
Molecular Docking and Structure-Based Drug Design Strategies

Abstract
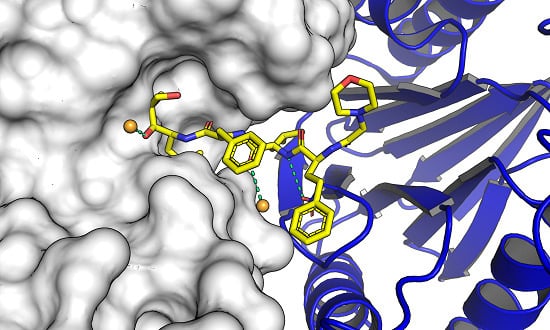
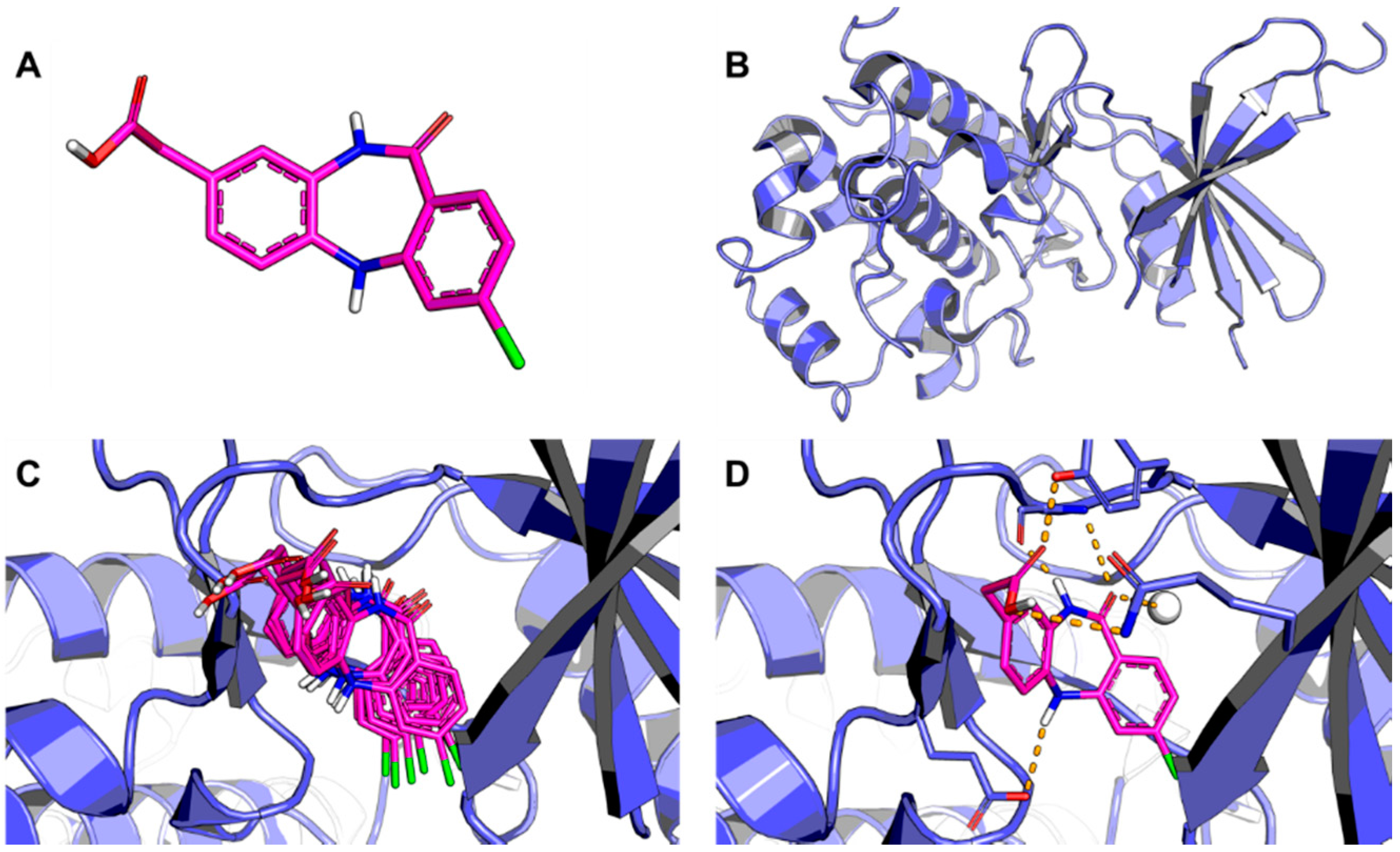
:
1. Introduction
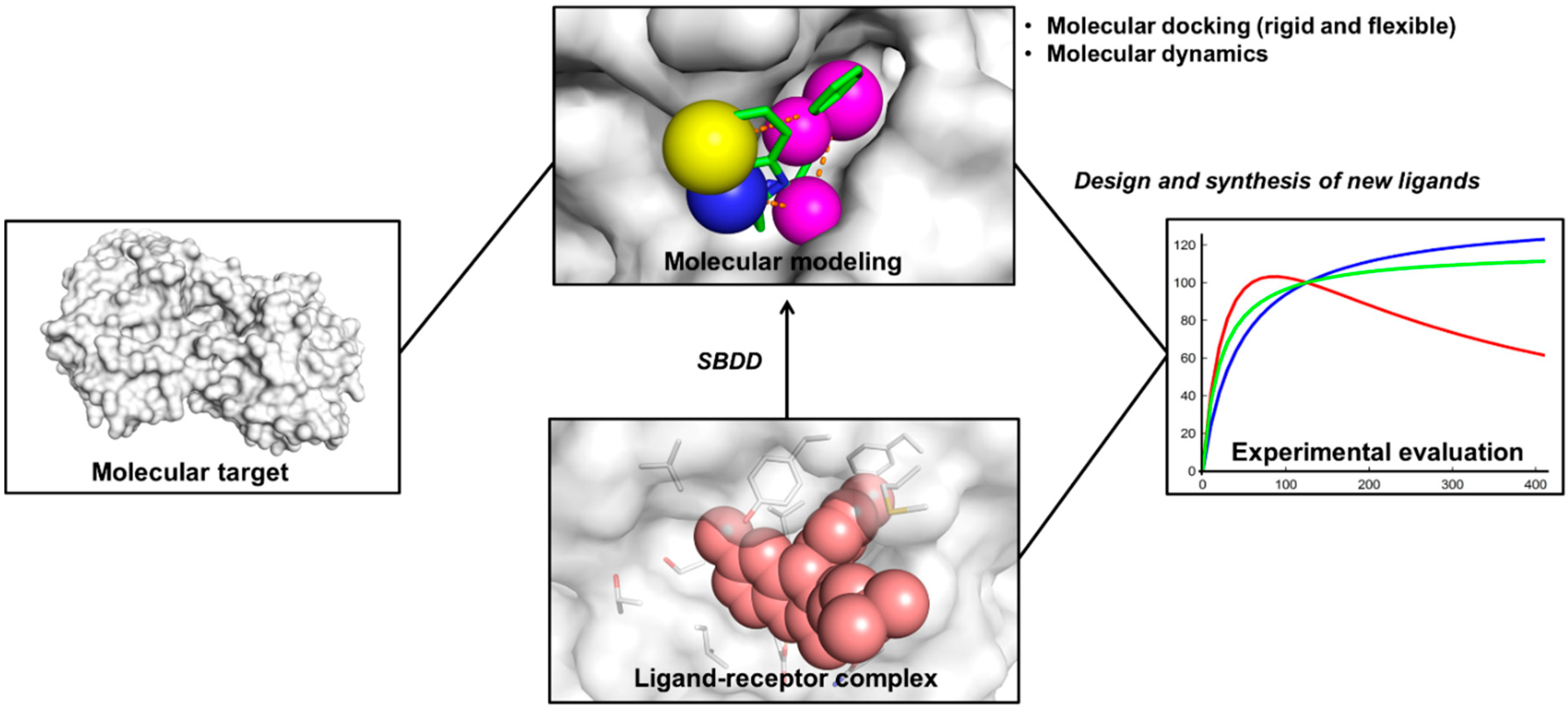
2. Structure-Based Drug Design (SBDD)

3. Molecular Docking

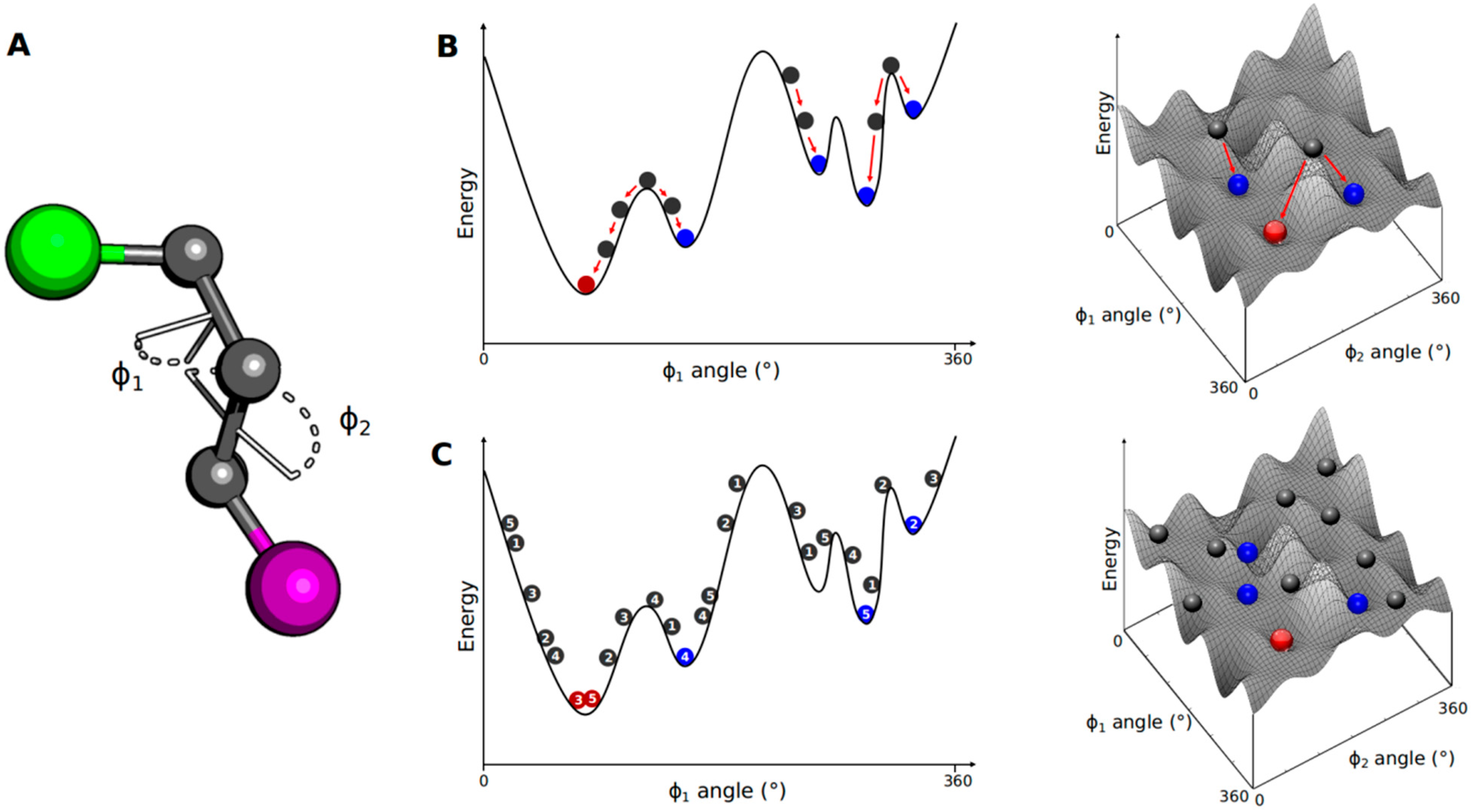
3.1. Conformational Search



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systematic Search | Random/Stochastic Search |
|---|---|
| eHiTS [28] | AutoDock [34] |
| FRED [30] | Gold [35] |
| Surflex-Dock [31] | PRO_LEADS [44] |
| DOCK [32] | EADock [45] |
| GLIDE [37] | ICM [46] |
| EUDOC [38] | LigandFit [47] |
| FlexX [39] | Molegro Virtual Docker [48] |
| Hammerhead [40] | CDocker [49] |
| Flog [41] | GlamDock [50] |
| SLIDE [42] | PLANTS [51] |
| ADAM [43] | MolDock [52] |
| MOE_Dock [53] |
3.2. Evaluation of Binding Energetics
| Force-Field-Based | Empirical | Knowledge-Based |
|---|---|---|
| DOCK [32] | AutoDock [34] | SMoG [82] |
| AutoDock [34] | GlideScore [37] | DrugScore [62] |
| GoldScore [35] | ChemScore [60] | PMF_Score [83] |
| ICM [46] | X_Score [66] | MotifScore [84] |
| LigandFit [47] | F_Score [73] | RF_Score [85] |
| Molegro Virtual Docker [48] | Fresno [75] | PESD_SVM [86] |
| SYBYL_G-Score [73] | SCORE [76] | PoseScore [87] |
| SYBYL_D-Score [73] | LUDI [77] | |
| MedusaScore [74] | SFCscore [78] | |
| HYDE [79] | ||
| LigScore [80] | ||
| PLP [81] |
3.3. Covalent Bonds in Molecular Docking
3.4. Molecular Dynamics
3.5. Structural Water
3.6. Protein-Protein Interaction Inhibitors and Molecular Docking
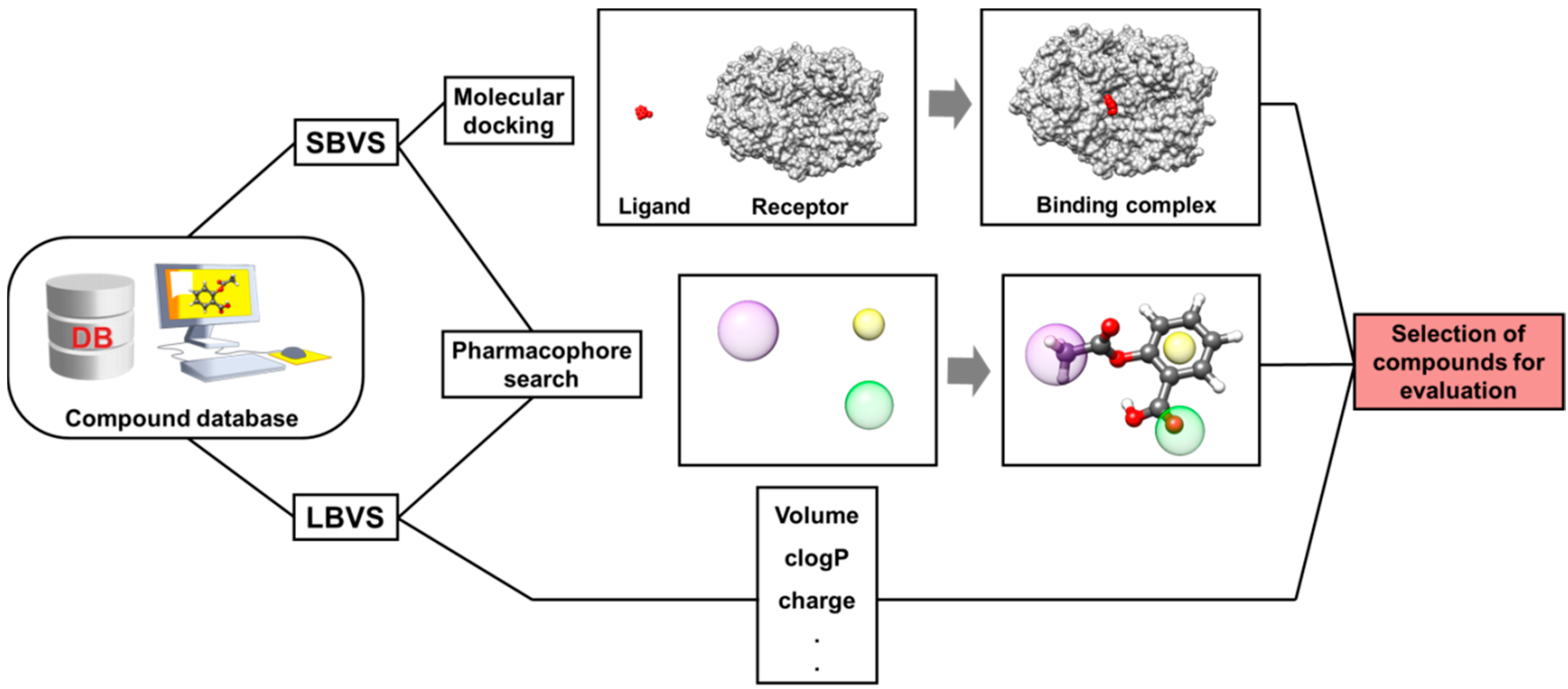
4. Virtual Screening (VS)

4.1. Ligand-Based Virtual Screening (LBVS)
| Software |
|---|
| Molinspiration [133,134] |
| OSIRIS Property Explorer [134,135] |
| Molsoft [136] |
| MoKa [137,138] |
| Commercial | Academic |
|---|---|
| Hiphop [141] | PharmaGist [154,155] |
| HypoGen [141] | ALADDIN [156] |
| HypoRefine [141] | RAPID [157] |
| GASP [142,143] | DANTE [158] |
| DiscoTech [143,144] | APOLLO [159] |
| GALAHAD [143,145] | CLEW [160] |
| LigandScout [146,147] | MPHIL [161] |
| MOE [148,149] | GAMMA [162] |
| PHASE [150,151] | SCAMPI [163] |
| XED [152,153] | Apex-3D [164] LigBuilder [165,166] |
4.2. Structure-Based Virtual Screening (SBVS)
| Database |
|---|
| Zinc [175,176] |
| PubChem [177,178] |
| ChemSpider [179,180] |
| ChEMBL [181,182] |
| NuBBE DB [183,184] |
| ChemBank [185,186] |
| eMolecules [187] |
| DrugBank [188,189] |
| Binding DB [190,191] |
5. Molecular Docking and Structure-Based Drug Design Studies
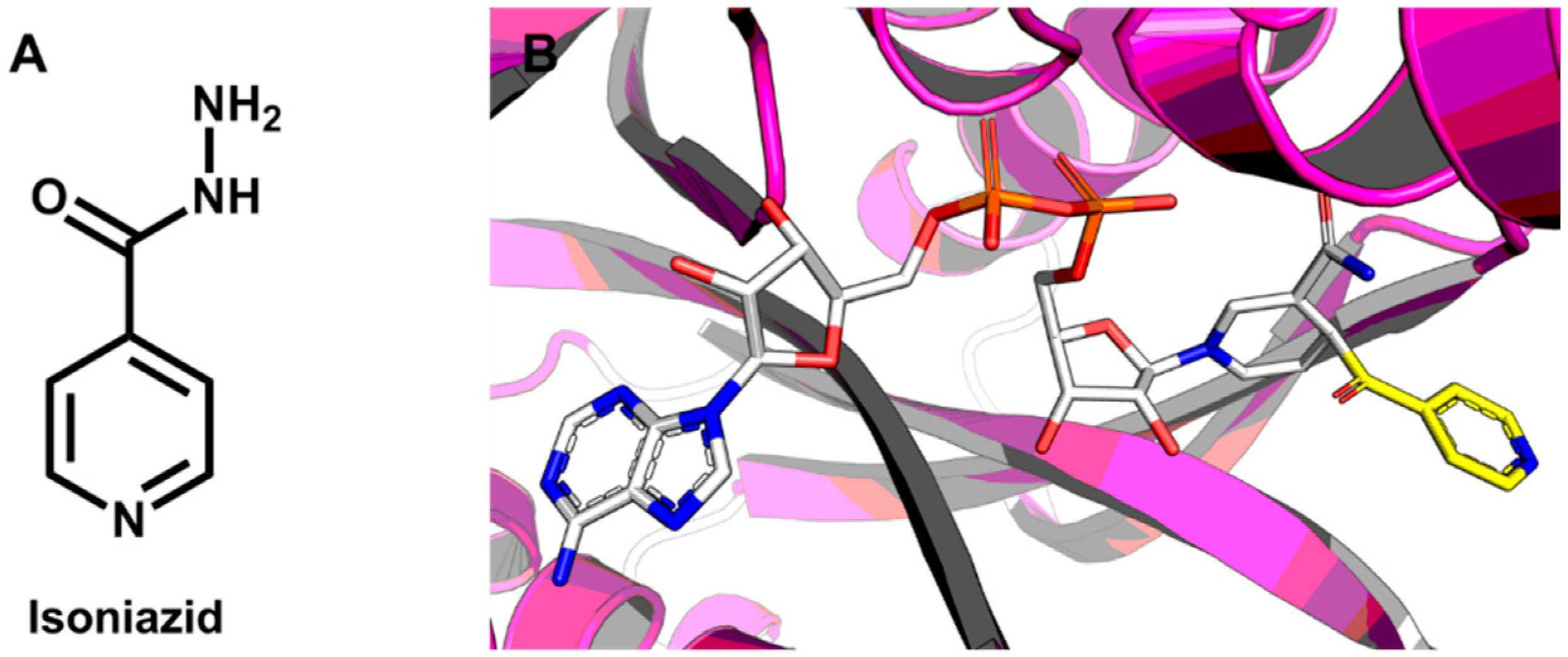
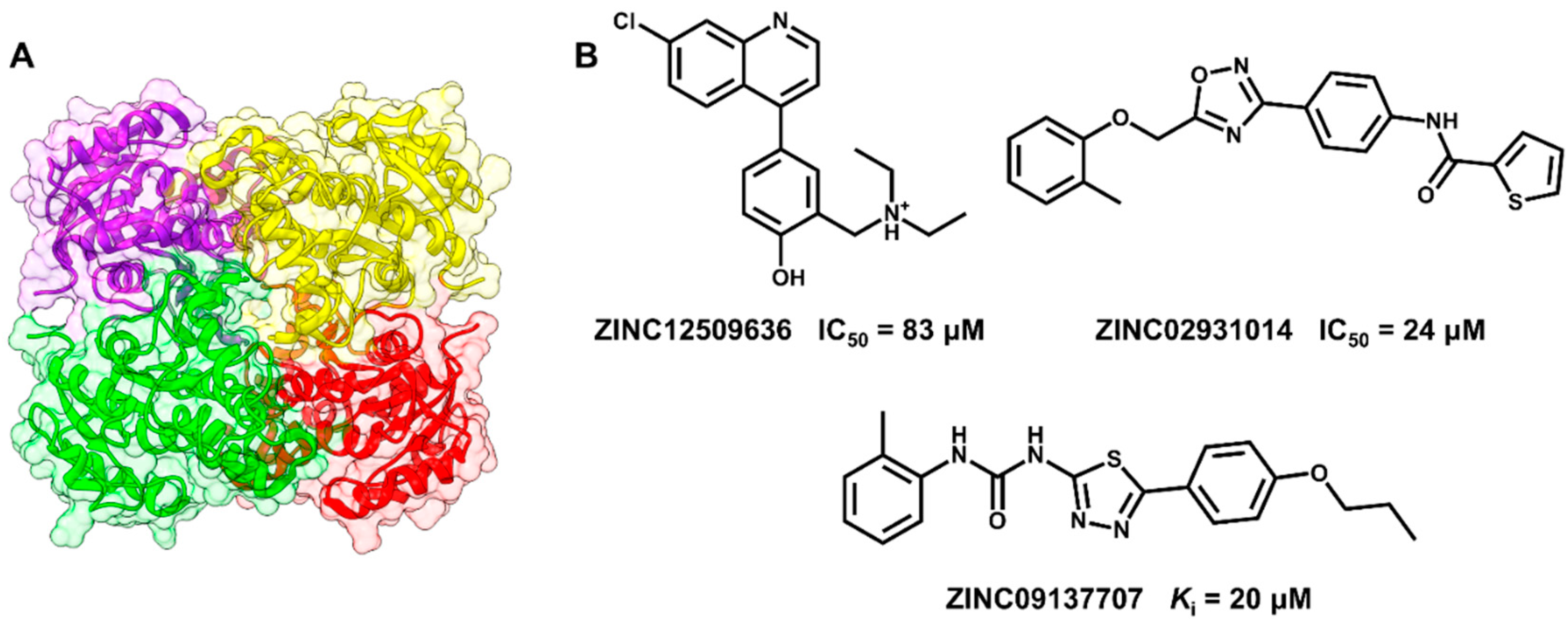
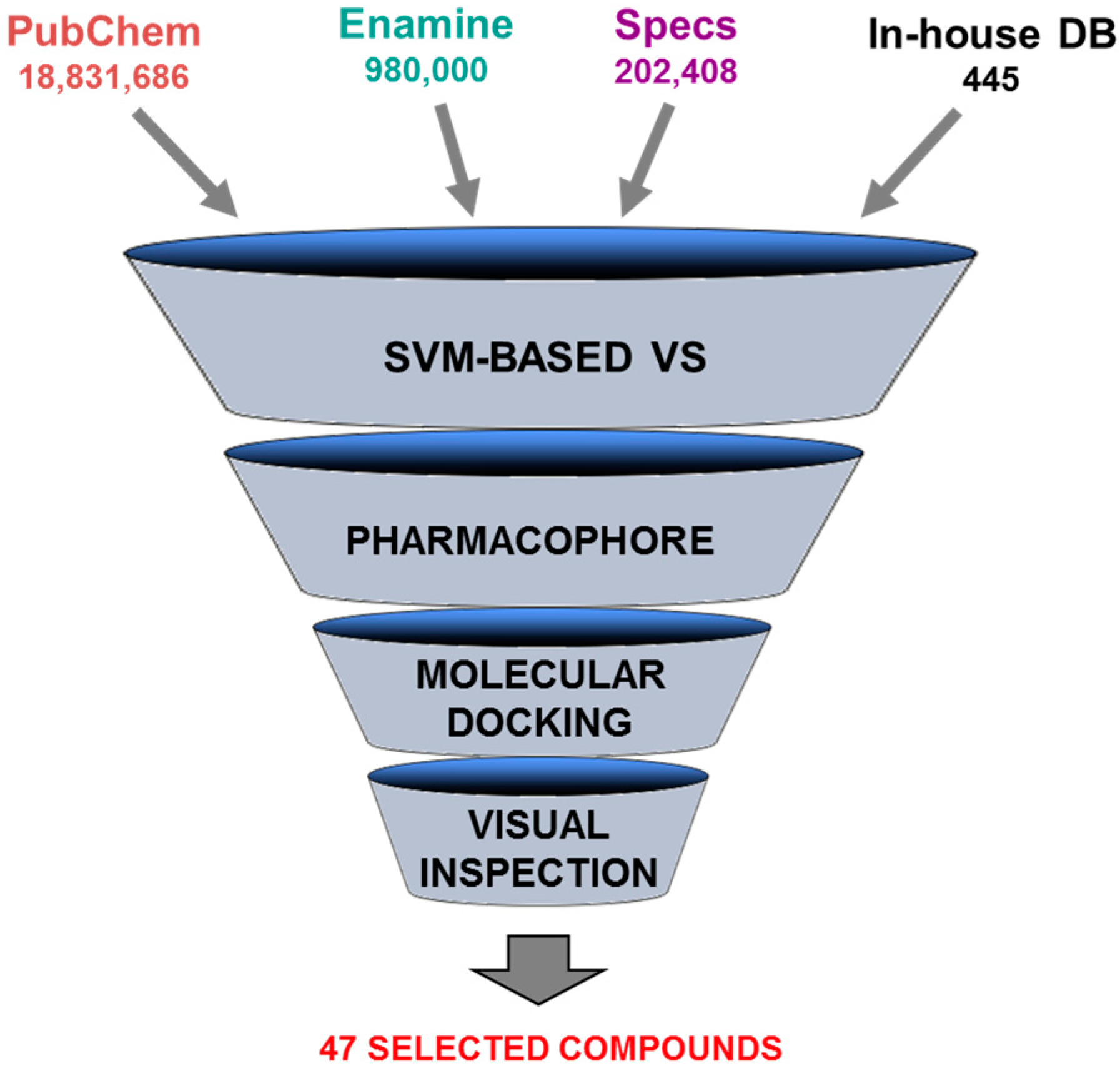
5.1. Discovery of Mycobacterium tuberculosis InhA Inhibitors Using SBVS and Pharmacophore Modeling


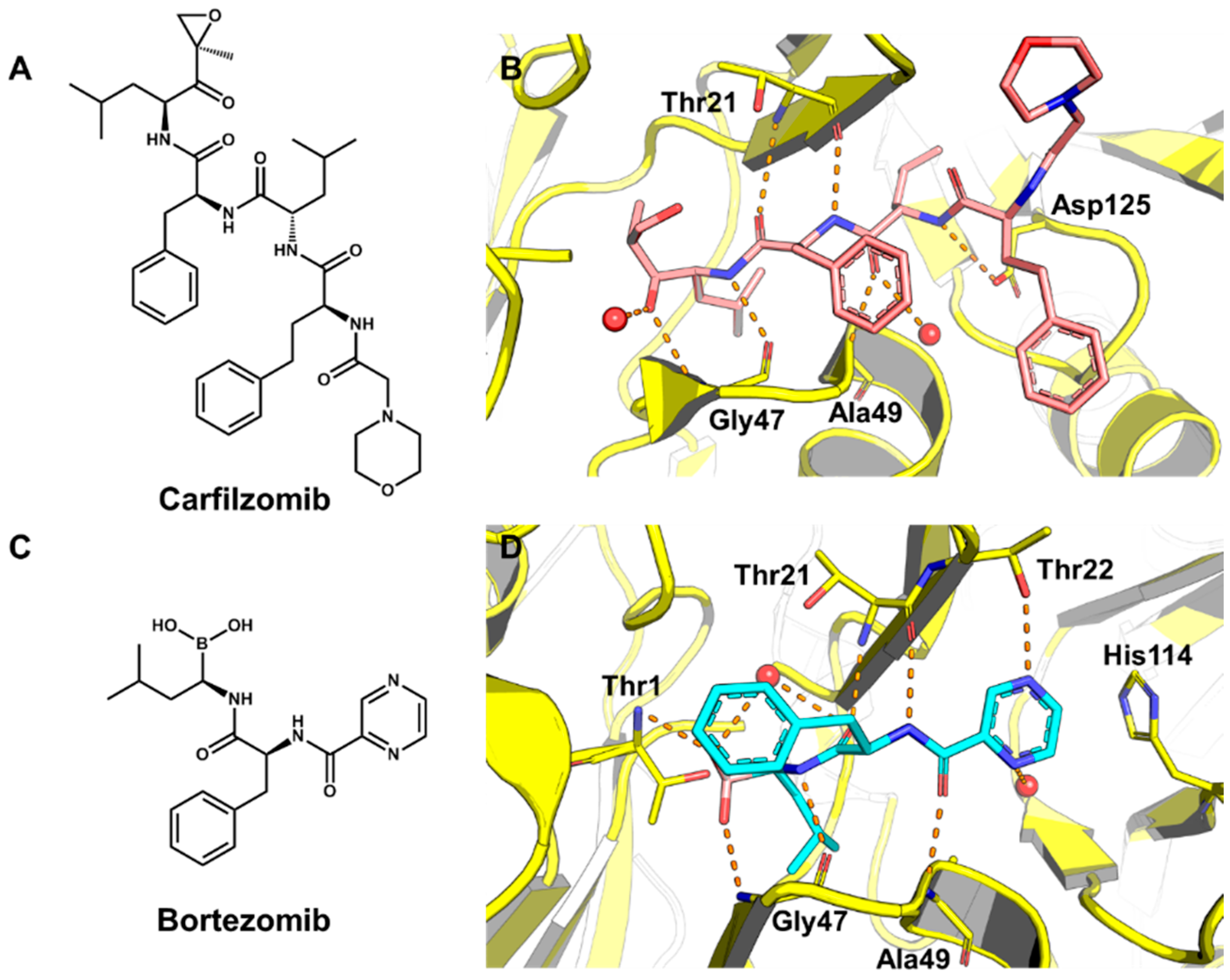
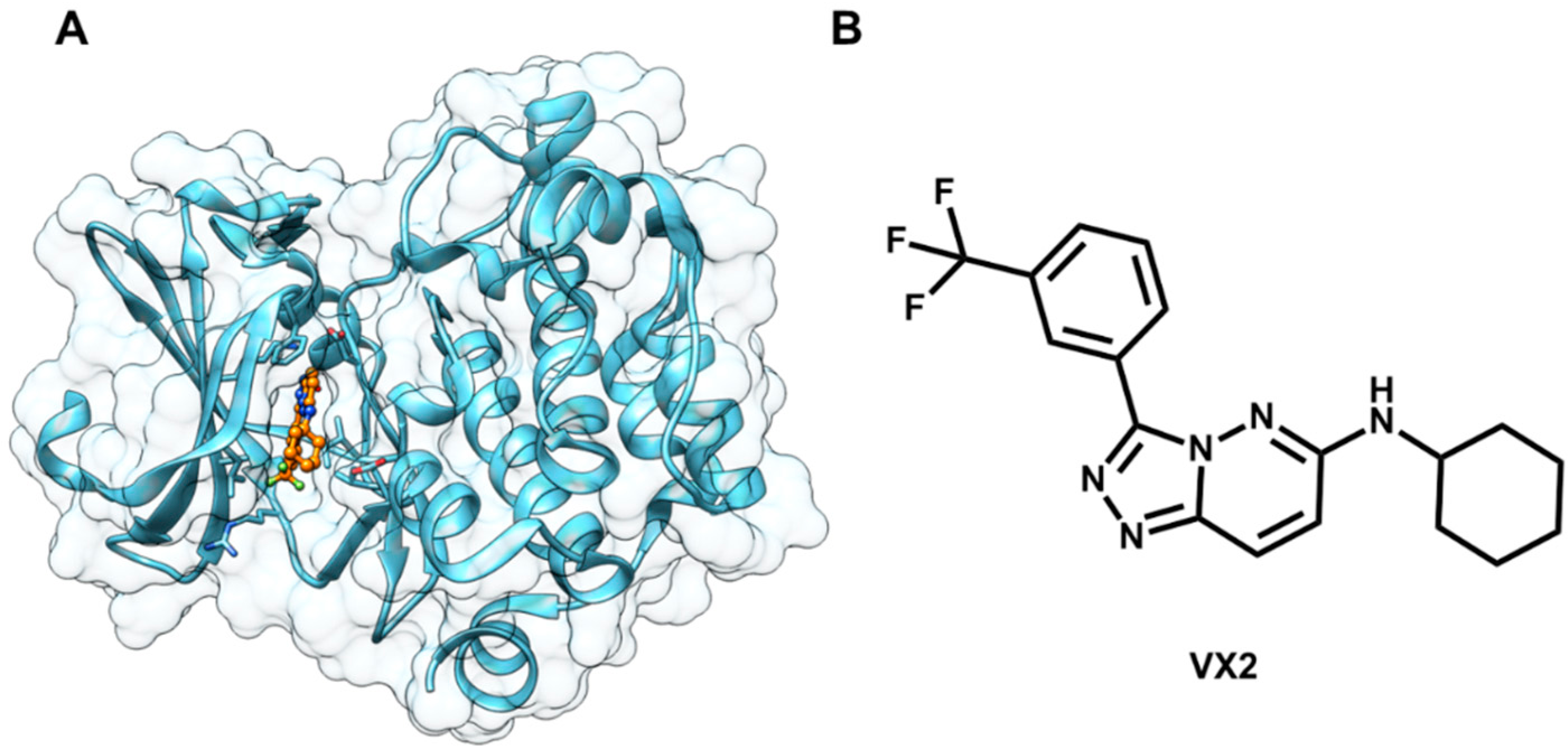
5.2. Discovery of Proteasome Inhibitors by SBVS


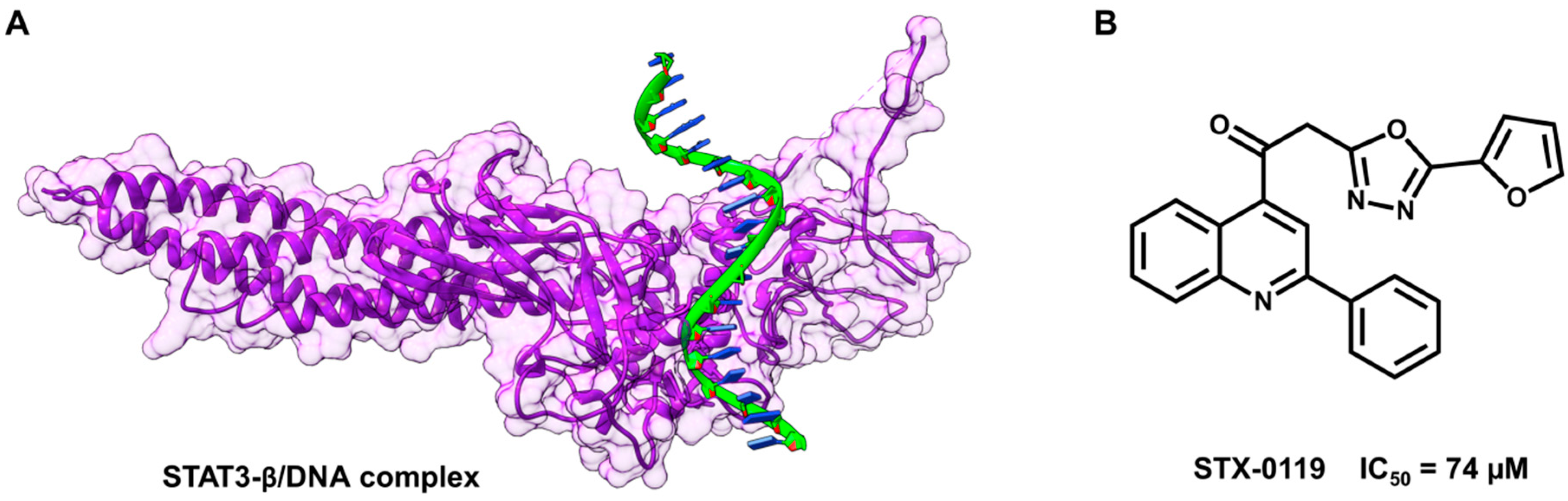
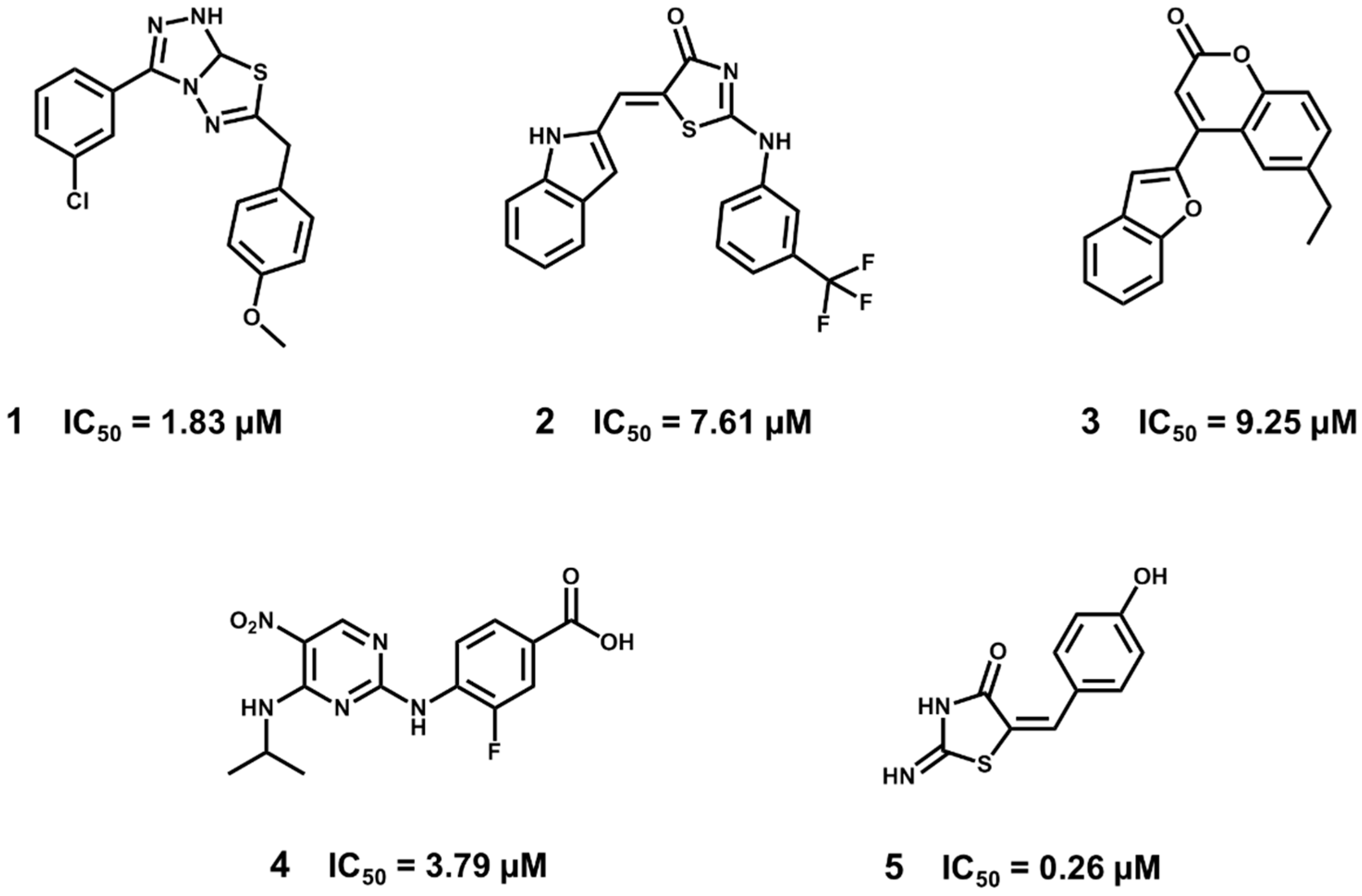
5.3. Identification of a New Series of STAT3 Inhibitors by VS

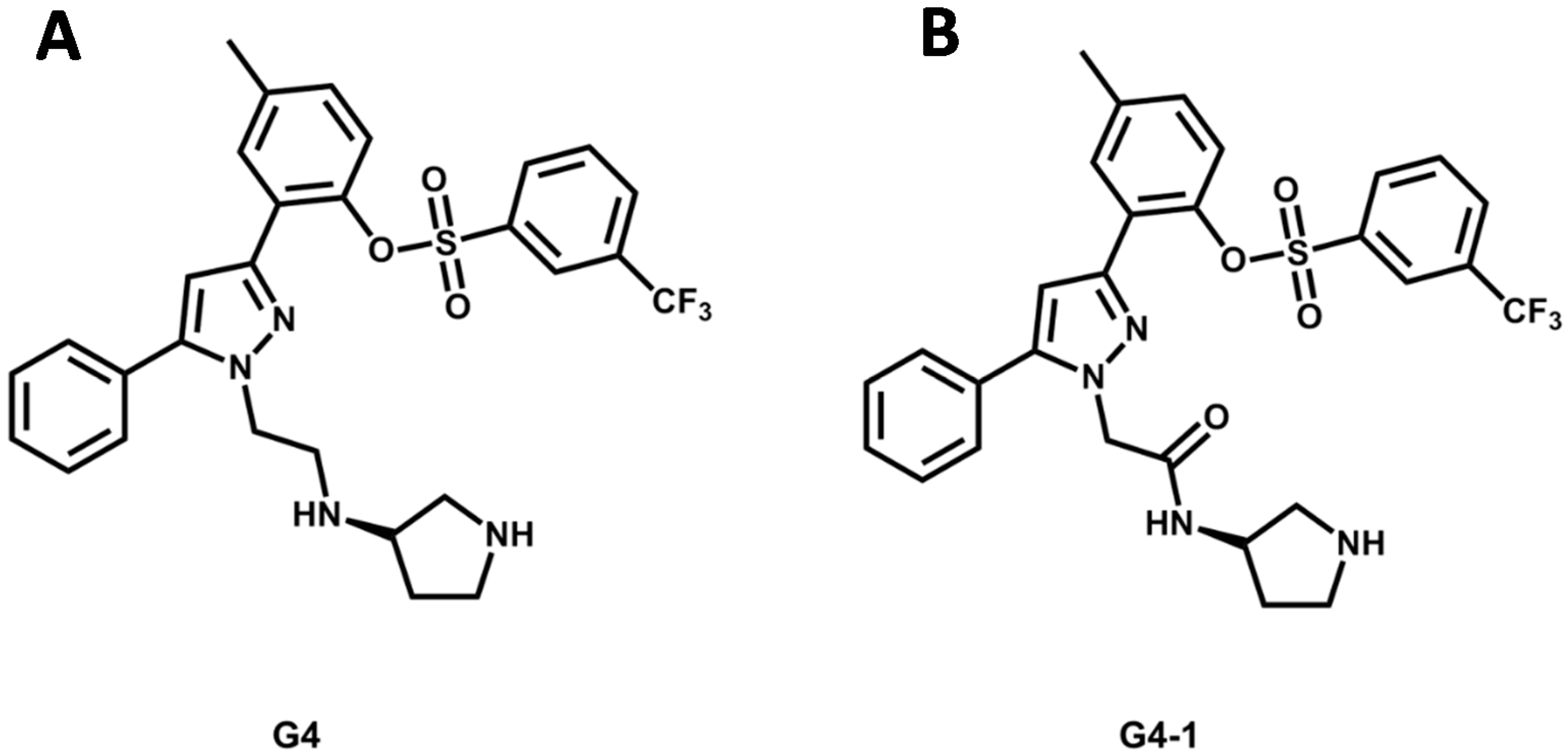
5.4. Discovery of Pim-1 Kinase Inhibitors by a Hierarchical Multistage VS



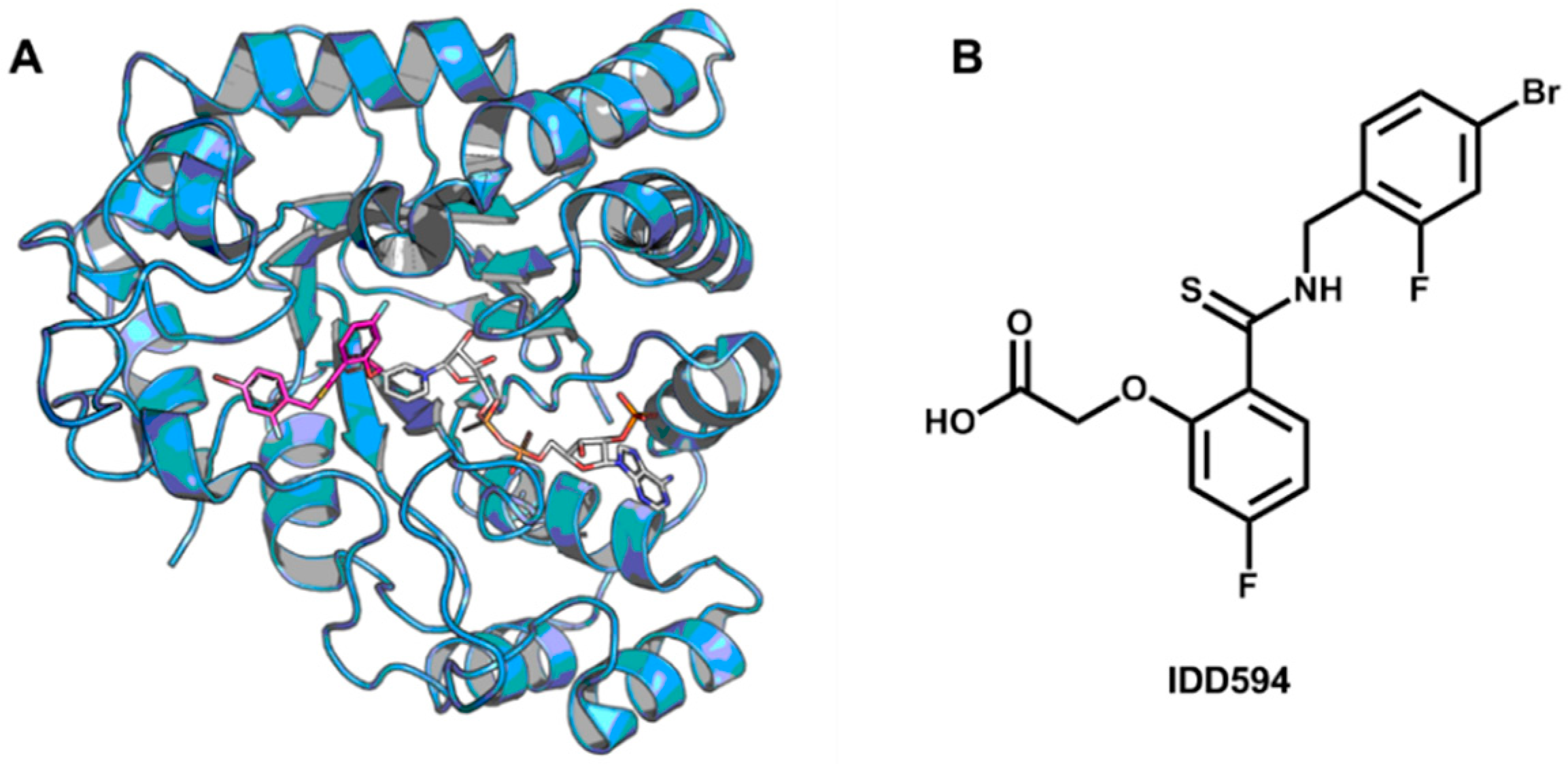
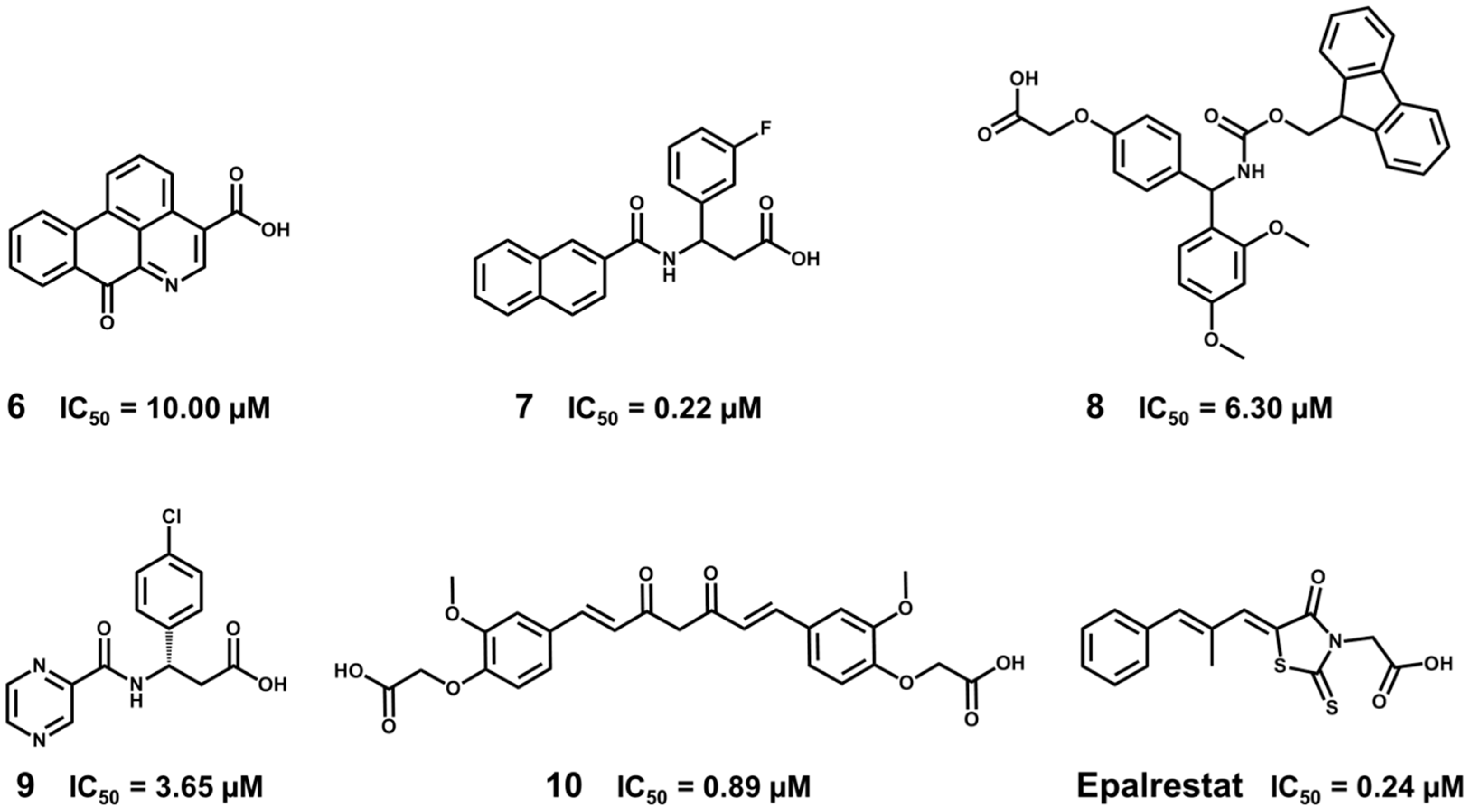
5.5. Identification of Aldose Reductase Inhibitors by MD and SBVS


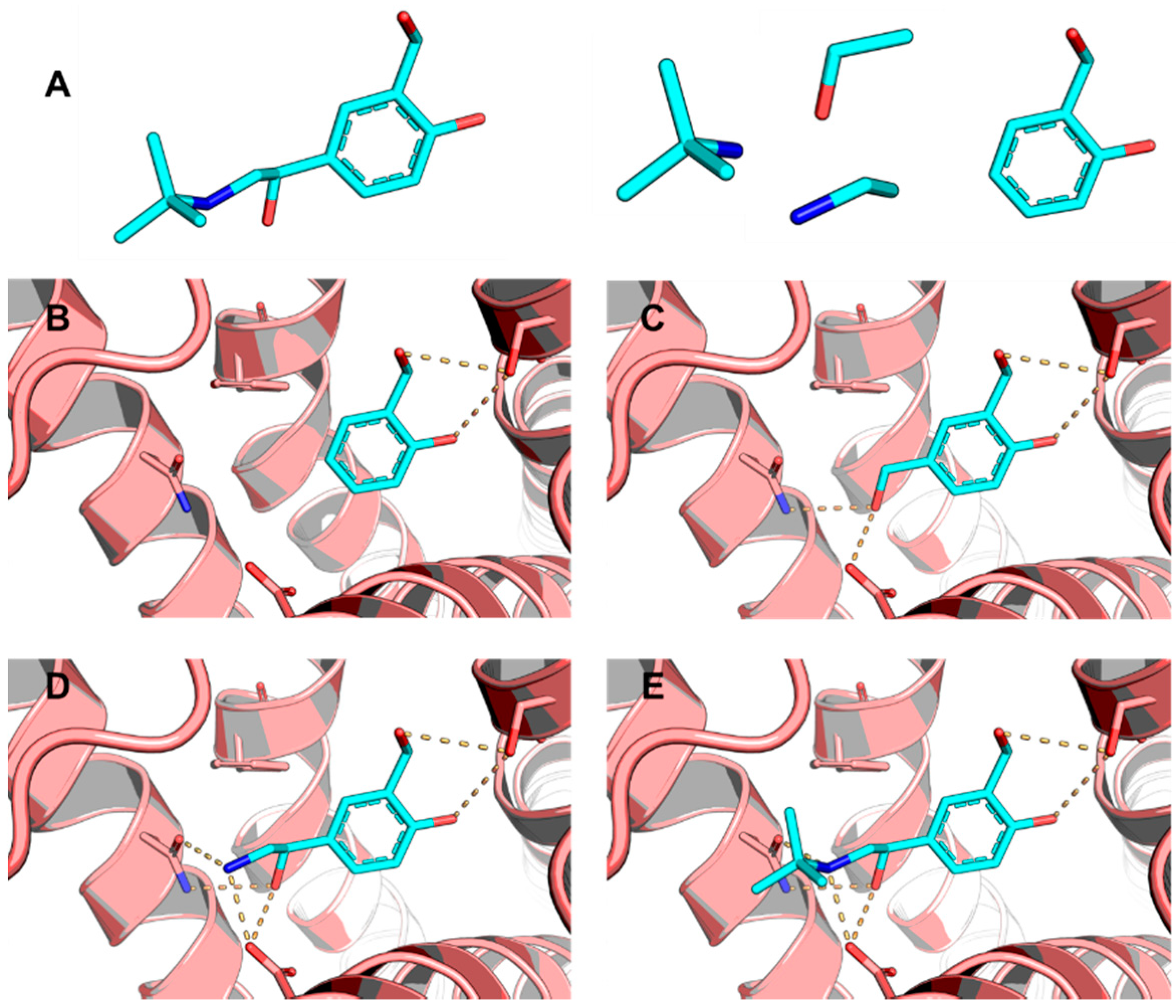
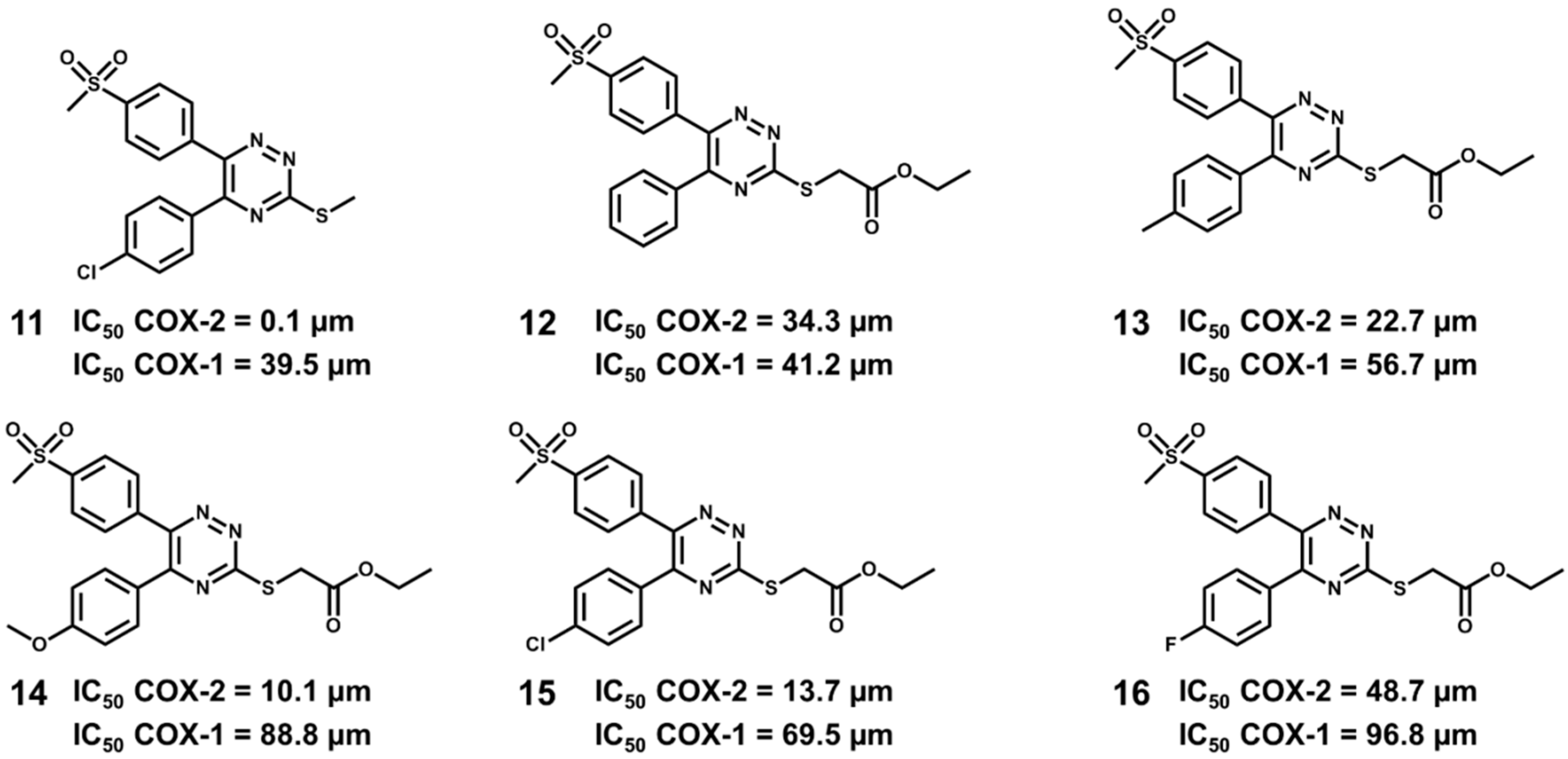
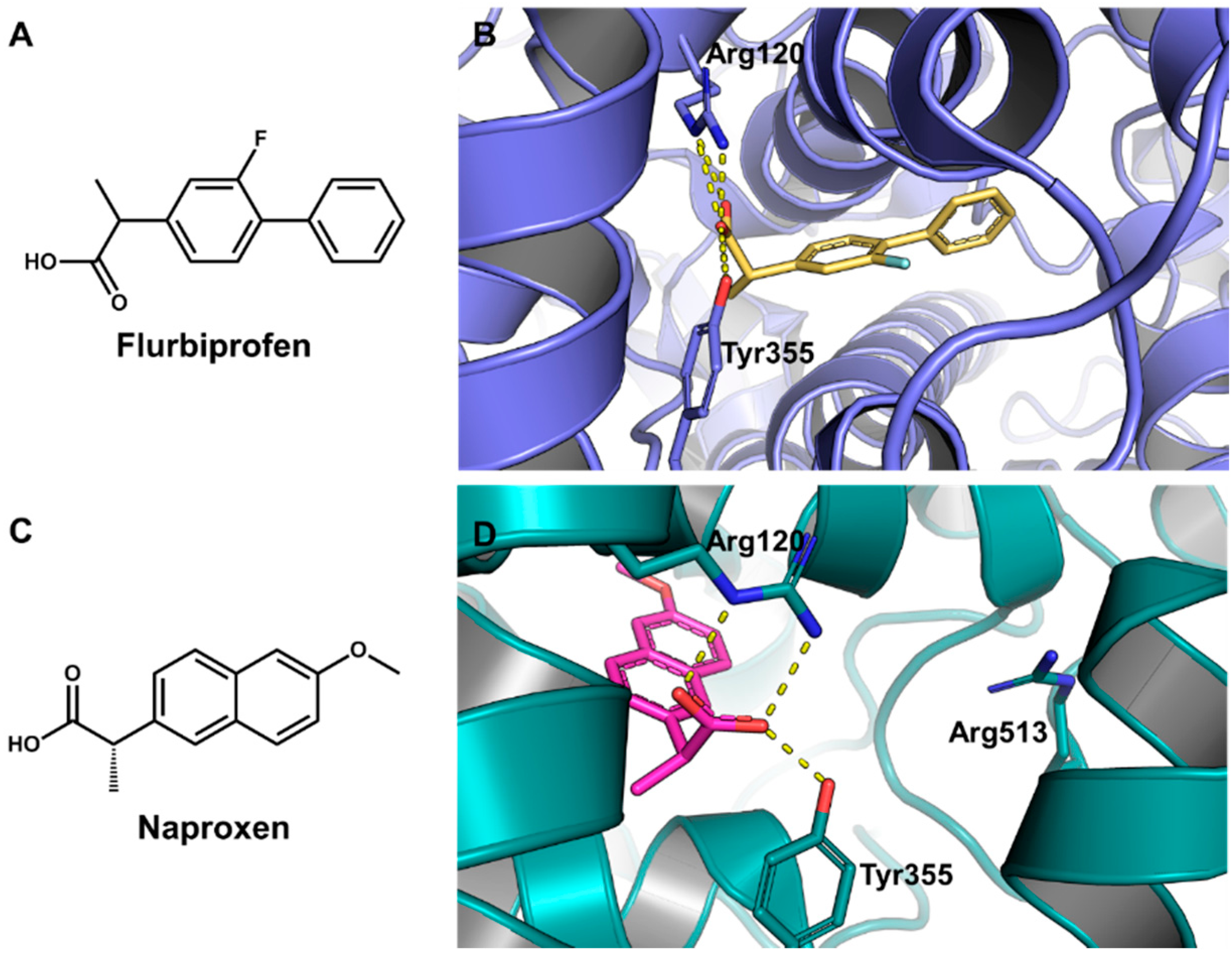
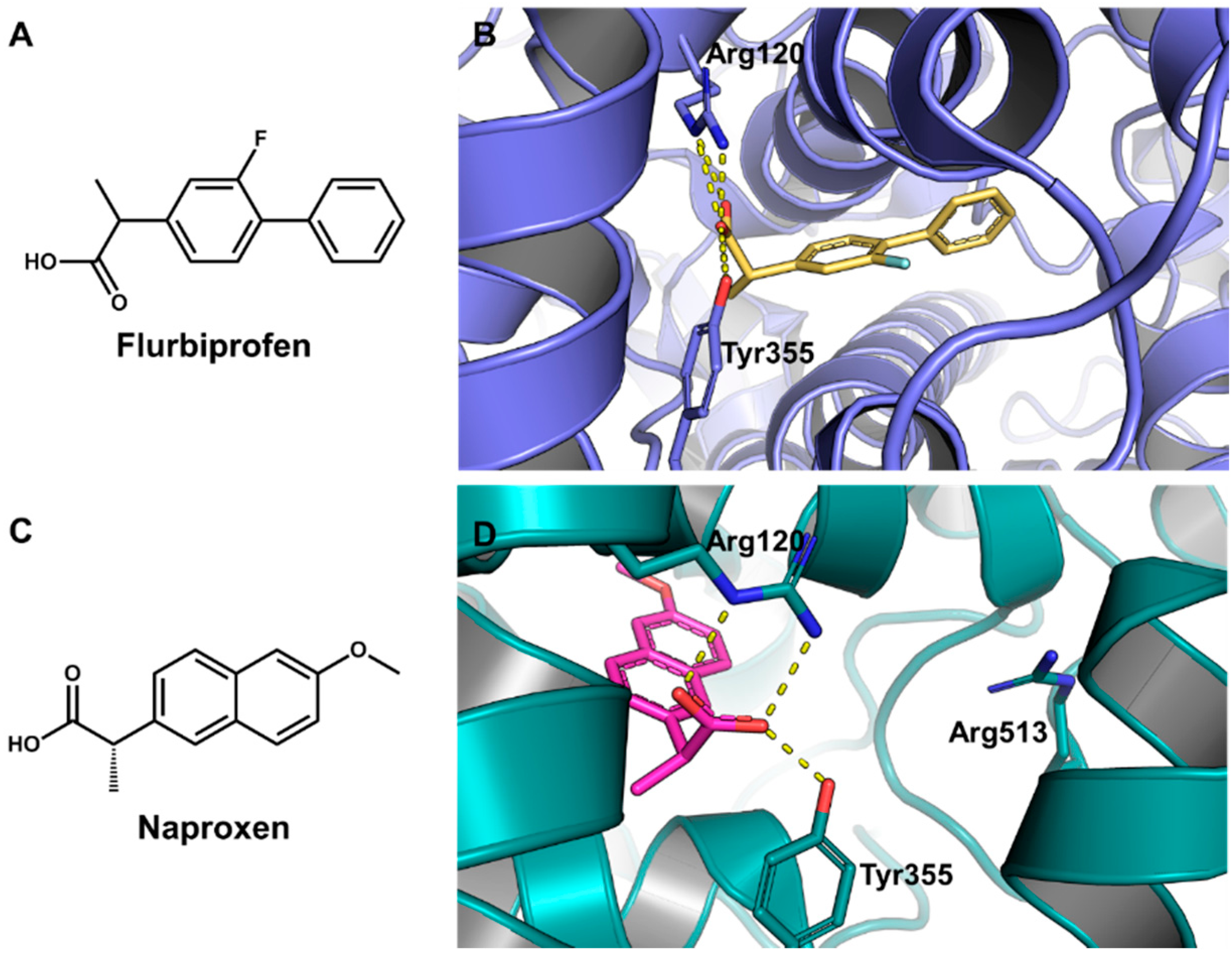
5.6. Design of Selective Cyclooxygenase-2 Inhibitors

