
Design and Synthesis of Imidazopyrazolopyridines as Novel Selective COX-2 Inhibitors


 ,
,
Abstract
:
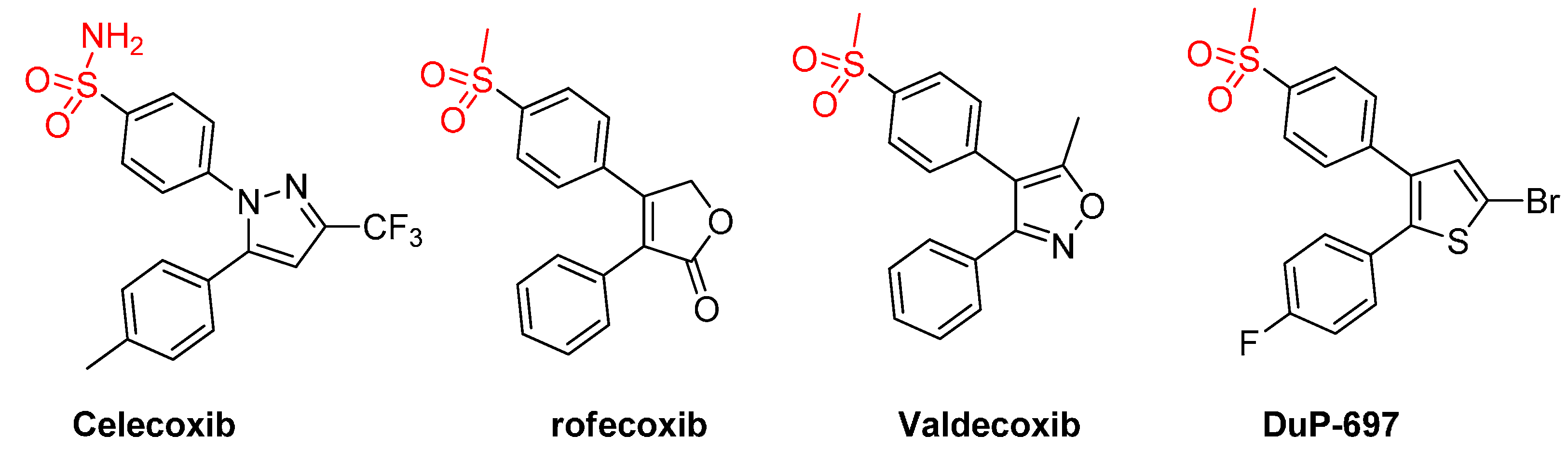
1. Introduction


2. Results and Discussion
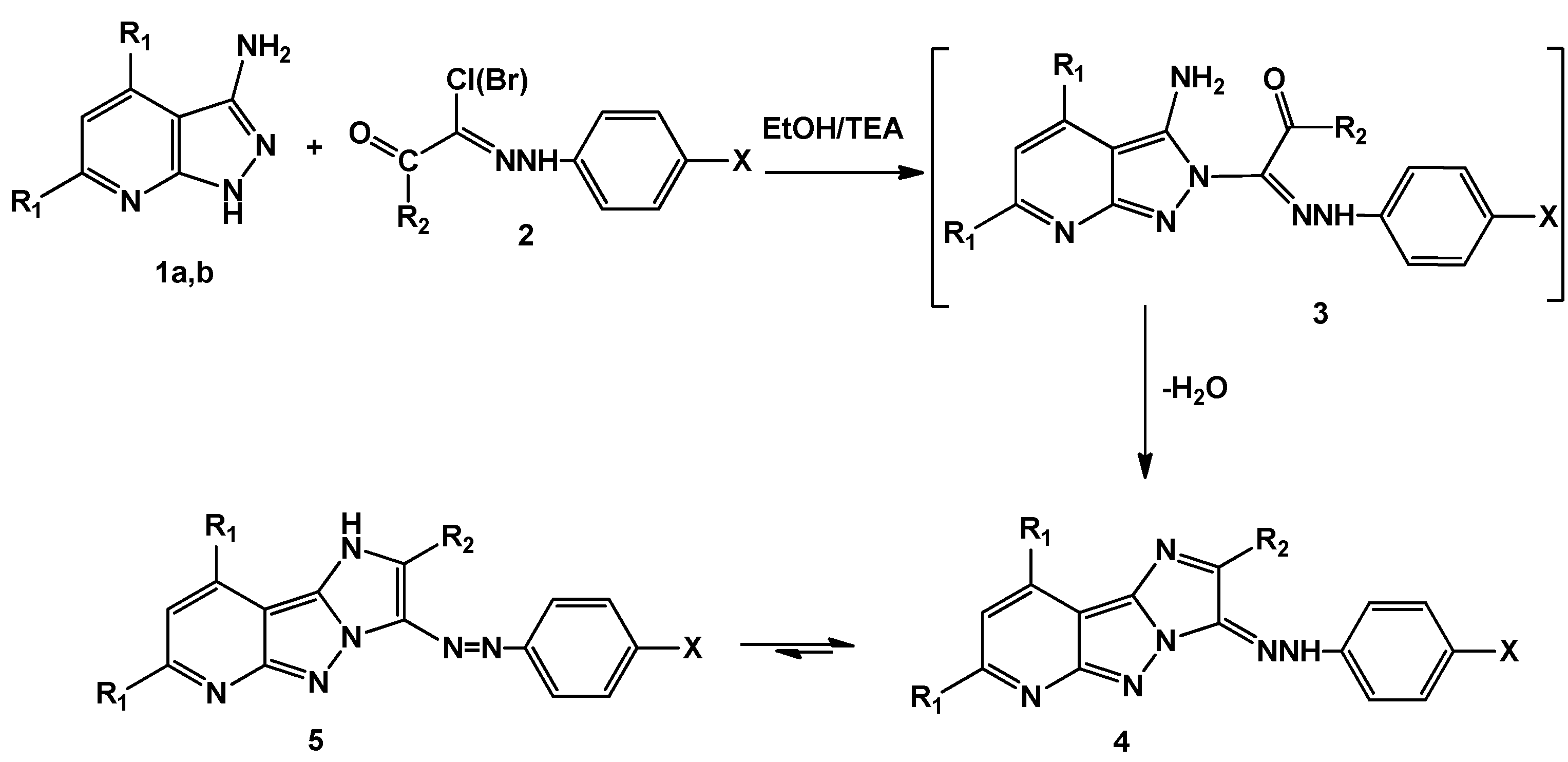
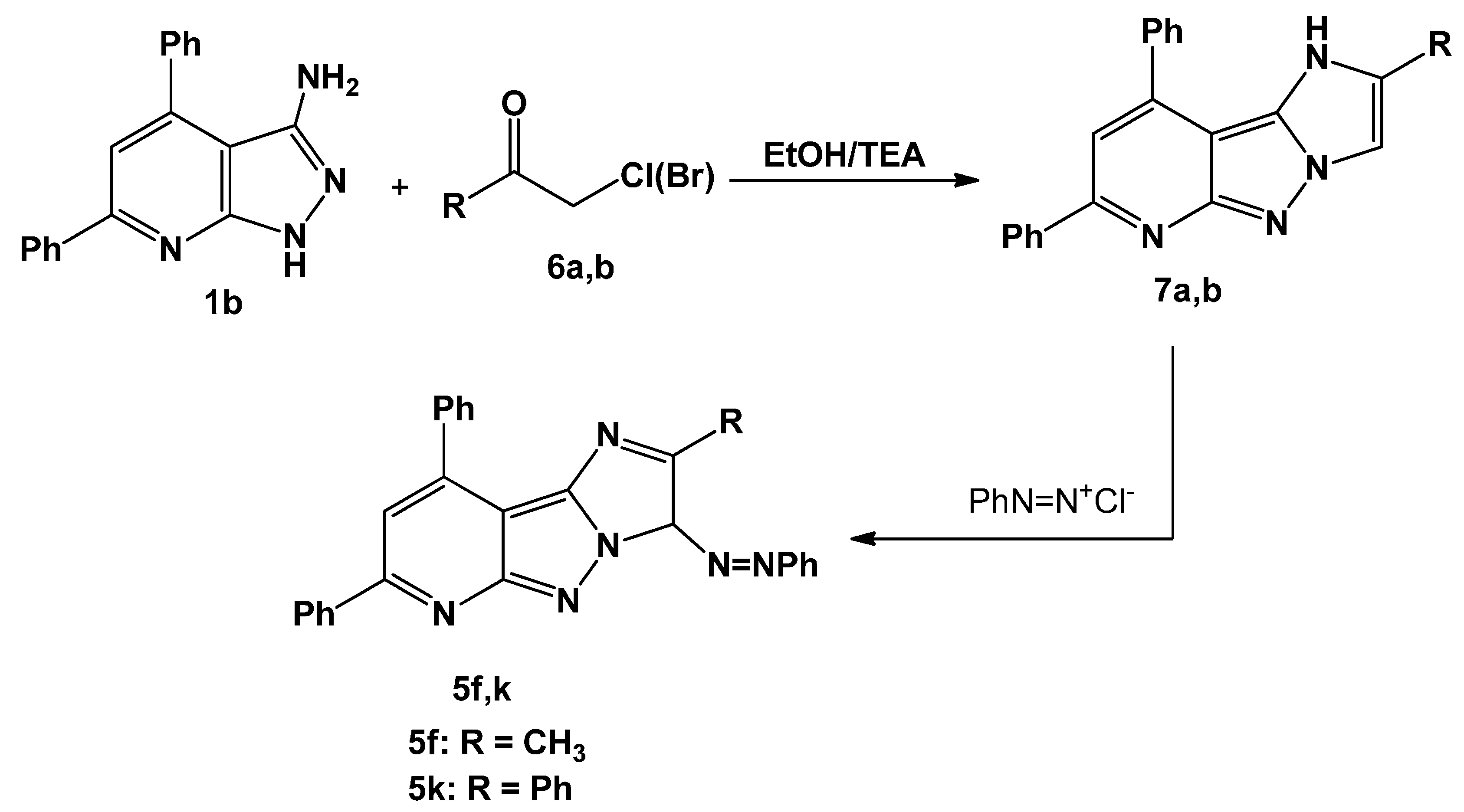
2.1. Chemistry

| Compd. No. | R1 | R2 | X | Compd. No. | R1 | R2 | X |
|---|---|---|---|---|---|---|---|
| a | Me | Me | H | g | Ph | Me | OMe |
| b | Me | Me | Cl | h | Ph | Me | Cl |
| c | Me | Me | OMe | i | Ph | Me | Br |
| d | Me | Me | Br | j | Ph | Me | NO2 |
| e | Me | Me | NO2 | k | Ph | Ph | H |
| f | Ph | Me | H | l | Ph |  | H |

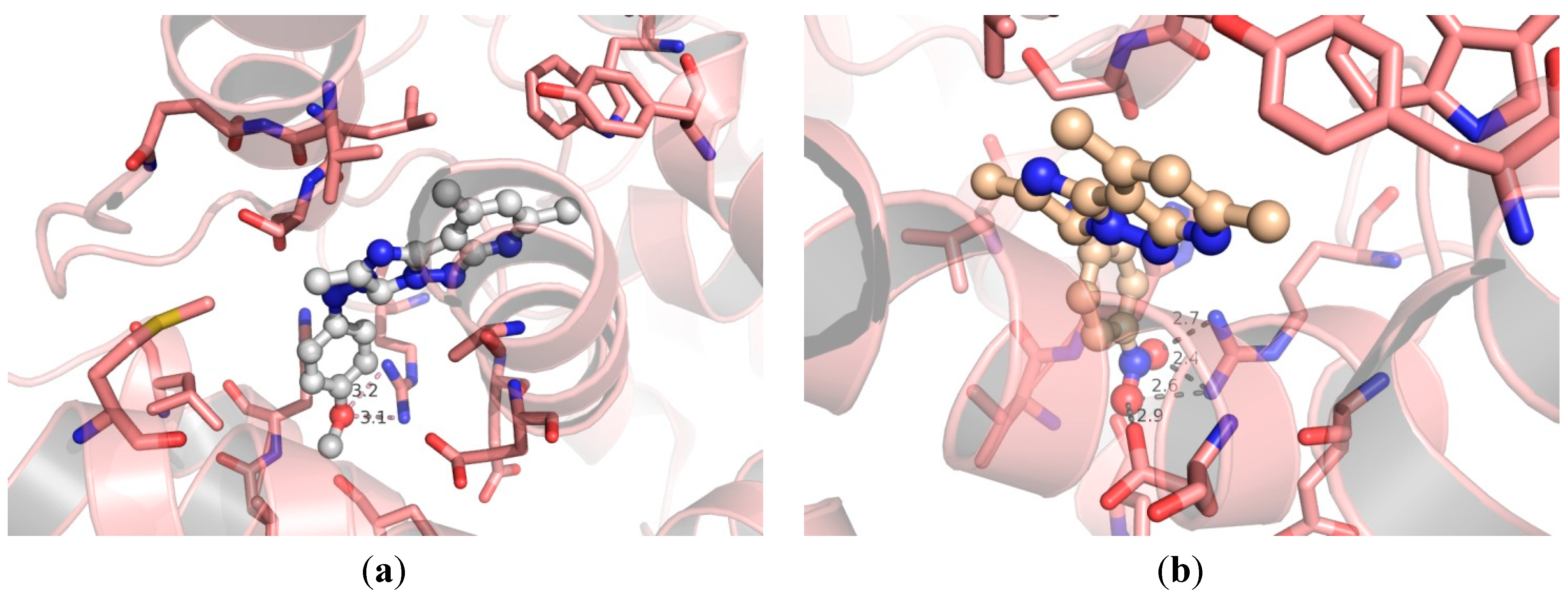
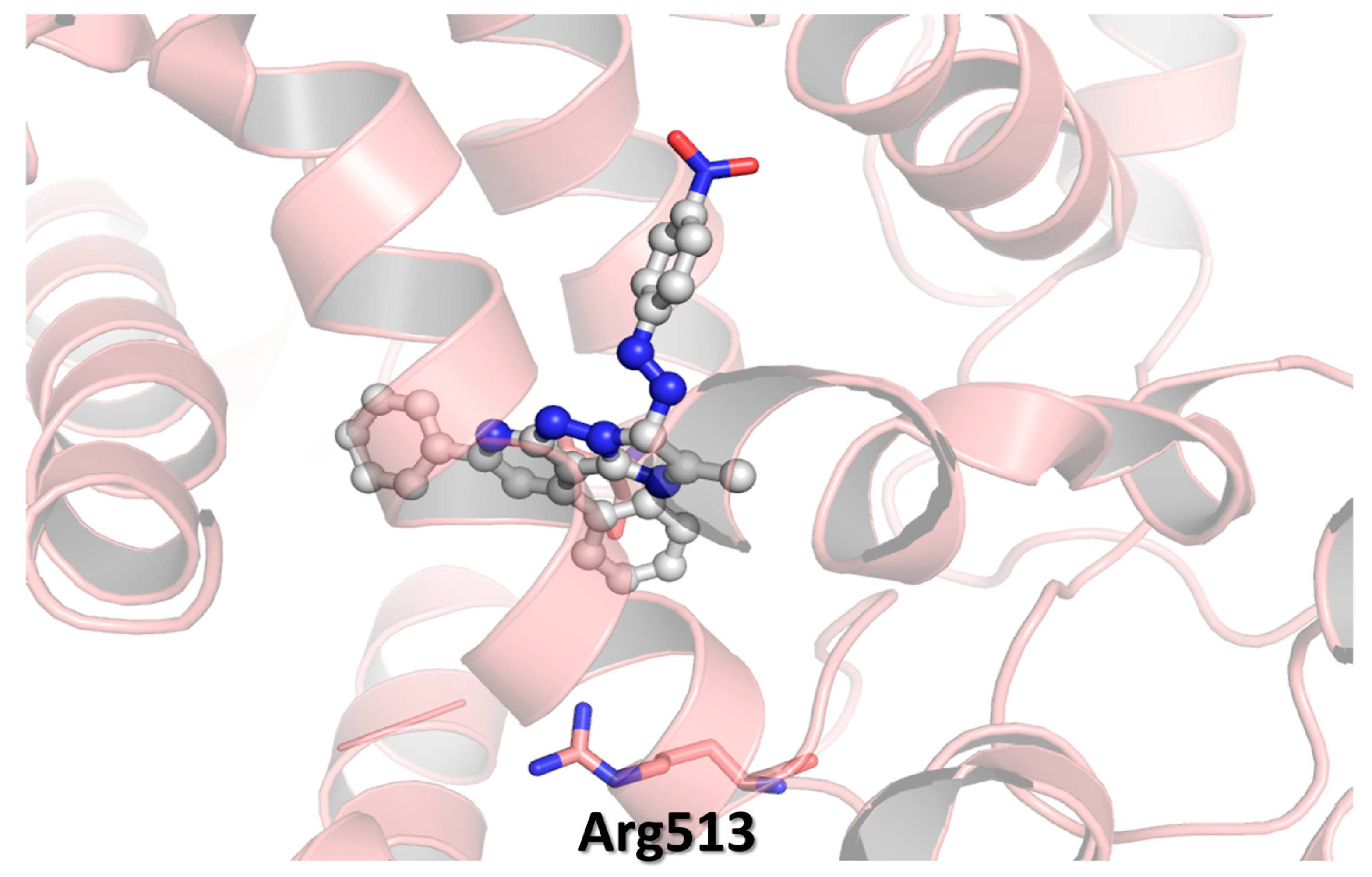
2.2. Biological Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. No. | Drug in μg/mL | Percentage Inhibition * | COX-2/COX-1 Selectivity | |
|---|---|---|---|---|
| COX-1 | COX-2 | |||
| 5a | 10 | 3.88 ± 0.0023 | 36.51 ± 0.35 | 9.4 |
| 5b | 10 | 3.84 ± 0.0034 | 59.11 ± 0.44 | 15.4 |
| 5c | 10 | 3.76 ± 0.0023 | 68.34 ± 0.53 | 18.2 |
| 5d | 10 | 3.80 ± 0.0032 | 67.72 ± 0.44 | 17.8 |
| 5e | 10 | 3.92 ± 0.0023 | 75.92 ± 0.36 | 19.4 |
| 5f | 10 | 3.96 ± 0.0034 | 25.31 ± 0.40 | 6.4 |
| 5g | 10 | 3.99 ± 0.0025 | 24.72 ± 0.59 | 6.2 |
| 5h | 10 | 3.62 ± 0.0036 | 20.85 ± 0.38 | 5.6 |
| 5i | 10 | 3.58 ± 0.0025 | 11.50 ± 0.47 | 3.2 |
| 5j | 10 | 3.54 ± 0.0034 | 22.15 ± 0.65 | 6.6 |
| 5k | 10 | 3.51 ± 0.0046 | 12.80 ± 0.74 | 3.6 |
| 5l | 10 | 3.69 ± 0.0027 | 9.58 ± 0.83 | 2.5 |
| Diclofenac | 10 | 56.78 | 7.88 | 0.13 |
