mickanin, C.; et al. Multiple cyclophilins involved in different cellular pathways mediate HCV replication. Virology 2010, 397, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Stauffer, S.; Berger, C.; Pertel, T.; Schmitt, J.; Kallis, S.; Lopez, M.Z.; Lohmann, V.; Luban, J.; Bartenschlager, R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 2009. [Google Scholar] [CrossRef]
- Hanoulle, X.; Badillo, A.; Wieruszeski, J.M.; Verdegem, D.; Landrieu, I.; Bartenschlager, R.; Penin, F.; Lippens, G. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyI cis/trans isomerase activity of cyclophilins A and B. J. Biol. Chem. 2009, 284, 13589–13601. [Google Scholar] [CrossRef] [PubMed]
- Coelmont, L.; Hanoulle, X.; Chatterji, U.; Berger, C.; Snoeck, J.; Bobardt, M.; Lim, P.; Vliegen, I.; Paeshuyse, J.; Vuagniaux, G. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Nag, A.; Robotham, J.M.; Tang, H. Suppression of viral RNA binding and the assembly of infectious hepatitis C virus particles in vitro by cyclophilin inhibitors. J. Virol. 2012, 86, 12616–12624. [Google Scholar] [CrossRef] [PubMed]
- Paeshuyse, J.; Kaul, A.; de Clercq, E.; Rosenwirth, B.; Dumont, J.M.; Scalfaro, P.; Bartenschlager, R.; Neyts, J. The non-immunosuppressive cyclosporine DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 2006, 43, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.; DiMassimo, B.; Rusnak, P.; Heuman, D.; Lalezari, J.; Sluder, A.; Scorneaux, B.; Mosier, S.; Kowalczyk, P.; Ribeill, Y.; et al. The cyclophilin inhibitor SCY-635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. J. Hepatol. 2012, 57, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Ahmed-Belkacem, A.; Colliandre, L.; Ahnou, N.; Lerat, H.; Bessin, Y.; Barthe, P.; Bourget, W.; Douguet, D.; Guichou, J.F.; Pawlotsky, J.M. New cyclophilin inhibitors unrelated to cyclosporine potently inhibit HCV replication and revert HCV-induced mitochondrial dysfunction. In Hepatology; Wiley-Blackwell: Hoboken, NJ, USA, 2012. [Google Scholar]
- Ni, S.; Yuan, Y.; Huang, J.; Mao, X.; Lv, M.; Zhu, J.; Shen, X.; Pei, J.; Lai, L.; Jiang, H.; Li, J. Discovering potent small molecule inhibitors of cyclophilin A using de novo drug design approach. J. Med. Chem. 2009, 52, 5295–5298. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Espiritu, C.; Bansal, S.; Steuer, H.M.M.; Niu, C.; Zennou, V.; Keilman, M.; Zhu, Y.; Lan, S.; Otto, M.J.; et al. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob. Agents Chemother. 2012, 56, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Prichard, M.N.; Shipman, C., Jr. A three-dimensional model to analyze drug-drug interactions. Antiviral Res. 1990, 14, 181–205. [Google Scholar] [CrossRef]
- Han, Q.; Xu, C.; Wu, C.; Zhu, W.; Yang, R.; Chen, X. Compensatory mutations in NS3 and NS5A proteins enhance the virus production capability of hepatitis C reporter virus. Virus Res. 2009, 145, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liao, Q.; Yang, R.; Chen, X.; Chen, X. A novel luciferase and GFP dual reporter virus for rapid and convenient evaluation of hepatitis C virus replication. Virus Res. 2011, 155, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Kofron, J.L.; Kuzmic, P.; Kishore, V.; Colon-Bonilla, E.; Rich, D.H. Determination of kinetic constants for peptidyl prolyl cis-trans isomerasesby an improved spectrophotometric assay. Biochemistry 1991, 30, 6127–6134. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, W.; Qing, J.; Mei, H.; Mao, F.; Huang, J.; Zhu, J.; Jiang, H.; Liu, L.; Zhang, L.; Li, J. Discovery of Novel Small Molecule Anti-HCV Agents via the CypA Inhibitory Mechanism Using O-Acylation-Directed Lead Optimization. Molecules 2015, 20, 10342-10359. https://doi.org/10.3390/molecules200610342
Yan W, Qing J, Mei H, Mao F, Huang J, Zhu J, Jiang H, Liu L, Zhang L, Li J. Discovery of Novel Small Molecule Anti-HCV Agents via the CypA Inhibitory Mechanism Using O-Acylation-Directed Lead Optimization. Molecules. 2015; 20(6):10342-10359. https://doi.org/10.3390/molecules200610342
Chicago/Turabian StyleYan, Wenzhong, Jie Qing, Hanbing Mei, Fei Mao, Jin Huang, Jin Zhu, Hualiang Jiang, Lei Liu, Linqi Zhang, and Jian Li. 2015. "Discovery of Novel Small Molecule Anti-HCV Agents via the CypA Inhibitory Mechanism Using O-Acylation-Directed Lead Optimization" Molecules 20, no. 6: 10342-10359. https://doi.org/10.3390/molecules200610342
APA StyleYan, W., Qing, J., Mei, H., Mao, F., Huang, J., Zhu, J., Jiang, H., Liu, L., Zhang, L., & Li, J. (2015). Discovery of Novel Small Molecule Anti-HCV Agents via the CypA Inhibitory Mechanism Using O-Acylation-Directed Lead Optimization. Molecules, 20(6), 10342-10359. https://doi.org/10.3390/molecules200610342