6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Berman, H.M. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Weigelt, J. Structural genomics-Impact on biomedicine and drug discovery. Exp. Cell Res. 2010, 316, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Salum, L.; Polikarpov, I.; Andricopulo, A.D. Structure-based approach for the study of estrogen receptor binding affinity and subtype selectivity. J. Chem. Inf. Model. 2008, 48, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Acharya, C.; Coop, A.; Polli, J.E.; Mackerell, A.D., Jr. Recent advances in ligand-based drug design: Relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aided Drug Des. 2011, 7, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Bacilieri, M.; Moro, S. Ligand-based drug design methodologies in drug discovery process an overview. Curr. Drug Discov. Technol. 2006, 3, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Griffith, R. Combination of ligand-and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef] [PubMed]
- Trossini, G.H.G.; Guido, R.V.C.; Oliva, G.; Ferreira, E.I.; Andricopulo, A.D. Quantitative structure-activity relationships for a series of inhibitors of cruzain from Trypanosoma cruzi: Molecular modeling, CoMFA and CoMSIA studies. J. Mol. Graph. Model. 2013, 28, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Valasani, K.R.; Vangavaragu, J.R.; Day, V.W.; Yan, S.S. Structure based design, synthesis, pharmacophore modeling, virtual screening, and molecular docking studies for identification of novel cyclophilin D inhibitors. J. Chem. Inf. Mod. 2014, 54, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Blaney, J. A very short history of structure-based design: How did we get here and where do we need to go? J. Comput. Aided Mol. Des. 2012, 26, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Moudgil, M.N.; Mandal, S.K. Rational drug design. Eur. J. Pharmacol. 2009, 625, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Urwyler, S. Allosteric modulation of family C G-protein-coupled receptors: From molecular insights to therapeutic perspectives. Pharmacol. Rev. 2011, 63, 59–126. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y. Ligand-receptor interaction platforms and their applications for drug discovery. Expert Opin. Drug Discov. 2012, 7, 969–988. [Google Scholar] [CrossRef] [PubMed]
- Kahsai, A.W.; Xiao, K.; Rajagopal, S.; Ahn, S.; Shukla, A.K.; Sun, J.; Oas, T.G.; Lefkowitz, R.J. Multiple ligand-specific conformations of the β2-adrenergic receptor. Nat. Chem. Biol. 2011, 7, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Kobilka, B.K. Structure-based drug screening for G-protein-coupled receptors. Trends Pharmacol. Sci. 2012, 33, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Chandrika, B.R.; Subramanian, J.; Sharma, S.D. Managing protein flexibility in docking and its applications. Drug Discov. Today 2009, 14, 394–400. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. Computer-aided drug-discovery techniques that account for receptor flexibility. Curr. Opin. Pharmacol. 2010, 10, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- López-Vallejo, F.; Caulfield, T.; Martínez-Mayorga, K.; Giulianotti, M.A.; Houghten, R.A.; Nefzi, A.; Medina-Franco, J.L. Integrating virtual screening and combinatorial chemistry for accelerated drug discovery. Comb. Chem. High Throughput Screen. 2011, 14, 475–487. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Agostino, M.; Ramsland, P.A. Challenges and advances in computational docking: 2009 in review. J. Mol. Recognit. 2011, 24, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Agrafiotis, D.K.; Gibbs, A.C.; Zhu, F.; Izrailev, S.; Martin, E. Conformational sampling of bioactive molecules: A comparative study. J. Chem. Inf. Model. 2007, 47, 1067–1086. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: Current status and future challenges. Proteins Struct. Funct. Bioinform. 