| Valdecoxib | 10 | 12.15 | 76.15 | 6.26 |
| Ibuprofen | 10 | 78.89 | 5.23 | 0.06 |

Spectrophotometric Assay of Recombinant Human COX-2

| Compd. No. | Dose [mg/kg] | % Protection against Edema | % Inhibition of Plasma PGE2 |
|---|---|---|---|
| 5a | 2.5 | 31.76 ± 0.66 | 72.19 ± 0.45 |
| 5 | 40.05 ± 0.77 | 73.78 ± 0.43 | |
| 5b | 2.5 | 69.10 ± 0.85 | 82.65 ± 0.56 |
| 5 | 71.42 ± 0.96 | 84.25 ± 0.45 | |
| 5c | 2.5 | 78.65 ± 0.87 | 83.59 ± 0.68 |
| 5 | 88.96 ± 0.78 | 85.19 ± 0.79 | |
| 5d | 2.5 | 67.32 ± 0.89 | 83.13 ± 0.80 |
| 5 | 70.60 ± 0.90 | 84.72 ± 0.97 | |
| 5e | 2.5 | 92.88 ± 0.97 | 91.73 ± 0.53 |
| 5 | 97.15 ± 0.86 | 93.31 ± 0.46 | |
| 5f | 2.5 | 31.36 ± 0.55 | 61.28 ± 0.43 |
| 5 | 33.74 ± 0.64 | 62.84 ± 0.56 | |
| 5g | 2.5 | 41.82 ± 0.75 | 80.82 ± 0.44 |
| 5 | 44.21 ± 0.66 | 82.38 ± 0.21 | |
| 5h | 2.5 | 42.28 ± 0.57 | 75.49 ± 0.45 |
| 5 | 54.68 ± 0.75 | 77.13 ± 0.89 | |
| 5i | 2.5 | 40.45 ± 0.63 | 75.96 ± 0.96 |
| 5 | 42.80 ± 0.54 | 77.61 ± 0.89 | |
| 5j | 2.5 | 26.46 ± 0.45 | 66.45 ± 0.83 |
| 5 | 28.71 ± 0.56 | 68.11 ± 0.83 | |
| 5k | 2.5 | 19.99 ± 0.67 | 66.93 ± 0.83 |
| 5 | 22.34 ± 0.78 | 68.60 ± 0.98 | |
| 5l | 2.5 | 38.10 ± 0.69 | 75.01 ± 0.65 |
| 5 | 40.29 ± 0.77 | 76.65 ± 0.34 | |
| Diclofenac sodium | 2.5 | 69.59 ± 0.44 | 55.49 ± 0.80 |
| 5 | 72.19 ± 0.54 | 70.59 ± 0.80 | |
| Valdicoxib | 2.5 | 78.89 ± 0.33 | 60.09 ± 0.86 |
| 5 | 87.19 ± 0.23 | 81.11 ± 0.21 |
3. Experimental Protocols
3.1. Chemistry
3.1.1. General
3.1.2. General Method for the Synthesis of 2,7,9-Trisubstituted-3-(aryldiazenyl)-3H-imidazo[1′,2′:1,5] pyrazolo[3,4-b]pyridine (5a–l)
