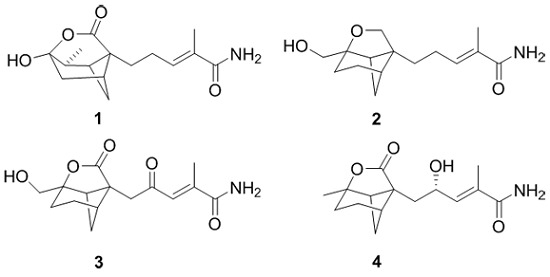
2. Results and Discussion
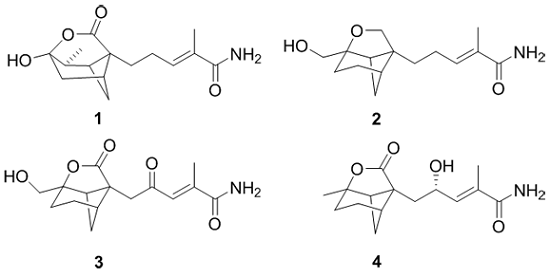
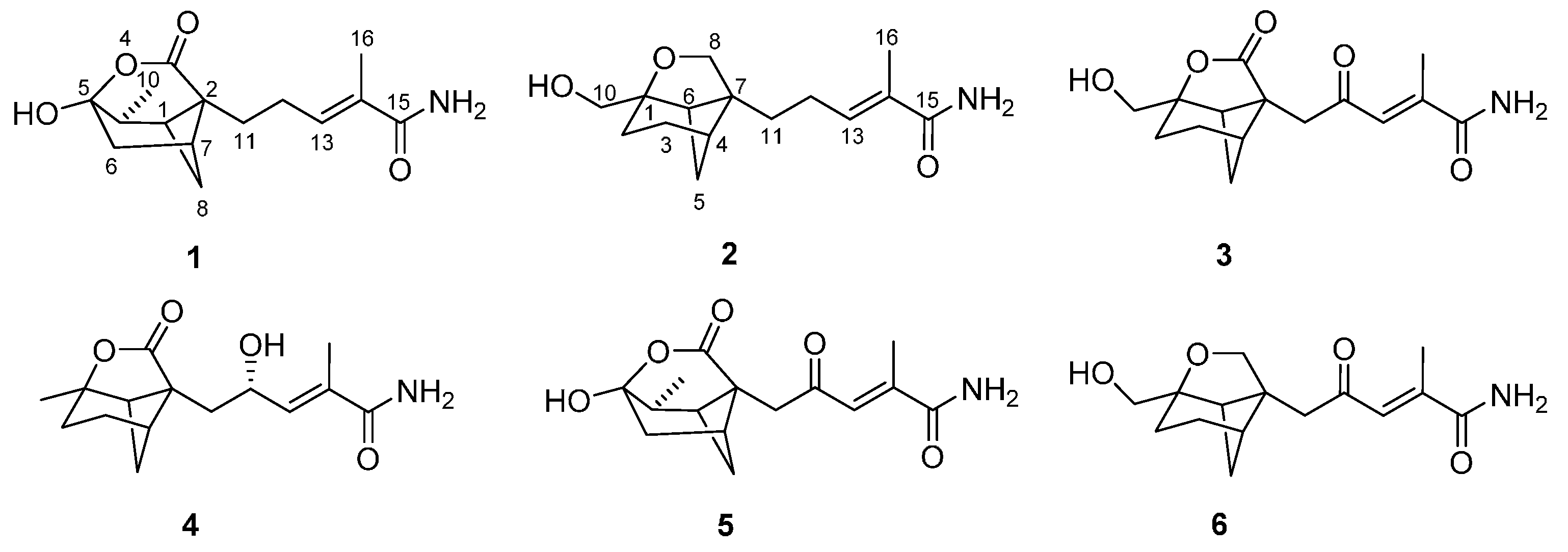
Brasilamide K (
1) gave a pseudomolecular ion [M + H]
+ peak by HRESIMS, corresponding to a molecular formula of C
15H
21NO
4 (six degrees of unsaturation). Analysis of its
1H- and
13C-NMR data (
Table 1) revealed the presence of three exchangeable protons (δ
H 6.74, 6.38, and 6.07, respectively), two methyl groups, four methylene units, three methines, two sp
3 quaternary carbons (one oxygenated), one trisubstituted olefin, and two carboxylic carbons (δ
C 172.9 and 170.9, respectively). The
1H-
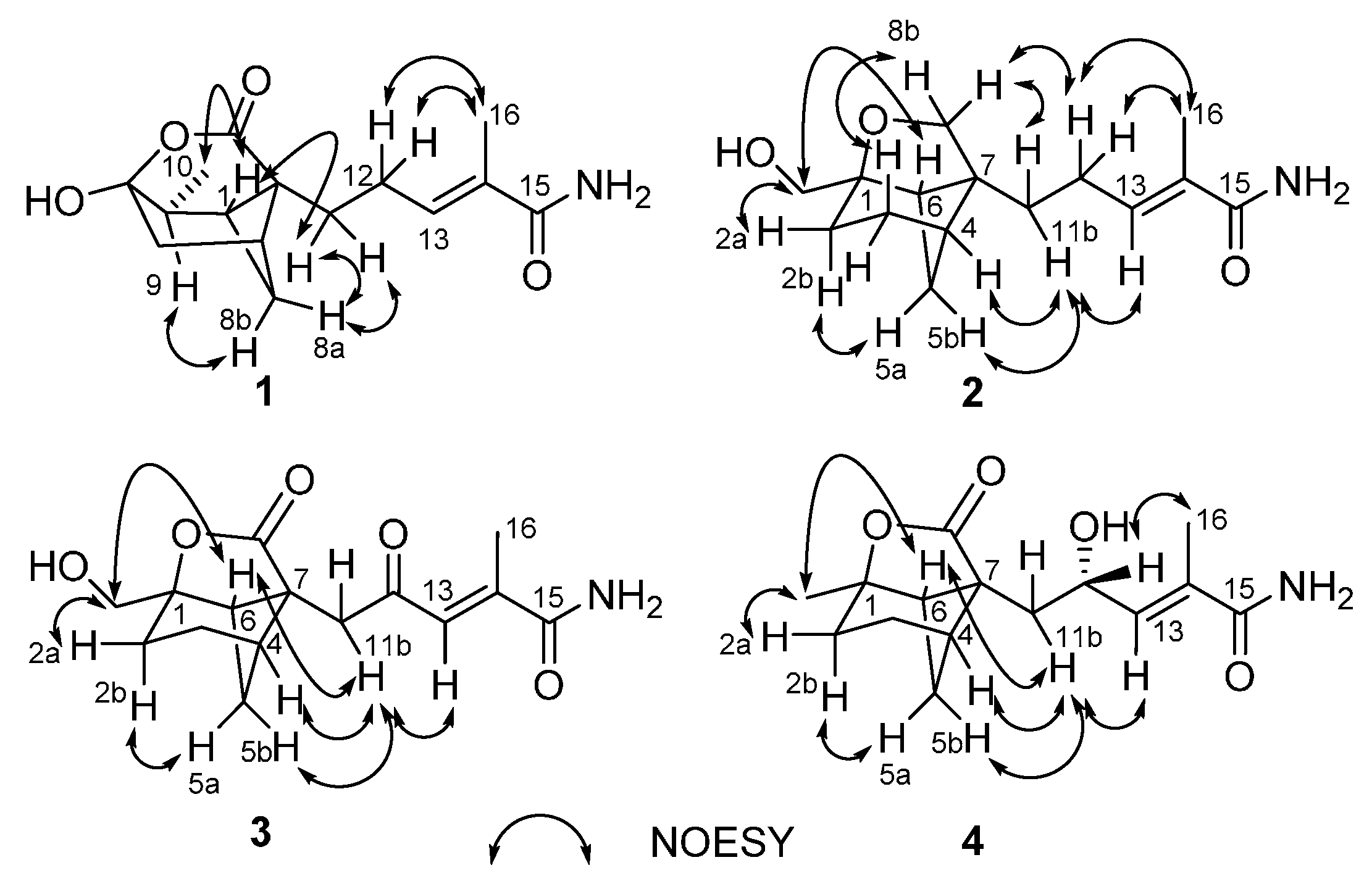
1H COSY NMR data showed two isolated spin-systems of C-6–C-10 (via C-7, C-8, C-1, and C-9) and C-11–C-13. HMBC correlations (