2006, 65, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, S.B.; Johnson, A.P. eHiTS: A new fast, exhaustive flexible ligand docking system. J. Mol. Graph. Model. 2007, 26, 198–212. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, B.; Goldblum, A. High quality binding modes in docking ligands to proteins. Proteins Struct. Funct. Bioinform. 2008, 71, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Ewing, T.J.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Dias, R.; de Azevedo, J.; Walter, F. Molecular docking algorithms. Curr. Drug Targets 2008, 9, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Huey, R.; Olson, A.J. Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4. J. Comput. Aided Mol. Des. 1996, 10, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Krovat, E.M.; Steindl, T.; Langer, T. Recent advances in docking and scoring. Curr. Comput. Aided Drug Des. 2005, 1, 93–102. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.P.; Perola, E.; Xu, K.; Prendergast, F.G. EUDOC: A computer program for identification of drug interaction sites in macromolecules and drug leads from chemical databases. J. Comput. Chem. 2001, 22, 1750–1771. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Welch, W.; Ruppert, J.; Jain, A.N. Hammerhead: Fast, fully automated docking of flexible ligands to protein binding sites. Chem. Biol. 1996, 3, 449–462. [Google Scholar] [CrossRef]
- Miller, M.D.; Kearsley, S.K.; Underwood, D.J.; Sheridan, R.P. FLOG: A system to select “quasi-flexible” ligands complementary to a receptor of known three-dimensional structure. J. Comput. Aided Mol. Des. 1994, 8, 153–174. [Google Scholar] [CrossRef] [PubMed]
- Schnecke, V.; Kuhn, L.A. Database screening for HIV protease ligands: The influence of binding-site conformation and representation on ligand selectivity. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1999, 242, 242–251. [Google Scholar]
- Mizutani, M.Y.; Tomioka, N.; Itai, A. Rational automatic search method for stable docking models of protein and ligand. J. Mol. Biol. 1994, 243, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Baxter, C.A.; Murray, C.W.; Clark, D.E.; Westhead, D.R.; Eldridge, M.D. Flexible docking using Tabu search and an empirical estimate of binding affinity. Proteins 1998, 33, 367–382. [Google Scholar] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. EADock: Docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Proteins 2007, 67, 1010–1025. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Sochacka, J. Docking of thiopurine derivatives to human serum albumin and binding site analysis with Molegro Virtual Docker. Acta Pol. Pharm. 2014, 71, 343–349. [Google Scholar] [PubMed]
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Tietze, S.; Apostolakis, J. GlamDock: Development and validation of a new docking tool on several thousand protein-ligand complexes. J. Chem. Inf. Model. 2007, 47, 1657–1672. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, N.; Hubbard, R. Towards predictive ligand design with free-energy based computational methods? Curr. Med. Chem. 2006, 13, 3583–3608. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Scoring functions for protein-ligand docking. Curr. Protein Pept. Sci. 2006, 7, 407–420. [Google Scholar] [PubMed]
- Huang, S.Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein-ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef] [PubMed]