3.1.3. Synthesis of 2-Substituted-7,9-diphenyl-3H-imidazo[1′,2′:1,5]pyrazolo[3,4-b]pyridine (7a,b)
3.1.4. Coupling of 7a,b with Benzenediazonium Chloride
3.2. Pharmacology
3.2.1. Cyclooxygenase Inhibition Activity
3.2.2. Anti-Inflammatory Activity: Carrageenan-Induced Edema (Rats Paw Test)
3.3. Statistical Analysis
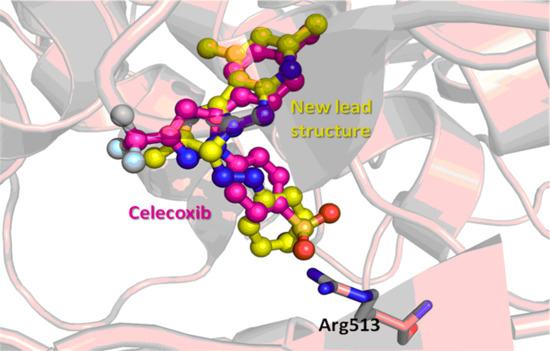
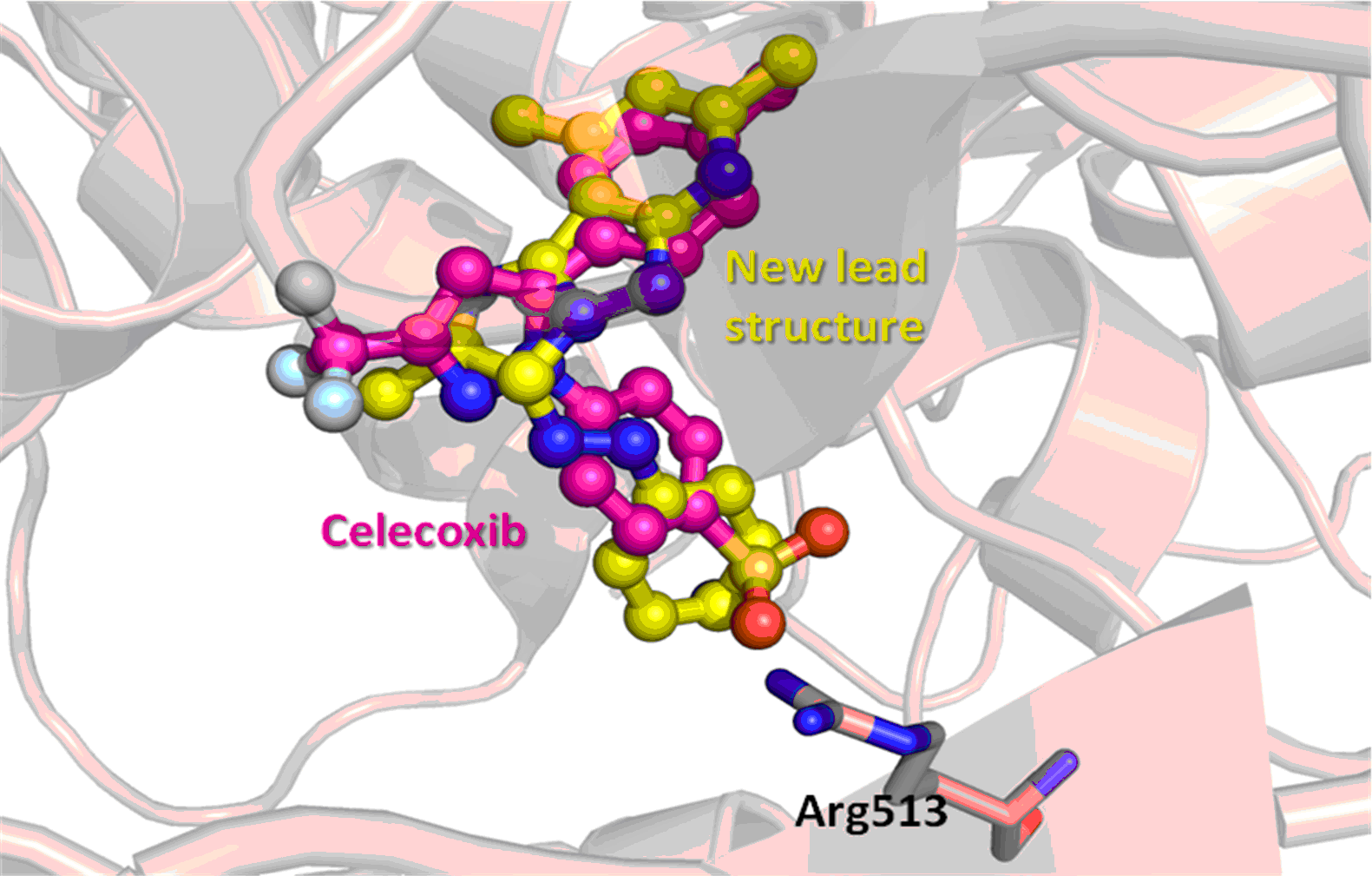
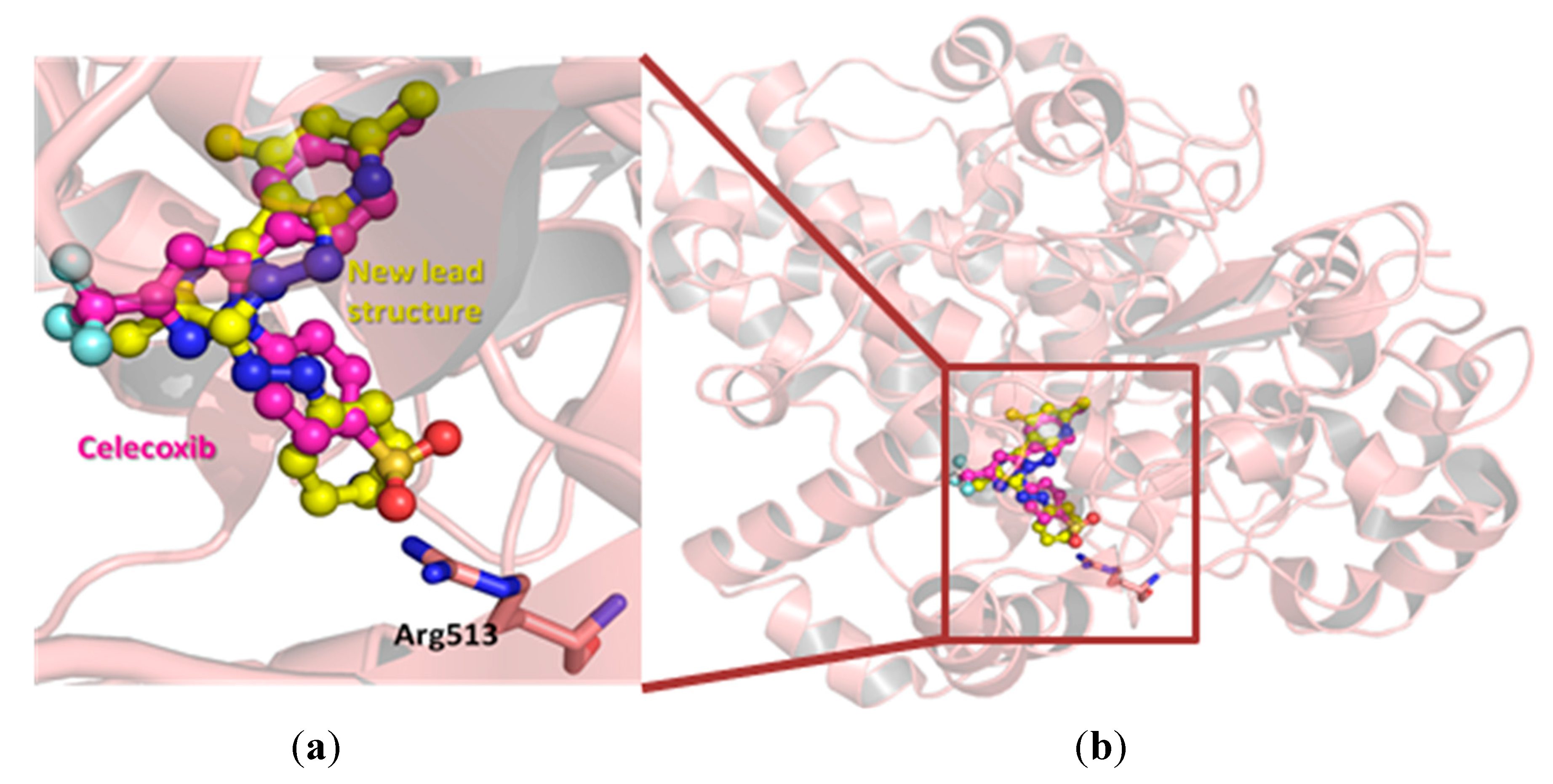
3.4. Molecular Modeling
4. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
References
- Misko, T.P.; Trotter, J.L.; Cross, A.H. Mediation of inflammation by encephalitogenic cells: Interferon gamma induction of nitric oxide synthase and cyclooxygenase 2. J. Neuroimmunol. 1995, 61, 195–204. [Google Scholar] [CrossRef]
- Seibert, K.; Masferrer, J.L. Role of inducible cyclooxygenase (COX-2) in inflammation. Receptor 1994, 4, 17–23. [Google Scholar] [PubMed]
- Seed, M.P.; Willoughby, D.A. COX-2, HO NO! Cyclooxygenase-2, heme oxygenase and nitric oxide synthase: Their role and interactions in inflammation. BIRAs Symposium, Saint Bartholomew’s Hospital, London, 26 April 1996. Inflamm. Res. 1997, 46, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Kogiso, M.; Shinohara, T.; Dorey, C.K.; Shibata, Y. Role of PPARgamma in COX-2 activation in mycobacterial pulmonary inflammation. Inflammation 2012, 35, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Renna, N.F.; Diez, E.R.; Lembo, C.; Miatello, R.M. Role of Cox-2 in vascular inflammation: An experimental model of metabolic syndrome. Mediat. Inflamm. 