
Synthesis, Biological Evaluation and Molecular Docking Studies of Piperidinylpiperidines and Spirochromanones Possessing Quinoline Moieties as Acetyl-CoA Carboxylase Inhibitors

Abstract
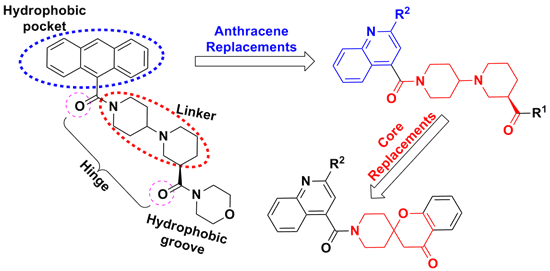
: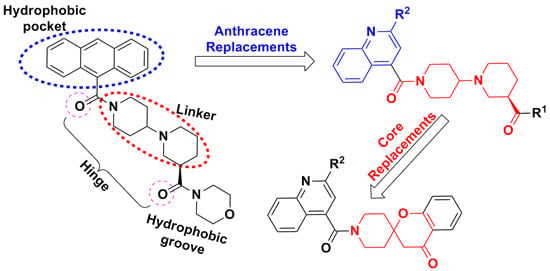
1. Introduction
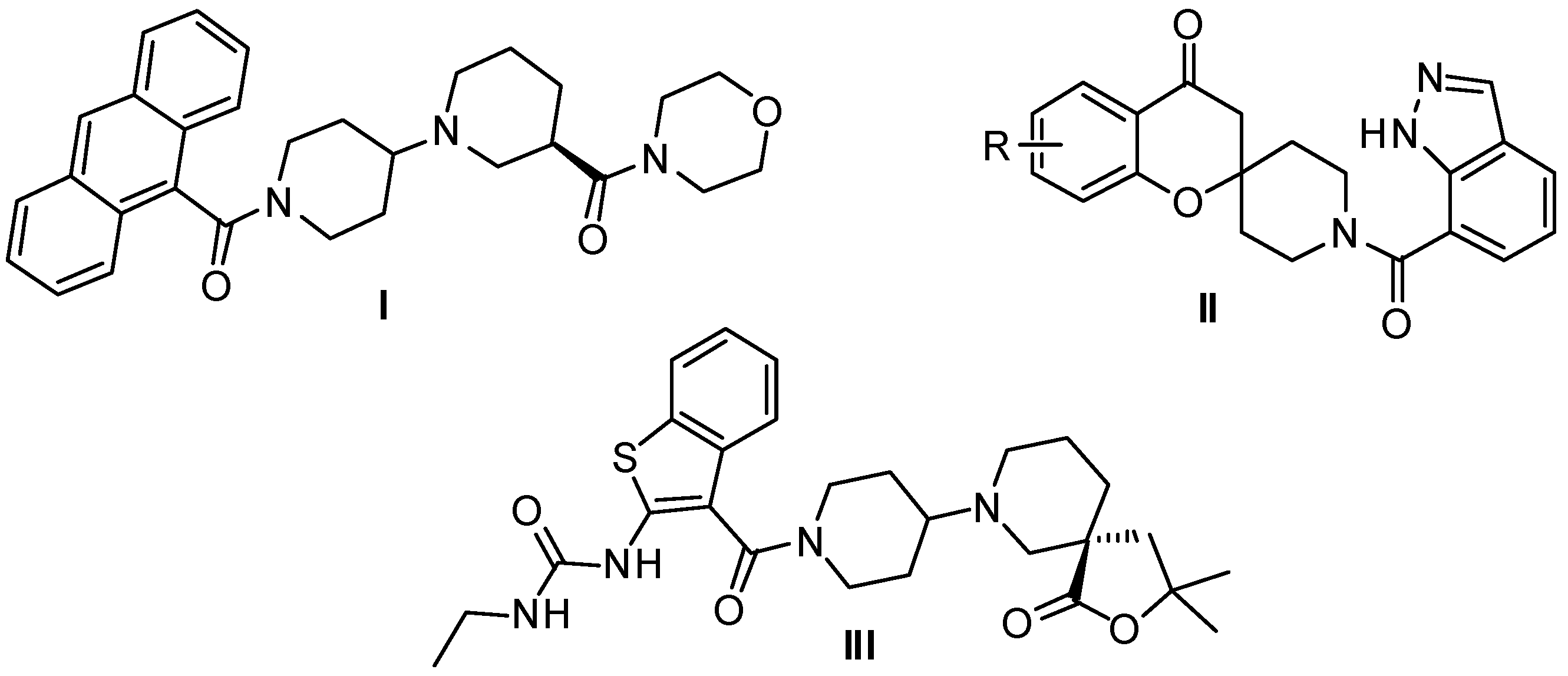


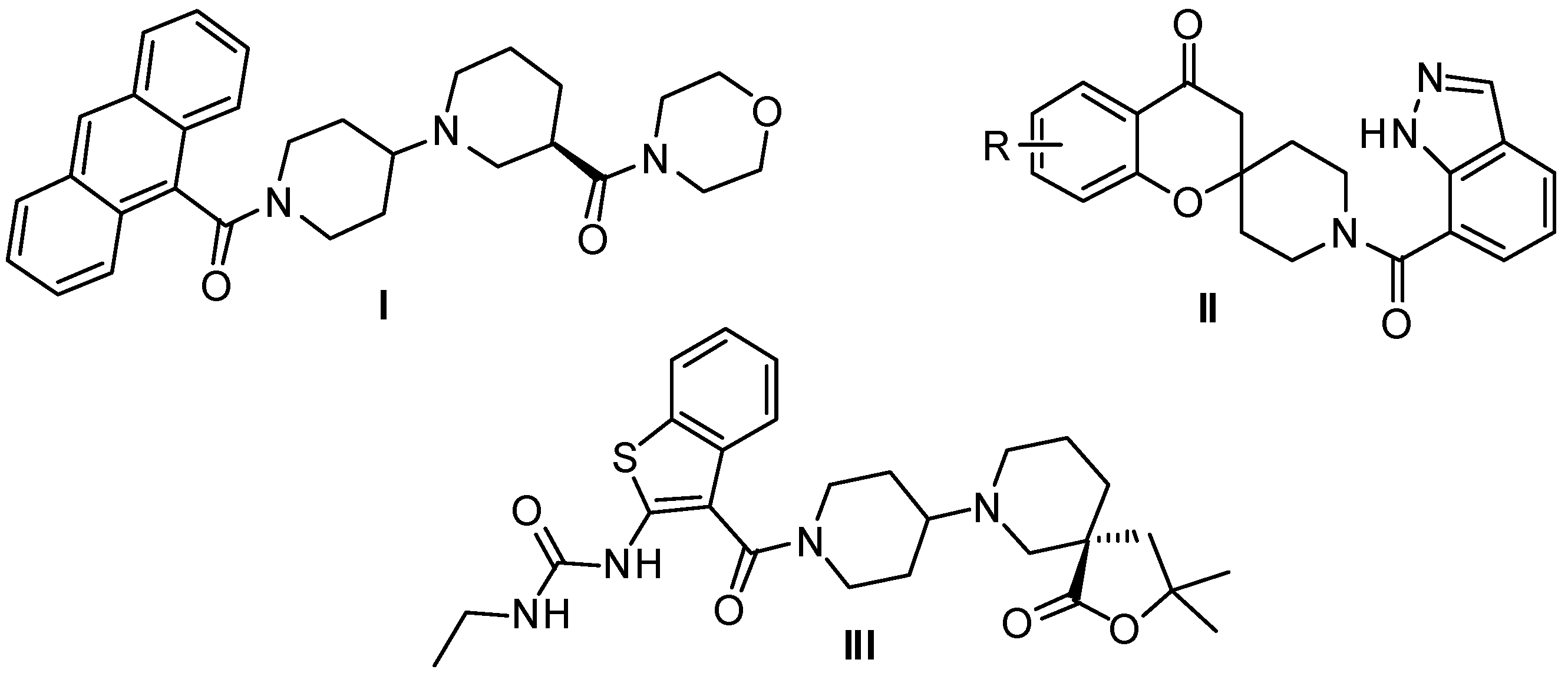