- Englebienne, P.; Moitessier, N. Docking ligands into flexible and solvated macromolecules. 5. Force-field-based prediction of binding affinities of ligands to proteins. J. Chem. Inf. Model. 2009, 49, 2564–2571. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.; Auton, T.R.; Eldridge, M.D. Empirical scoring functions. II. The testing of an empirical scoring function for the prediction of ligand-receptor binding affinities and the use of Bayesian regression to improve the quality of the model. J. Comput. Aided Mol. Des. 1998, 12, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. An iterative knowledge-based scoring function to predict protein-ligand interactions: I. Derivation of interaction potentials. J. Comput. Chem. 2006, 27, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Hendlich, M.; Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109. [Google Scholar] [CrossRef] [PubMed]
- Feher, M. Consensus scoring for protein-ligand interactions. Drug Discov. Today 2006, 11, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Terp, G.E.; Johansen, B.N.; Christensen, I.T.; Jørgensen, F.S. A new concept for multidimensional selection of ligand conformations (MultiSelect) and multidimensional scoring (MultiScore) of protein-ligand binding affinities. J. Med. Chem. 2001, 44, 2333–2343. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Betzi, S.; Suhre, K.; Chétrit, B.; Guerlesquin, F.; Morelli, X. GFscore: A general nonlinear consensus scoring function for high-throughput docking. J. Chem. Inf. Model. 2006, 46, 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, R.; Fukunishi, H. Supervised consensus scoring for docking and virtual screening. J. Chem. Inf. Model. 2007, 47, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Bar-Haim, S.; Aharon, A.; Ben-Moshe, T.; Marantz, Y.; Senderowitz, H. SeleX-CS: A new consensus scoring algorithm for hit discovery and lead optimization. J. Chem. Inf. Model. 2009, 49, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Masuda, Y.; Muroya, A.; Yasuno, K.; Takahashi, O.; Furuya, T. Evaluation of docking calculations on X-ray structures using CONSENSUS-DOCK. Chem. Pharm. Bull. Tokyo 2010, 58, 1655–1657. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Shoichet, B.K. Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model. 2010, 50, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Ruvinsky, A.M. Role of binding entropy in the refinement of protein-ligand docking predictions: Analysis based on the use of 11 scoring functions. J. Comput. Chem. 2007, 28, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lu, Y.; Wang, S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Biedermannova, L.; Vondrasek, J.; Dokholyan, N.V. MedusaScore: An accurate force field-based scoring function for virtual drug screening. J. Chem. Inf. Model. 2008, 48, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Rognan, D.; Lauemoller, S.L.; Holm, A.; Buus, S.; Tschinke, V. Predicting binding affinities of protein ligands from three-dimensional models: Application to peptide binding to class I major histocompatibility proteins. J. Med. Chem. 1999, 42, 4650–4658. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, L.; Lai, L.; Tang, Y. SCORE: A new empirical method for estimating the binding affinity of a protein-ligand complex. Mol. Model. Annu. 1998, 4, 379–394. [Google Scholar] [CrossRef]
- Böhm, H.J. The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. J. Comput. Aided Mol. Des. 1994, 8, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Sotriffer, C.A.; Sanschagrin, P.; Matter, H.; Klebe, G. SFCscore: Scoring functions for affinity prediction of protein-ligand complexes. Proteins Struct. Funct. Bioinform. 2008, 73, 395–419. [Google Scholar] [CrossRef] [PubMed]
- Reulecke, I.; Lange, G.; Albrecht, J.; Klein, R.; Rarey, M. Towards an integrated description of hydrogen bonding and dehydration: Decreasing false positives in virtual screening with the HYDE scoring function. ChemMedChem 2008, 3, 885–897. [Google Scholar] [CrossRef] [PubMed]