2013. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Lee, I.T.; Yang, Y.L.; Lee, C.W.; Kou, Y.R.; Yang, C.M. Induction of COX-2/PGE(2)/IL-6 is crucial for cigarette smoke extract-induced airway inflammation: Role of TLR4-dependent NADPH oxidase activation. Free Radic. Biol. Med. 2010, 48, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Zarrilli, R.; Tuccillo, C.; Santangelo, M.; Nardone, G.; Romano, M. Increased COX-2, but not COX-1, mRNA expression in Helicobacter pylori gastritis. Am. J. Gastroenterol. 1999, 94, 3376–3378. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R. N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Reddy, G.V.; Reddanna, P. Eicosanoids in inflammation and cancer: The role of COX-2. Expert Rev. Clin. Immunol. 2009, 5, 145–165. [Google Scholar] [CrossRef] [PubMed]
- Hahm, K.B.; Lim, H.Y.; Sohn, S.; Kwon, H.J.; Lee, K.M.; Lee, J.S.; Surh, Y.J.; Kim, Y.B.; Joo, H.J.; Kim, W.S.; et al. In vitro evidence of the role of COX-2 in attenuating gastric inflammation and promoting gastric carcinogenesis. J. Environ. Pathol. Toxicol. Oncol. 2002, 21, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Misra, U.K.; Pizzo, S.V. Evidence for a pro-proliferative feedback loop in prostate cancer: The role of Epac1 and COX-2-dependent pathways. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Clemente, M.; Sanchez-Archidona, A.R.; Sardon, D.; Diez, L.; Martin-Ruiz, A.; Caceres, S.; Sassi, F.; Pérez-Alenza, M.D.; Lllera, J.C.; Dunner, S.; et al. Different role of COX-2 and angiogenesis in canine inflammatory and non-inflammatory mammary cancer. Vet. J. 2013, 197, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Serna-Marquez, N.; Villegas-Comonfort, S.; Galindo-Hernandez, O.; Navarro-Tito, N.; Millan, A.; Salazar, E.P. Role of LOXs and COX-2 on FAK activation and cell migration induced by linoleic acid in MDA-MB-231 breast cancer cells. Cell. Oncol. 2013, 36, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Nadda, N.; Setia, S.; Vaish, V.; Sanyal, S.N. Role of cytokines in experimentally induced lung cancer and chemoprevention by COX-2 selective inhibitor, Etoricoxib. Mol. Cell. Biochem. 2013, 372, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Geis, G.S. Update on clinical developments with celecoxib, a new specific COX-2 inhibitor: what can we expect. J. Rheumatol. 1999, 56, 31–36. [Google Scholar] [CrossRef]
- Folco, G.C. New NSAIDs and gastroduodenal damage. Ital. J. Gastroenterol. 1996, 28, 28–29. [Google Scholar] [PubMed]
- Dammann, H.G. Preferential COX-2 inhibition: Its clinical relevance for gastrointestinal non-steroidal anti-inflammatory rheumatic drug toxicity. Z. Gastroenterol. 1999, 37, 45–58. [Google Scholar] [PubMed]
- Dannhardt, G.; Laufer, S. Structural approaches to explain the selectivity of COX-2 inhibitors: Is there a common pharmacophore. Curr. Med. Chem. 2000, 7, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. 2011, 10, 655–683. [Google Scholar] [PubMed]