Figure 2) from H-13 to C-15 and C-16, and from H
3-16 to C-13, C-14, and C-15 enabled both methyl carbon C-16 and carboxylic carbon C-15 attached to the C-14 of C-13/C-14 olefin. HMBC cross-peaks from H-1, H
2-6, and H-9 to C-2 and C-5, plus H
2-8 to C-2 indicated that the sp
3 quaternary carbon C-2 is located between C-1 and C-7, whereas the C-5 oxygenated sp
3 quaternary carbon is attached to both C-6 and C-9, completing the bicyclo(3.1.1)heptane ring, while those from H
2-11 to C-1, C-2, C-3, and C-7 led to the connection of C-2 to C-3 and C-11. Considering the doubly-oxygenated nature of C-5 (δ
C 103.0) and the unsaturation requirement of
1, the C-3 carboxylic carbon must acylate one of the oxygen atoms attached to C-5 to form a δ-lactone moiety, thereby completing the 4-oxatricyclo(3.3.1.0
2,7)nonane skeleton in
1. The remaining two exchangeable protons were assigned as 15-NH
2, by default. Collectively, these data permitted assignment of the planar structure of
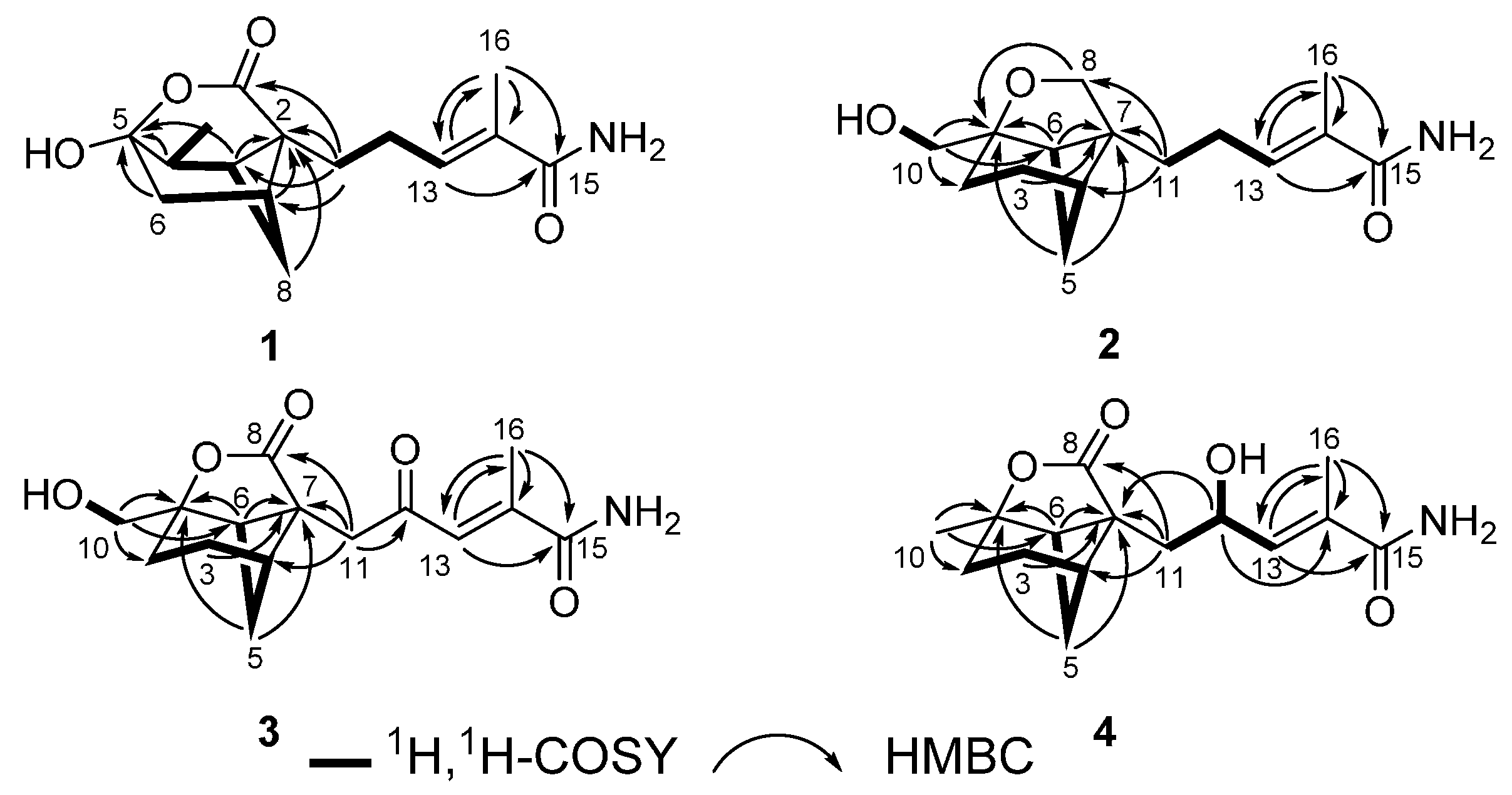
1. The relative configuration of
1 was deduced to be the same as brasilamide A (
5) by comparison of its NOESY data (
Figure 3) with those of
5 [
13]. NOESY correlations of H-1 withH
3-10 and H
2-11, H-8a with H
2-11, and H-8b with H-9 revealed their proximity in space. The C-13/C-14 olefin is assigned the
E-geometry based on NOESY correlations of H
2-12 with H
3-16. The absolute configuration of
1 was proposed as 1
S,2
S,5
R,7
R,9
S by analogy to those of
5, which was secured by X-ray crystallography [
13].
Table 1.
NMR Data for 1 and 2 (Acetone-d6).
Table 1.