- Krammer, A.; Kirchhoff, P.D.; Jiang, X.; Venkatachalam, C.M.; Waldman, M. LigScore: A novel scoring function for predicting binding affinities. J. Mol. Graph. Model. 2005, 23, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Gehlhaar, D.K.; Verkhivker, G.M.; Rejto, P.A.; Sherman, C.J.; Fogel, D.B.; Fogel, L.J.; Freer, S.T. Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: Conformationally flexible docking by evolutionary programming. Chem. Biol. 1995, 2, 317–324. [Google Scholar] [CrossRef]
- DeWitte, R.S.; Shakhnovich, E.I. SMoG: De novo design method based on simple, fast, and accurate free energy estimates. 1. Methodology and supporting evidence. J. Am. Chem. Soc. 1996, 118, 11733–11744. [Google Scholar] [CrossRef]
- Muegge, I.; Martin, Y.C.; Hajduk, P.J.; Fesik, S.W. Evaluation of PMF scoring in docking weak ligands to the FK506 binding protein. J. Med. Chem. 1999, 42, 2498–2503. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.R.; Hwang, M.J. An interaction-motif-based scoring function for protein-ligand docking. BMC Bioinform. 2010, 11, 298. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P.J.; Mitchell, J.B. A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Krein, M.P.; Breneman, C.M. Binding affinity prediction with property-encoded shape distribution signatures. J. Chem. Inf. Model. 2010, 50, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Schneidman-Duhovny, D.; Irwin, J.J.; Dong, G.; Shoichet, B.K.; Sali, A. Statistical potential for modeling and ranking of protein-ligand interactions. J. Chem. Inf. Model. 2011, 51, 3078–3092. [Google Scholar] [CrossRef] [PubMed]
- Kumalo, H.M.; Bhakat, S.; Soliman, M.E. Theory and applications of covalent docking in drug discovery: Merits and pitfalls. Molecules 2015, 20, 1984–2000. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Zhou, S.; Su, C.T.T.; Ge, Z.; Li, R.; Kwoh, C.K. CovalentDock: Automated covalent docking with parameterized covalent linkage energy estimation and molecular geometry constraints. J. Comput. Chem. 2013, 34, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Moitessier, N.; Englebienne, P.; Lee, D.; Lawandi, J.; Corbeil, C.R. Towards the development of universal, fast and highly accurate docking/scoring methods: A long way to go. Br. J. Pharmacol. 2008, 153, S7–S26. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lill, M.A. Induced fit docking, and the use of QM/MM methods in docking. Drug Discov. Today Technol. 2013, 10, e411–e418. [Google Scholar] [CrossRef] [PubMed]
- Menikarachchi, L.C.; Gascón, J.A. QM/MM approaches in medicinal chemistry research. Curr. Top. Med. Chem. 2010, 10, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Miller, R.M.; Krishnan, S.; Uchida, K.; Irwin, J.J.; Eidam, O.; Cimermančič, P.; Gibold, L.; Bonnet, R.; Shoichet, B.K.; et al. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol. 2014, 10, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Bianco, G.; Forli, S.; Goodsell, D.S.; Olson, A.J. Covalent docking using Autodock: Two-point attractor and flexible sidechain methods. Protein Sci. 2015. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H. Accommodating protein flexibility for structure-based drug design. Curr. Top. Med. Chem. 2011, 11, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Salsbury, F.R., Jr. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr. Opin. Pharmacol. 2010, 10, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Harvey, M.J.; de Fabritiis, G. High-throughput molecular dynamics: The powerful new tool for drug discovery. Drug Discov. Today 2012, 17, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Alonso, H.; Bliznyuk, A.A.; Gready, J.E. Combining docking and molecular dynamic simulations in drug design. Med. Res. Rev. 2006, 26, 531–568. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Nichols, S.E.; Baron, R.; Ivetac, A.; McCammon, J.A. Predictive power of molecular dynamics receptor structures in virtual screening. J. Chem. Inf. Model. 2011, 51, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Christen, M.; Hünenberger, P.H.; Bakowies, D.; Baron, R.; Bürgi, R.; Geerke, D.P.; Heinz, T.N.; Kastenholz, M.A.; Kräutler, V.; Oostenbrink, C.; et al. The GROMOS software for biomolecular simulation: GROMOS05. J. Comput. Chem. 2005, 26, 1719–1751. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Ribeiro, A.J.; Coimbra, J.; Neves, R.P.; Martins, S.A.; Moorthy, N.S.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking in the new millennium—A retrospective of 10 years in the field. Curr. Med. Chem. 2013, 20, 2296–2314. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G. Virtual ligand screening: Strategies, perspectives and limitations. Drug Discov. Today 2006, 11, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.; Tirado-Rives, J.; Jorgensen, W.L. Energetics of displacing water molecules from protein binding sites: Consequences for ligand optimization. J. Am. Chem. Soc. 2009, 131, 15403–15411. [Google Scholar] [CrossRef] [PubMed]
- Amadasi, A.; Surface, J.A.; Spyrakis, F.; Cozzini, P.; Mozzarelli, A.; Kellogg, G.E. Robust classification of “relevant” water molecules in putative protein binding sites. J. Med. Chem. 2008, 51, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Amadasi, A.; Spyrakis, F.; Cozzini, P.; Abraham, D.J.; Kellogg, G.E.; Mozzarelli, A. Mapping the energetics of water-protein and water-ligand interactions with the “natural” HINT forcefield: Predictive tools for characterizing the roles of water in biomolecules. J. Mol. Biol. 2006, 358, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Kellogg, G.E.; Chen, D.L. The importance of being exhaustive. Optimization of bridging structural water molecules and water networks in models of biological systems. Chem. Biodivers. 2004, 1, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.W.; Kann, M.G. Chapter 4: Protein interactions and disease. PLoS Comput. Biol. 2012, 8, e1002819. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, Z.; Rew, Y.; Gribble, M.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chen, X.; Chow, D.; et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 2014, 57, 1454–1472. [Google Scholar] [CrossRef] [PubMed]
- Kuritzkes, D.; Kar, S.; Kirkpatrick, P. Maraviroc. Nat. Rev. Drug Discov. 2008, 7, 15–16. [Google Scholar] [CrossRef]
- Fuller, J.C.; Burgoyne, N.J.; Jackson, R.M. Predicting druggable binding sites at the protein-protein interface. Drug Discov. Today 2009, 14, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Laurie, A.T.; Jackson, R.M. Q-SiteFinder: An energy-based method for the prediction of protein-ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Meireles, L.M.; Dömling, A.S.; Camacho, C.J. ANCHOR: A web server and database for analysis of protein-protein interaction binding pockets for drug discovery. Nucleic Acids Res. 2010, 38, W407–W411. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.J.; Zhang, C. FastContact: Rapid estimate of contact and binding free energies. Bioinformatics 2005, 21, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Moreira, I.S.; Fernandes, P.A.; Ramos, M.J. Protein-protein docking dealing with the unknown. J. Comput. Chem. 2010, 31, 317–342. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Lyne, P.D. Structure-based virtual screening an overview. Drug Discov. Today 2002, 7, 649–657. [Google Scholar] [CrossRef]