- Mason, R.P.; Walter, M.F.; McNulty, H.P.; Lockwood, S.F.; Byun, J.; Day, C.A.; Charles, A.B.S.; Jacob, R.F. Rofecoxib increases susceptibility of human LDL and membrane lipids to oxidative damage: A mechanism of cardiotoxicity. J. Cardiovasc. Pharmacol. 2006, 47, 7–14. [Google Scholar] [CrossRef]
- Park, S.J.; Buschmann, H.; Bolm, C. Bioactive sulfoximines: Syntheses and properties of Vioxx analogs. Bioorg. Med. Chem. Lett. 2011, 21, 4888–4890. [Google Scholar] [CrossRef] [PubMed]
- Baxter, I. Pyrazolopyridine Derivatives as Selective COX-2 Inhibitors. E.P. 1127058 A1, 1 September 2001. [Google Scholar]
- Campbell, I.B.; Naylor, A. Cyclooxygenase-2 Inhibitors (COX-2); 4-[2-(3-Fluoro-phenyl)-6-trifluoromethyl-pyrazolo[1,5-a]pyridin-3-yl]benzenesulfonamide for Example; Fever, Pain, Inflammation, Neurodegenerative Disorders, Oral Diseases, Influenza, Arthritis, Allergies. U.S. Patent 7223772 B1, 29 May 2007. [Google Scholar]
- Alberti, M.J. Antiinflammatory Agents; Autoimmune Disease Treatments; Pyrazolo[1,5-a] Pyridine Derivatives. U.S. Patent 7166597 B2, 23 January 2007. [Google Scholar]
- Cavalli, A.; Poluzzi, E.; de Ponti, F.; Recanatini, M. Toward a Pharmacophore for Drugs Inducing the Long Qt Syndrome: Insights from a Comfa Study of Herg K(+) Channel Blockers. J. Med. Chem. 2002, 45, 3844–3853. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Limburg, D.; Graneto, M.J.; Springer, J.; Hamper, J.R.; Liao, S.; Pawlitz, J.L.; Kurumbail, R.G.; Maziasz, T.; Talley, J.J.; et al. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 2010, 20, 7159–7163. [Google Scholar] [CrossRef] [PubMed]