NMR Data for 1 and 2 (Acetone-d6).
| Pos. | 1 | 2 |
|---|
| δC a | δH b (J in Hz) | HMBC | δC a | δH b (J in Hz) | HMBC |
|---|
| 1 | 43.3 | 2.00, t (6.0) | 2, 5, 7, 8, 10, 11 | 89.8 | | |
| 2 | 47.0 | | | 28.8 | 1.48, m; 1.90, m | 1, 3, 4, 6, 10 |
| 3 | 172.9 | | | 23.3 | 1.76, m; 1.83, m | 1, 2, 4, 5, 7 |
| 4 | | | | 40.5 | 2.22, m | 2, 6, 11 |
| 5 | 103.0 | | | 23.8 | 1.47, d (11.0); 2.10, ddd (11.0, 6.0, 5.5) | 1, 3, 4, 6, 7, 8 |
| 6 | 41.9 | 2.09, m | 2, 5, 8 | 48.0 | 2.27, dd (5.5, 5.0) | 1, 2, 4, 7, 10, 11 |
| 7 | 36.2 | 2.36, m | 1, 8 | 56.0 | | |
| 8 | 37.1 | 1.31, d (11.0); 2.70, ddd (11.0, 7.0, 6.0) | 1, 2, 3, 6, 7, 9 | 71.5 | 3.46, d (9.0); 3.80, d (9.0) | 1, 4, 6, 7, 11 |
| 9 | 44.4 | 2.24, q (7.0) | 1, 2, 5, 8, 10 | | | |
| 10 | 12.9 | 0.89, d (7.0) | 1, 5, 9 | 67.2 | 3.40, d (12.0); 3.43, d (12.0) | 1, 2, 6 |
| 11 | 30.1 | 1.95, m | 1, 2, 3, 7, 12, 13 | 33.6 | 1.68, m; 1.85, m | 4, 6, 7, 8, 12, 13 |
| 12 | 25.5 | 2.34, m | 2, 11, 13, 14 | 25.2 | 2.04, m; 2.16, m | 11, 13, 14 |
| 13 | 136.0 | 6.45, t (7.5) | 12, 15, 16 | 136.1 | 6.40, t (7.5) | 11, 12, 15, 16 |
| 14 | 132.0 | | | 131.8 | | |
| 15 | 170.9 | | | 170.9 | | |
| 16 | 12.7 | 1.82, s | 13, 14, 15 | 12.6 | 1.80, s | 13, 14, 15 |
| OH-5 | | 6.38, s | | | | |
| OH-10 | | | | | 3.32, brs | |
| NH2-15 | | 6.74, brs; 6.07, brs | | | 6.73, brs; 6.09, brs | |
Figure 2.
The 1H-1H-COSY and selected HMBC (H→C) correlations for 1–4.
Figure 2.
The 1H-1H-COSY and selected HMBC (H→C) correlations for 1–4.
Brasilamide L (
2) was assigned the molecular formula C
15H
23NO
3 (five degrees of unsaturation) by HRESIMS (266.1743). Interpretation of its
1H- and
13C-NMR data (
Table 1) revealed the presence of three exchangeable protons (δ
H 6.73, 6.09, and 3.32, respectively), one methyl group, seven methylene units (two oxygenated), two methines, two sp
3 quaternary carbons (one oxygenated), one trisubstituted olefin, and one carboxylic carbons (δ
C 170.9). The
1H-
1H COSY NMR data showed three isolated spin-systems of C-2–C-6, C-10–OH, and C-11–C-13. HMBC cross-peaks (
Figure 2) from H-13 to C-15 and C-16, and from H
3-16 to C-13, C-14, and C-15 enabled both methyl carbon C-16 and carboxylic carbon C-15 attached to the C-14 of C-13/C-14 olefin. HMBC correlations from H
2-3, H
2-5, and H-6 to C-1 and C-7, plus H
2-10 to C-1, C-2, and C-6 indicated that the sp
3 quaternary carbon C-7 is located between C-4 and C-6, and C-2, C-6, and C-10 are all connected to the C-1 oxygenated sp
3 quaternary carbon, completing the bicyclo(3.1.1)heptane ring. While those from H
2-11 to C-4, C-6, C-7, and C-8 led to the connection of C-7 to C-8 and C-11. A key correlation from H
2-8 to C-1 established the furan ring, thereby completing the 9-oxatricyclo(4.3.0.0
4,7)nonane skeleton in
2. The remaining two exchangeable protons were also assigned as 15-NH
2 by default. On the basis of these data, the gross structure of
2 was established as shown. NOESY correlations (
Figure 3) of H-3b with H-8b, H-4 and H-5b with H-11b, and H-6 with H
2-10 established the relative configuration of
1, which is the same as that of brasilamide C (
6) by comparison of their NOESY data with those of
6 [
13]. The C-13/C-14 olefin is also assigned the
E-geometry based on NOESY correlations of H
2-12 with H
3-16. The absolute configuration of
2 could be deduced as shown by analogy to
6.
Figure 3.
NOESY correlations for 1–4.
Figure 3.
NOESY correlations for 1–4.