- Stahura, F.L.; Bajorath, J. Virtual screening methods that complement HTS. Comb. Chem. High Throughput Screen. 2004, 7, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Kodadek, T. The rise, fall and reinvention of combinatorial chemistry. Chem. Commun. 2011, 47, 9757–9763. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Matter, H. Integrating virtual screening in lead discovery. Curr. Opin. Chem. Biol. 2004, 8, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Geppert, H.; Vogt, M.; Bajorath, J. Current trends in ligand-based virtual screening: Molecular representations, data mining methods, new application areas, and performance evaluation. J. Chem. Inf. Model. 2010, 50, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Ripphausen, P.; Nisius, B.; Bajorath, J. State-of-the-art in ligand-based virtual screening. Drug Discov. Today 2011, 16, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2013, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Inglese, J.; Auld, D.S.; Jadhav, A.; Johnson, R.L.; Simeonov, A.; Yasgar, A.; Zheng, W.; Austin, C.P. Quantitative high-throughput screening: A titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. USA 2006, 103, 11473–11478. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration Cheminformatics. Available online: http://www.molinspiration.com/ (accessed on 9 May 2015).
- Jarrahpour, A.; Fathi, J.; Mimouni, M.; Hadda, T.B.; Sheikh, J.; Chohan, Z.; Parvez, A. Petra, Osiris and Molinspiration (POM) together as a successful support in drug design: Antibacterial activity and biopharmaceutical characterization of some azo Schiff bases. Med. Chem. Res. 2012, 21, 1984–1990. [Google Scholar] [CrossRef]
- Organic Chemistry Portal. Available online: http://www.organic-chemistry.org/prog/peo/ (accessed on 9 May 2015).
- Molsoft. Available online: http://molsoft.com/ (accessed on 9 May 2015).
- Molecular Discovery. Available online: http://www.moldiscovery.com/software/moka/ (accessed on 9 May 2015).
- Milletti, F.; Storchi, L.; Sforna, G.; Cruciani, G. New and original pKa prediction method using grid molecular interaction fields. J. Chem. Inf. Model. 2007, 47, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, G.M.; Heiss, M.; Mangold, M.; Markt, P.; Kirchmair, J.; Wolber, G.; Liedl, K.R. One concept, three implementations of 3D pharmacophore-based virtual screening: Distinct coverage of chemical search space. J. Chem. Inf. Model. 2010, 50, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Seidel, T.; Bendix, F.; Langer, T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov. Today 2008, 13, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Pharmacophore and ligand-based design with Biovia Discovery Studio® datasheet. Available online: http://accelrys.com/products/datasheets/pharmacophore-modeling.pdf (accessed on 9 May 2015).
- Jones, G.; Willett, P.; Glen, R.C. A genetic algorithm for flexible molecular overlay and pharmacophore elucidation. J. Comput. Aided Mol. Des. 1995, 9, 532–549. [Google Scholar] [CrossRef] [PubMed]
- Certara. Available online: http://www.certara.com/ (accessed on 9 May 2015).
- Martin, Y.C.; Bures, M.G.; Danaher, E.A.; DeLazzer, J.; Lico, I.; Pavlik, P.A. A fast new approach to pharmacophore mapping and its application to dopaminergic and benzodiazepine agonists. J. Comput. Aided. Mol. Des. 1993, 7, 83–102. [Google Scholar] [CrossRef] [PubMed]
- Richmond, N.J.; Abrams, C.A.; Wolohan, P.R.N.; Abrahamian, E.; Willett, P.; Clark, R.D. GALAHAD: 1. Pharmacophore identification by hypermolecular alignment of ligands in 3D. J. Comput. Aided Mol. Des. 2006, 20, 567–587. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2004, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Inteligand. Available online: http://www.inteligand.com/ (accessed on 9 May 2015).
- Chemical Computing Group. Available online: https://www.chemcomp.com/ (accessed on 9 May 2015).