- Michaux, C.; Charlier, C. Structural approach for COX-2 inhibition. Mini-Rev. Med. Chem. 2004, 4, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J.; Kalgutkar, A.S. Cyclooxygenase 2 inhibitors: Discovery, selectivity and the future. Trends Pharmacol. Sci. 1999, 20, 465–469. [Google Scholar] [CrossRef]
- Shi, D.Q.; Shi, J.W.; Yao, H.; Jiang, H.; Wang, X.S. An Efficient Synthesis of Pyrazolo[3,4-b] pyridine Derivatives in Aqueous Media. J. Chin. Chem. Soc. 2007, 54, 1341–1345. [Google Scholar] [CrossRef]
- El-Gendy, M.S.; Abdel-Aziem, A.; Abdelhamid, A.O. Reactions and Antimicrobial activity of (3-(3-(4-Methoxyphenyl)acryloyl)-2H-Chromen-2-one. Int. J. Adv. Res. 2013, 1, 557–568. [Google Scholar]
- Copeland, R.A.; Williams, J.M.; Giannaras, J.; Nurnberg, S.; Covington, M.; Pinto, D.; Pick, S.; Trzaskos, J.M. Mechanism of selective inhibition of the inducible isoform or prostaglandin G/H Synthase. Proc. Natl. Acad. Sci. USA 1994, 91, 11202–11206. [Google Scholar]
- Gouda, A.M. Utility of 3-amino-4,6-dimethyl-1H-pyrazolo[3,4-b]pyridine in heterocyclic synthesis. J. Heterocycl. Chem. 2011, 48, 1–10. [Google Scholar] [CrossRef]
- Deeb, A.; Essawy, A.; El-Gendy, A.M.; Shaban, A. Heterocyclic synthesis with 3-cyano-2(1H) pyridinethione: Synthesis of 3-oxo-2,3-dihydroisothiazolo[5,4-b]pyridine and related compounds. Monatsh. Chem. 1990, 121, 281–287. [Google Scholar] [CrossRef]