The elemental composition of brasilamideM (
3) was established as C
15H
19NO
5 (seven degrees of unsaturation) by HRESIMS (
m/
z 294.1332 [M + H]
+), 28 mass units higher than
2. The
1H-, and
13C-NMR spectra (
Table 2) of
3 displayed signals for structural features similar to
2, except that two methylene units (δ
H/C 3.46, 3.80/71.5; 2.04, 2.16/25.2) in
2 are replaced by one carboxy carbon atom (δ
C 177.8) and one α,β-unsaturated ketone carbon atom (δ
C 199.5) in the spectra of
3, respectively. It was confirmed by HMBC correlations (
Figure 2) from H
2-11 to C-8 and C-12. Analysis of NOESY data (
Figure 3) of
3 revealed the same relative configuration as
2, implying that its absolute configuration could be deduced as shown by analogy to
2.
The molecular formula of brasilamideN (
4) was determined to be C
15H
21NO
4 (six degrees of unsaturation) on the basis of its HRESIMS (
m/
z 280.1546 [M + H]
+). Interpretation of its NMR data (
Table 2) revealed structural features similar to those presented in
3, except that the oxygenated methylene unit (δ
H/C 3.70/66.4) and the C-10 carboxy carbon (δ
C 199.5) are replaced by the methyl (δ
H/C 1.42/25.1) and the oxygenated methine unit (δ
H/C 4.57/67.1), respectively. This observations were confirmed by
1H-
1H COSY and HMBC correlations (
Figure 2) from H
3-10 to C-1, C-2 and C-6, H-12 to C-7, C-11, C-13, and C-14. The relative configuration of
4 was deduced to be the same as those of
3 on the basis of its NOESY data (
Figure 3) except the stereogenic center C-12. The absolute configuration of C-12 in
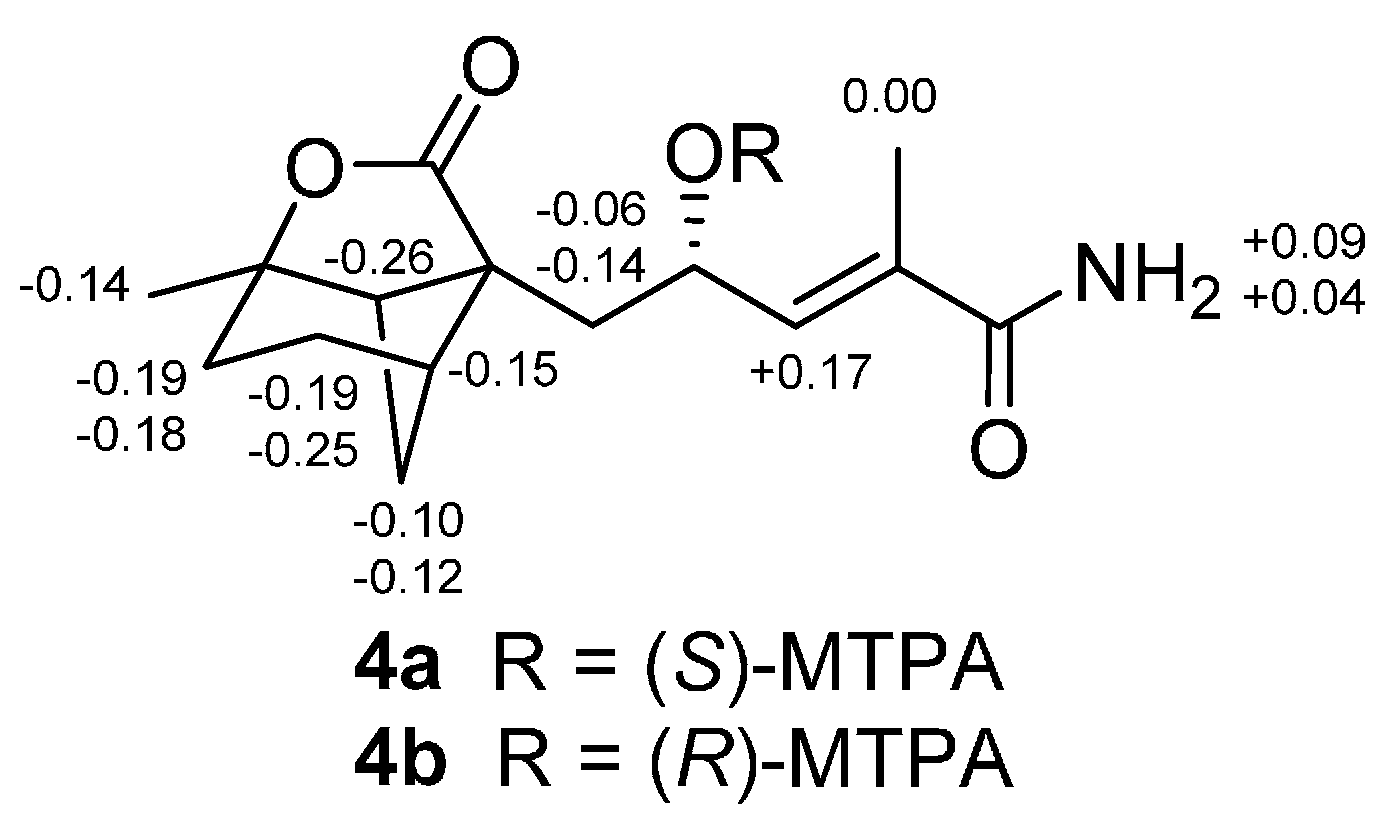
4 was assigned by application of the modified Mosher method [
15]. Treatment of
4 with (
S)-MTPA Cl and (
R)-MTPA Cl afforded the
R-MTPA ester (
4a) and
S-MTPA ester (
4b), respectively. The difference in chemical shift values (∆δ = δ
S − δ
R) for the diastereomeric
S-MTPA (
4b) and
R-MTPA (
4a) esters (
Figure 4) was calculated to assign the 12
S absolute configuration. The absolute configuration of
4 was deduced as shown by analogy to
3.