- Labute, P.; Williams, C.; Feher, M.; Sourial, E.; Schmidt, J.M. Flexible alignment of small molecules. J. Med. Chem. 2001, 44, 1483–1490. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 10–11. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger. Available online: http://www.schrodinger.com/ (accessed on 9 May 2015).
- Cresset. Available online: http://www.cresset-group.com/ (accessed on 9 May 2015).
- Vinter, J.G. Extended electron distributions applied to the molecular mechanics of some intermolecular interactions. J. Comput. Aided Mol. Des. 1994, 8, 653–668. [Google Scholar] [CrossRef] [PubMed]
- PharmaGist. Available online: http://bioinfo3d.cs.tau.ac.il/PharmaGist/ (accessed on 9 May 2015).
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [PubMed]
- Van Drie, J.H.; Weininger, D.; Martin, Y.C. ALADDIN: An integrated tool for computer-assisted molecular design and pharmacophore recognition from geometric, steric, and substructure searching of three-dimensional molecular structures. J. Comput. Aided Mol. Des. 1989, 3, 225–251. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.M.; Finn, P.W.; Kavraki, L.E.; Latombe, J.C.; Motwani, R.; Venkatasubramanian, S.; Shelton, C.; Yao, A. RAPID: Randomized pharmacophore identification for drug design. Comp. Geom. 1998, 4, 263–272. [Google Scholar]
- Van Drie, J.H. Strategies for the determination of pharmacophoric 3D database queries. J. Comput. Aided Mol. Des. 1997, 11, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Arauzo, H.; Koehler, K.F.; Hagen, T.J.; Cook, J.M. Synthetic and computer assisted analysis of the pharmacophore for agonists at benzodiazepine receptors. Life Sci. 1991, 49, 207–216. [Google Scholar] [CrossRef]
- Dolata, D.P.; Parrill, A.L.; Walters, W.P. CLEW: The generation of pharmacophore hypotheses through machine learning. SAR QSAR Environ. Res. 1998, 9, 53–81. [Google Scholar] [CrossRef]
- Holliday, J.D.; Willett, P. Using a genetic algorithm to identify common structural features in sets of ligands. J. Mol. Graph. Model. 1997, 15, 221–232. [Google Scholar] [CrossRef]
- Handschuh, S.; Wagener, M.; Gasteiger, J. Superposition of three-dimensional chemical structures allowing for conformational flexibility by a hybrid method. J. Chem. Inf. Comput. Sci. 1998, 38, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Rusinko, A., 3rd; Tropsha, A.; Young, S.S. Automated pharmacophore identification for large chemical data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Talele, T.T.; Kulkarni, V.M. Three-dimensional quantitative structure-activity relationships (QSAR) and receptor mapping of cytochrome P-45014αDM inhibiting azole antifungal agents. J. Chem. Inf. Comput. Sci. 1999, 39, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Gao, Y.; Lai, L. LigBuilder: A multi-purpose program for structure-based drug design. J. Mol. Model. 2000, 6, 498–516. [Google Scholar] [CrossRef]
- LigBuilder. Available online: ligbuilder.org (accessed on 18 June 2015).
- Lionta, E.; Spyrou, G.; Vassilatis, D.K.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Gangwal, R.P.; Damre, M.V.; Das, N.R.; Dhoke, G.V.; Bhadauriya, A.; Varikoti, R.A.; Sharma, S.S.; Sangamwar, A.T. Structure based virtual screening to identify selective phosphodiesterase 4B inhibitors. J. Mol. Graph. Model. 2015, 57, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar]
- Cavasotto, C.N.; Orry, W.; Andrew, J. Ligand docking and structure-based virtual screening in drug discovery. Curr. Top. Med. Chem. 2007, 7, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Markt, P.; Distinto, S.; Wolber, G.; Langer, T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection—What can we learn from earlier mistakes? J. Comput. Aided Mol. Des. 2008, 22, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N.; Nicholls, A. Recommendations for evaluation of computational methods. J. Comput. Aided Mol. Des. 2008, 22, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Moura Barbosa, A.J.; del Rio, A. Freely accessible databases of commercial compounds for high- throughput virtual screenings. Curr. Top. Med. Chem. 2012, 12, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Sastry, M.; Lowrie, J.F.; Dixon, S.L.; Sherman, W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- ZINC. Available online: zinc.docking.org (accessed on 9 May 2015).
- PubChem. Available online: pubchem.ncbi.nlm.nih.gov (accessed on 9 May 2015).
- Li, Q.; Cheng, T.; Wang, Y.; Bryant, S.H. PubChem as a public resource for drug discovery. Drug Discov. Today 2010, 15, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- ChemSpider. Available online: www.chemspider.com (accessed on 9 May 2015).
- Pence, H.E.; Williams, A. ChemSpider: An online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- ChEMBL. Available online: www.ebi.ac.uk/chembl (accessed on 9 May 2015).
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [PubMed]
- Núcleo de Bioensaios Biossíntese e Ecofisiologia de Produtos Naturais. Available online: http://nubbe.iq.unesp.br/portal/ (accessed on 9 May 2015).
- Valli, M.; dos Santos, R.N.; Figueira, L.D.; Nakajima, C.H.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. Development of a natural products database from the biodiversity of Brazil. J. Nat. Prod. 2013, 76, 439–444. [Google Scholar] [CrossRef] [PubMed]
- ChemBank. Available online: chembank.broadinstitute.org (accessed on 9 May 2015).
- Seiler, K.P.; George, G.A.; Happ, M.P.; Bodycombe, N.E.; Carrinski, H.A.; Norton, S.; Brudz, S.; Sullivan, J.P.; Muhlich, J.; Serrano, M.; et al. ChemBank: A small-molecule screening and cheminformatics resource database. Nucleic Acids Res. 2008, 36, D351–D359. [Google Scholar] [CrossRef] [PubMed]
- eMolecules. Available online: www.emolecules.com (accessed on 9 May 2015).
- DrugBank. Available online: drugbank.ca (accessed on 9 May 2015).
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- The Binding Database. Available online: www.bindingdb.org/bind/index.jsp (accessed on 9 May 2015).
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Akdemir, A.; Rucktooa, P.; Jongejan, A.; van Elk, R.; Bertrand, S.; Sixma, T.K.; de Esch, I.J. Acetylcholine binding protein (AChBP) as template for hierarchical in silico screening procedures to identify structurally novel ligands for the nicotinic receptors. Bioorganic Med. Chem. 2011, 19, 6107–6119. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- UCSF Chimera. Available online: http://www.cgl.ucsf.edu/chimera/ (accessed on 9 May 2015).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- VMD Visual Molecular Dynamics. Available online: http://www.ks.uiuc.edu/Research/vmd (accessed on 9 May 2015).
- Pymol. Available online: http://www.pymol.org/ (accessed on 9 May 2015).
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Ball Biochemical Algorithms Library. Available online: http://www.ball-project.org/ (accessed on 9 May 2015).
- Moll, A.; Hildebrandt, A.; Lenhof, H.P.; Kohlbacher, O. BALLView: A tool for research and education in molecular modeling. Bioinformatics 2006, 22, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Home Page for RasMol and OpenRasMol. Available online: http://www.rasmol.org/ (accessed on 9 May 2015).
- Sayle, R.A.; Milner-White, E.J. RASMOL: Biomolecular graphics for all. Trends Biochem. Sci. 1995, 20, 374–376. [Google Scholar] [CrossRef]
- Jmol: An open-source Java viewer for chemical structures in 3D. Available online: http://www.jmol.org/ (accessed on 18 June 2015).
- JSmol. Available online: http://wiki.jmol.org/index.php/JSmol (accessed on 18 June 2015).
- Molle, V.; Gulten, G.; Vilchèze, C.; Veyron-Churlet, R.; Zanella-Cléon, I.; Sacchettini, J.C.; Jacobs, W.R., Jr.; Kremer, L. Phosphorylation of InhA inhibits mycolic acid biosynthesis and growth of Mycobacterium tuberculosis. Mol. Microbiol. 2010, 78, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Marrakchi, H.; Lanéelle, G.; Quémard, A. InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiology 2000, 146, 289–296. [Google Scholar] [PubMed]
- Pauli, I.; dos Santos, R.N.; Rostirolla, D.C.; Martinelli, L.K.; Ducati, R.G.; Timmers, L.F.S.M.; Basso, L.A.; Santos, D.S.; Guido, R.V.C.; Andricopulo, A.D.; et al. Discovery of new inhibitors of Mycobacterium tuberculosis InhA enzyme using virtual screening and a 3D-pharmacophore-based approach. J. Chem. Inf. Model. 2013, 53, 2390–2401. [Google Scholar] [CrossRef] [PubMed]
- Abrahamian, E.; Fox, P.C.; Naerum, L.; Christensen, I.T.; Thøgersen, H.; Clark, R.D. Efficient generation, storage, and manipulation of fully flexible pharmacophore multiplets and their use in 3-D similarity searching. J. Chem. Inf. Comput. Sci. 2003, 43, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The proteasome: Structure, function, and role in the cell. Cancer Treat. Rev. 2003, 29, 3–9. [Google Scholar] [CrossRef]
- Miller, Z.; Kim, K.S.; Lee, D.M.; Kasam, V.; Baek, S.E.; Lee, K.H.; Zhang, Y.Y.; Ao, L.; Carmony, K.; Lee, N.R.; et al. Proteasome inhibitors with pyrazole scaffolds from structure-based virtual screening. J. Med. Chem. 2015, 58, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.J.; John, S. Signal transducer and activator of transcription (STAT) signalling and T-cell lymphomas. Immunology 2005, 114, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Timofeeva, O.A.; Tarasova, N.I.; Zhang, X.; Chasovskikh, S.; Cheema, A.K.; Wang, H.; Brown, M.L.; Dritschilo, A. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc. Natl. Acad. Sci. USA 2013, 110, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Masuda, Y.; Uehara, Y.; Sato, H.; Muroya, A.; Takahashi, O.; Yokotagawa, T.; Furuya, T.; Okawara, T.; Otsuka, M.; Ogo, N.; et al. Identification of a new series of STAT3 inhibitors by virtual screening. ACS Med. Chem. Lett. 2010, 1, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Möröy, T. The serine/threonine kinase Pim-1. Int. J. Biochem. Cell Biol. 2005, 37, 726–730. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Aparicio, C.; Carnero, A. Pim kinases in cancer: Diagnostic, prognostic and treatment opportunities. Biochem. Pharmacol. 2013, 85, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.X.; Li, L.L.; Zheng, R.L.; Xie, H.Z.; Cao, Z.X.; Feng, S.; Pan, Y.L.; Chen, X.; Wei, Y.Q.; Yang, S.Y. Discovery of novel Pim-1 kinase inhibitors by a hierarchical multistage virtual screening approach based on SVM model, pharmacophore, and molecular docking. J. Chem. Inf. Model. 2011, 51, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Ramana, K.V.; Bhatnagar, A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocr. Rev. 2005, 26, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gu, Q.; Zheng, X.; Ye, J.; Liu, Z.; Li, J.; Hu, X.; Hagler, A.; Xu, J. Discovery of new selective human aldose reductase inhibitors through virtual screening multiple binding pocket conformations. J. Chem. Inf. Model. 2013, 53, 2409–2422. [Google Scholar] [CrossRef] [PubMed]
- Perrone, M.G.; Scilimati, A.; Simone, L.; Vitale, P. Selective COX-1 inhibition: A therapeutic target to be reconsidered. Curr. Med. Chem. 2010, 17, 3769–3805. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Guan, P.P.; Wang, T.; Yu, X.; Guo, J.J; Wang, Z.Y. Aggravation of Alzheimer’s disease due to the COX-2-mediated reciprocal regulation of IL-1β and Aβ between glial and neuron cells. Aging Cell 2014, 13, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Dadashpour, S.; Tuylu Kucukkilinc, T.; Unsal Tan, O.; Ozadali, K.; Irannejad, H.; Emami, S. Design, synthesis and in vitro study of 5,6-diaryl-1,2,4-triazine-3-ylthioacetate derivatives as COX-2 and β-amyloid aggregation inhibitors. Arch. Pharm. 2015, 348, 179–187. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384-13421. https://doi.org/10.3390/molecules200713384
Ferreira LG, Dos Santos RN, Oliva G, Andricopulo AD. Molecular Docking and Structure-Based Drug Design Strategies. Molecules. 2015; 20(7):13384-13421. https://doi.org/10.3390/molecules200713384
Chicago/Turabian StyleFerreira, Leonardo G., Ricardo N. Dos Santos, Glaucius Oliva, and Adriano D. Andricopulo. 2015. "Molecular Docking and Structure-Based Drug Design Strategies" Molecules 20, no. 7: 13384-13421. https://doi.org/10.3390/molecules200713384
APA StyleFerreira, L. G., Dos Santos, R. N., Oliva, G., & Andricopulo, A. D. (2015). Molecular Docking and Structure-Based Drug Design Strategies. Molecules, 20(7), 13384-13421. https://doi.org/10.3390/molecules200713384