- Eweiss, N.F.; Osman, A. Synthesis of heterocycles. Part II new routes to acetylthiadiazolines and alkylazothiazoles. J. Heterocycl. Chem. 1980, 17, 1713–1717. [Google Scholar] [CrossRef]
- Shawali, A.S.; Abdelhamid, A.O. Reaction of dimethylphenacylsulfonium bromide with N-nitrosoacetarylamides and reactions of the products with nucleophiles. Bull. Chem. Soc. Jpn. 1976, 49, 321–327. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the synthesized compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badrey, M.G.; Abdel-Aziz, H.M.; Gomha, S.M.; Abdalla, M.M.; Mayhoub, A.S. Design and Synthesis of Imidazopyrazolopyridines as Novel Selective COX-2 Inhibitors. Molecules 2015, 20, 15287-15303. https://doi.org/10.3390/molecules200815287
Badrey MG, Abdel-Aziz HM, Gomha SM, Abdalla MM, Mayhoub AS. Design and Synthesis of Imidazopyrazolopyridines as Novel Selective COX-2 Inhibitors. Molecules. 2015; 20(8):15287-15303. https://doi.org/10.3390/molecules200815287
Chicago/Turabian StyleBadrey, Mohamed G., Hassan M. Abdel-Aziz, Sobhi M. Gomha, Mohamed M. Abdalla, and Abdelrahman S. Mayhoub. 2015. "Design and Synthesis of Imidazopyrazolopyridines as Novel Selective COX-2 Inhibitors" Molecules 20, no. 8: 15287-15303. https://doi.org/10.3390/molecules200815287
APA StyleBadrey, M. G., Abdel-Aziz, H. M., Gomha, S. M., Abdalla, M. M., & Mayhoub, A. S. (2015). Design and Synthesis of Imidazopyrazolopyridines as Novel Selective COX-2 Inhibitors. Molecules, 20(8), 15287-15303. https://doi.org/10.3390/molecules200815287