Table 2.
NMR Data for 3 and 4 (Acetone-d6).
Table 2.
NMR Data for 3 and 4 (Acetone-d6).
| Pos. | 3 | 4 |
|---|
| δC a | δH b (J in Hz) | HMBC a | δC c | δH d (J in Hz) | HMBC c |
|---|
| 1 | 90.7 | | | 88.7 | | |
| 2 | 26.0 | 1.84, m; 2.12, m | 1, 3, 4, 6 | 30.5 | 1.78, m; 2.06, m | 1, 3, 4, 6 |
| 3 | 23.2 | 1.77, m; 1.96, m | 1, 2, 4, 5, 7 | 23.8 | 1.77, m; 1.94, m | 1, 2, 4, 5, 7 |
| 4 | 41.6 | 2.43, m | 2, 3, 5, 6, 7, 11 | 42.7 | 2.38, m | 2, 6, 7, 11 |
| 5 | 23.5 | 1.72, d (11.0); 2.30, ddd (11.0, 6.0, 5.5) | 1, 3, 4, 6, 7, 8 | 23.2 | 1.76, d (11.0); 2.35, ddd (11.0, 6.0, 5.5) | 1, 3, 4, 6, 7, 8 |
| 6 | 44.3 | 2.82, dd (5.5, 5.0) | 1, 2, 4, 7, 10, 11 | 48.7 | 2.90, dd (6.7, 5.5) | 1, 2, 4, 5, 10, 11 |
| 7 | 53.6 | | | 55.9 | | |
| 8 | 177.8 | | | 179.1 | | |
| 10 | 66.4 | 3.70, brs | 1, 2, 6 | 25.1 | 1.42, s | 1, 2, 6 |
| 11 | 44.5 | 3.09, d (19.0); 3.15, d (19.0) | 4, 6, 7, 8, 12 | 37.3 | 1.89, m; 1.99, m | 4, 6, 7, 8, 12, 13 |
| 12 | 199.5 | | | 67.1 | 4.57, ddd (10.7, 10.6, 5.2) | 7, 11, 13, 14 |
| 13 | 128.2 | 6.82, q (1.5) | 12, 14, 15, 16 | 139.1 | 6.25, dq (10.6, 1.5) | 11, 14, 15, 16 |
| 14 | 146.2 | | | 131.3 | | |
| 15 | 170.6 | | | 171.3 | | |
| 16 | 14.9 | 2.15, d (1.5) | 13, 14, 15 | 13.2 | 1.83, d (1.5) | 13, 14, 15 |
| OH-10 | | 4.10, brs | | | | |
| OH-12 | | | | | 4.15, brs | 11 |
| NH2-15 | | 7.18, brs; 6.62, brs | | | 6.73, brs; 6.19, brs | |
Figure 4.
∆δ Values (in ppm) = δS − δR obtained for (S)- and (R)-MTPA esters 4b and 4a.
Figure 4.
∆δ Values (in ppm) = δS − δR obtained for (S)- and (R)-MTPA esters 4b and 4a.
Compounds 1–6 were tested for cytotoxicity against a panel of eight human tumor cell lines A549 (human lung adenocarcinoma cells), A375 (human malignant melanoma cells), MCF-7 (human breast cancer cells), CNE1-LMP1 (stable oncoprotein LMP1 integrated nasopharyngeal carcinoma cells), EC109 (human esophageal cancer cells), MGC (human gastric cancer cells), PANC-1 (human pancreatic carcinoma cells), and Hep3B-2 (human hepatoma carcinoma cells). Unfortunately, compounds 1–6 did not show detectable cytotoxicity at 50 µM.
Bergamotane sesquiterpenoids incorporating 4-oxatricyclo(3.3.1.0
2,7)nonane skeletonare rare. Precedents include brasilamides A and B, which were isolated from
P. brasiliense in our previous study [
13]. Structurally, brasilamide K (
1) is a hydrogenated analogue of brasilamide A (
5) [
13], with a tetrahydro-2
H-pyrone moiety attached to the bicyclo(3.1.1)heptane ring at C-2 and C-5, completing 4-oxatricyclo (3.3.1.0
2,7)nonane skeleton. BrasilamidesL-N (
2–
4) are new additions of bergamotane sesquiterpenoids, possessing the unique 9-oxatricyclo(4.3.0.0
4,7)nonane skeleton. Brasilamides L (
2) and M (
3) are the hydrogenated and oxygenated products of brasilamide C (
6) [
13], respectively. Whereas brasilamide N (
4) is structurally related to
6, but differs by having a methyl group at C-1, a carboxylic carbon at C-8, and a hydroxy group at C-12 instead of an oxygenated methlene unit, a methylene unit, and a ketone carbon, respectively.
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations were measured on a Perkin-Elmer 241 polarimeter, and UV data were obtained on a Shimadzu Biospec-1601 spectrophotometer. IR data were recorded using a Nicolet Magna-IR 750 spectrophotometer. 1H- and 13C-NMR data were acquired with Varian Mercury-400, -500, and -600 spectrometers using solvent signals (acetone-d6: δH 2.05/δC 29.8, 206.1) as references. The HMQC and HMBC experiments were optimized for 145.0 and 8.0 Hz, respectively. ESIMS and HRESIMS data were obtained using an Agilent Accurate-Mass-Q-TOF LC/MS 6520 instrument equipped with an electrospray ionization (ESI) source. The fragmentor and capillary voltages were kept at 125 and 3500 V, respectively. Nitrogen was supplied as the nebulizing and drying gas. The temperature of the drying gas was set at 300 °C. The flow rate of the drying gas and the pressure of the nebulizer were 10 L/min and 10 psi, respectively. All MS experiments were performed in positive ion mode. Full-scan spectra were acquired over a scan range of m/z 100−1000 at 1.03 spectra/s. HPLC separations were performed on an Agilent 1260 instrument (Agilent, Waldbronn, Germany) equipped with a variable-wavelength UV detector.
3.2. Fungal Material
The culture of P. brasiliense Verkley was isolated from branches of Acer truncatum Bunge on Dongling Mountain, Beijing, in March, 2005. The isolate was identified and assigned the accession number M3-3341 in L.G.’s culture collection at the Institute of Microbiology, Chinese Academy of Sciences, Beijing. The fungal strain was cultured on slants of potato dextrose agar at 25 °C for 10 days. Agar plugs were cut into small pieces (about 0.5 × 0.5 ×0.5 cm3) under aseptic conditions, 15 pieces were used to inoculate in three Erlenmeyer flasks (250 mL), each containing 50 mL of media (0.4% glucose, 1% malt extract, and 0.4% yeast extract), and the final pH of the media was adjusted to 6.5. After sterilization, three flasks of the inoculated media were incubated at 25 °C on a rotary shaker at 170 rpm for five days to prepare the seed culture. Spore inoculum was prepared by suspending the seed culture in sterile, distilled H2O to give a final spore/cell suspension of 1 × 106/mL. Fermentation was carried out in 12 Fernbach flasks (500 mL), each containing 80 g of rice. Distilled H2O (120 mL) was added to each flask, and the contents were soaked overnight before autoclaving at 15 psi for 30 min. After cooling to room temperature, each flask was inoculated with 5.0 mL of the spore inoculum and incubated at 25 °C for 40 days.
3.3. Extraction and Isolation
The fermented material was extracted repeatedly with EtOAc (4 × 1.0 L), and the organic solvent was evaporated to dryness under vacuum to afford the crude extract (9.0 g), which was fractionated by silica gel VLC using petroleum ether-EtOAc gradient elution. The fraction (100 mg) eluted with 72% EtOAc was separated by Sephadex LH-20 column chromatography (CC) eluting with 1:1 CH2Cl2/MeOH. The resulting subfractions were combined and further purified by semipreparative RP HPLC (Agilent Zorbax SB-C18 column; 5 μm; 9.4 mm × 250 mm; 25% MeOH in H2O for 2 min, followed by 25%–60% over 33 min; 2 mL/min) to afford 1 (1.2 mg, tR 25.58 min), and 4 (9.0 mg, tR 24.82 min). Fractions (190 mg) eluted with 80% and 95% EtOAc were fractionated again by Sephadex LH-20 CC eluting with 1:1 CH2Cl2/MeOH as eluents. Purification of the resulting subfractions afforded 2 (4.0 mg, tR 28.40 min; 30% MeOH in H2O for 2 min, followed by 30%–60% over 35 min; 2 mL/min), and 3 (7.0 mg, tR 13.21 min; 25% MeOH in H2O for 2 min, followed by 25%–38% over 30 min; 2 mL/min).
Brasilamide K (
1), colorless oil;
−9.0 (
c 0.1, MeOH); UV (MeOH) λ
max (log ε) 208 (3.76) nm; IR (neat) ν
max 3349 (br), 2932, 1713, 1666, 1637, 1591, 1332, 1211, 1109 cm
−1;
1H-,
13C-NMR and HMBC data see
Table 1; HRESIMS
m/
z 280.1534 (calcd for C
15H
22NO
4, 280.1543).
Brasilamide L (
2), colorless powder;
+14.0 (
c 0.1, MeOH); UV (MeOH) λ
max (log ε) 221 (3.70) nm; IR (neat) ν
max 3331 (br), 2921, 2866, 1666, 1598, 1379, 1198, 1023 cm
−1;
1H-,
13C-NMR and HMBC data see
Table 1; HRESIMS
m/
z 266.1743 (calcd for C
15H
24NO
3, 266.1751).
Brasilamide M (
3), colorless oil;
+37.0 (
c 0.1, MeOH); UV (MeOH) λ
max (log
ε) 226 (3.58) nm; IR (neat) ν
max 3359 (br), 2934, 2870, 1749, 1667, 1603, 1347, 1156, 1078 cm
−1;
1H-,
13C-NMR and HMBC data see
Table 2; HRESIMS
m/
z 294.1332 (calcd for C
15H
20NO
5, 294.1336).
Brasilamide N (
4), colorless oil;
+34.7 (
c 0.5, MeOH); UV (MeOH) λ
max (log
ε) 214 (3.70) nm; IR (neat)
νmax 3350 (br), 2937, 2868, 1739, 1674, 1642, 1600, 1382, 1197, 1028 cm
−1;
1H-,
13C-NMR and HMBC data see
Table 2; HRESIMS
m/
z 280.1546 (calcd for C
15H
22NO
4, 280.1543).
3.4. Preparation of (R)-MTPA (4a) and (S)-MTPA (4b) Esters
A sample of 4 (1.0 mg, 0.004 mmol), (S)-MTPA Cl (2.0 μL, 0.011 mmol), and pyridine-d5 (0.5 mL) were allowed to react in an NMR tube at ambient temperature for 24 h. The mixture was evaporated to dryness and purified by RP HPLC (from 70% to 100% MeOH in 20 min) to afford 4a (0.8 mg, tR 12.01 min): colorless oil; 1H-NMR (acetone-d6, 500 MHz) δ 6.72 (1H, brs, NH2-15), 6.32 (1H, brs, NH2-15), 6.09 (1H, dd, J = 9.4, 1.5 Hz, H-13), 5.91 (1H, ddd, J = 9.4, 8.0, 5.5 Hz, H-12), 2.65 (1H, dd, J = 5.5, 5.2 Hz, H-6), 2.44 (1H, m, H-4), 2.37 (1H, m, H-5b), 2.34 (1H, m, H-2b), 2.22 (1H, m, H-11b), 2.17 (1H, m, H-3b), 2.01 (3H, d, J = 1.5 Hz, H3-16), 1.98 (1H, m, H-11a), 1.93 (1H, m, H-2a), 1.91 (1H, m, H-3a), 1.78 (1H, d, J = 10.3 Hz, H-5a), 1.39 (3H, s, H3-10); ESIMS m/z 496 [M + H]+.
Another sample of 4 (1.0 mg, 0.004 mmol), (R)-MTPA Cl (2.0 μL, 0.011 mmol), and pyridine-d5 (0.5 mL) was processed as described above for 4a to afford 4b, which was purified by RP HPLC (from 70% to 100% MeOH in 20 min) to afford 4b (0.6 mg, tR 12.61 min): colorless oil; 1H-NMR (acetone-d6, 500 MHz) δ 6.81 (1H, brs, NH2-15), 6.36 (1H, brs, NH2-15), 6.26 (1H, dd, J = 9.4, 1.5 Hz, H-13), 5.82 (1H, ddd, J = 9.4, 8.0, 5.5 Hz, H-12), 2.39 (1H, dd, J = 5.5, 5.2 Hz, H-6), 2.29 (1H, m, H-4), 2.25 (1H, m, H-5b), 2.16 (1H, m, H-2b), 2.08 (1H, m, H-11b), 1.92 (1H, m, H-3b), 2.01 (3H, d, J = 1.5 Hz, H3-16), 1.92 (1H, m, H-11a), 1.74 (1H, m, H-2a), 1.72 (1H, m, H-3a), 1.68 (1H, d, J = 10.3 Hz, H-5a), 1.26 (3H, s, H3-10); ESIMS m/z 496 [M + H]+.
3.5. MTS Assay
The MTS assay [
9] was run in triplicate. In a 96-well plate, each well was plated with 2–5 × 10
3 cells (depending on the cell multiplication rate). After cell attachment overnight, the medium was removed, and each well was treated with 100 μL of medium containing 0.1% DMSO, or appropriate concentrations of the test compounds and the positive control paclitaxel (Sigma, St. Louis, MO, USA) (100 mM as stock solution of a compound in DMSO and serial dilutions; the test compounds showed good solubility in DMSO and did not precipitate when added to the cells). The plate was incubated for 72 h at 37 °C in a humidified, 5% CO
2 atmosphere. Proliferation assessed by adding 20 μL of MTS (Promega, Madison, WI, USA) to each well in the dark, followed by a 90 min incubation at 37 °C. The assay plate was read at 490 nm using a microplate reader.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