2. Results and Discussion
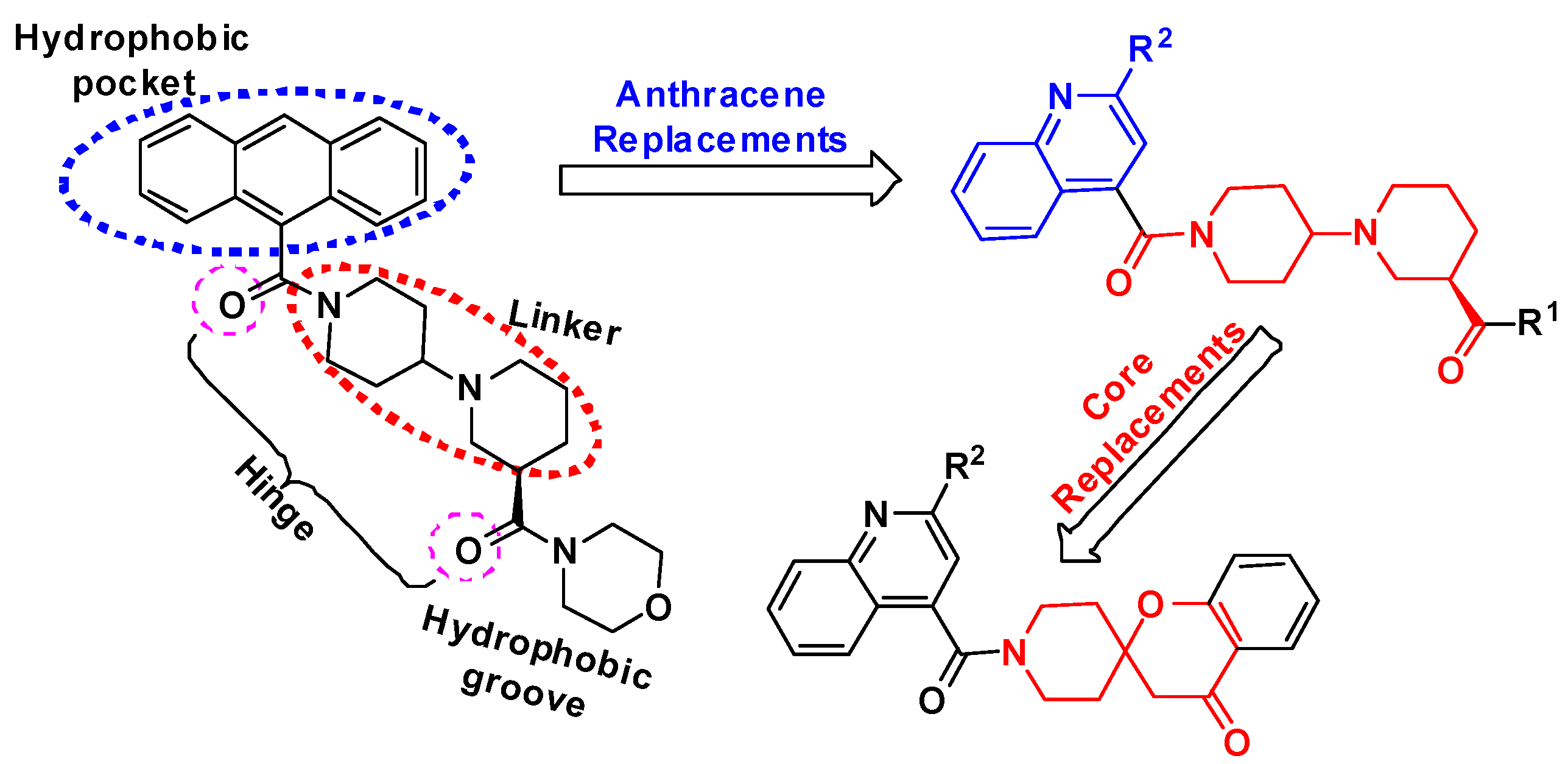
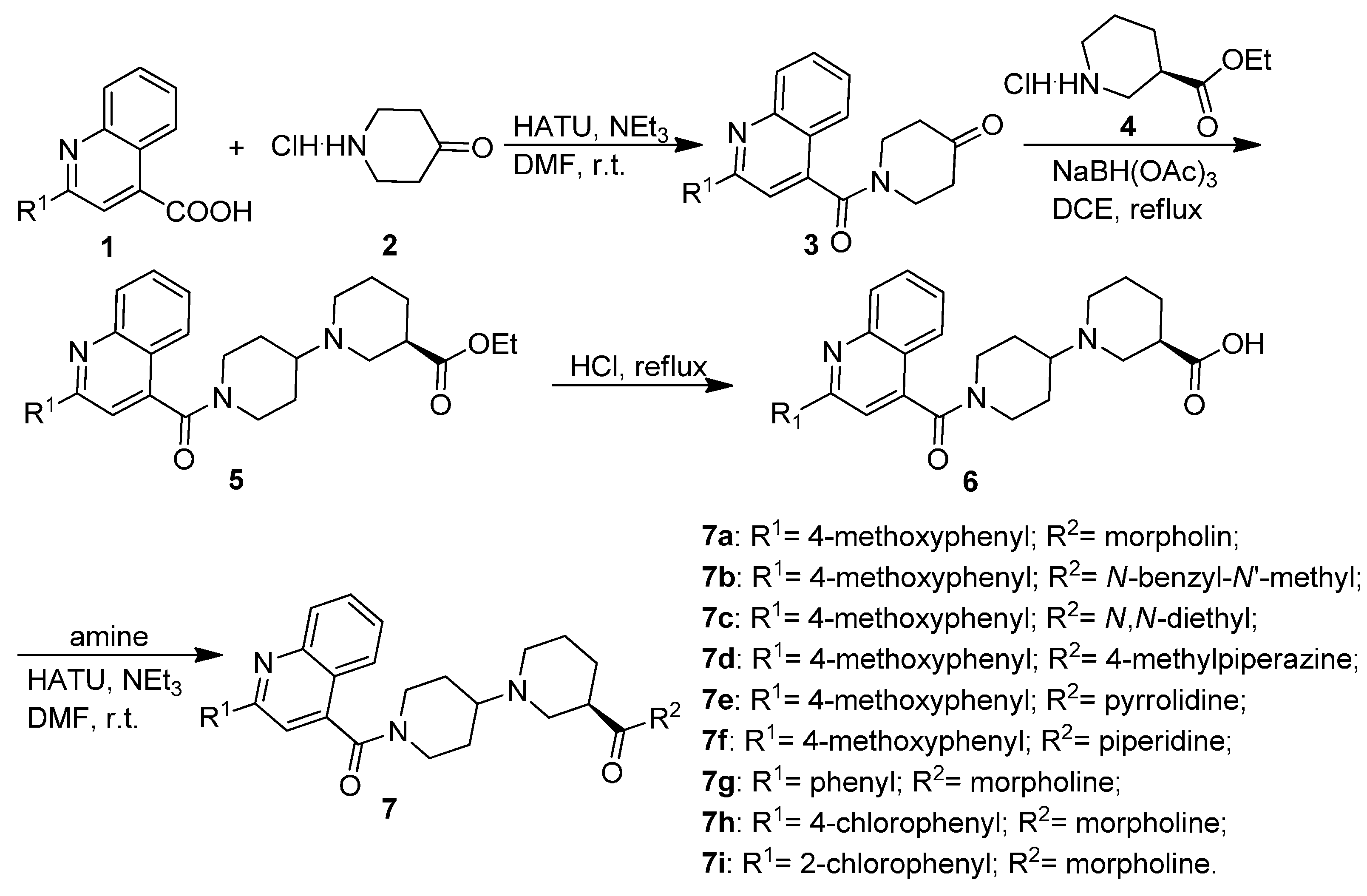
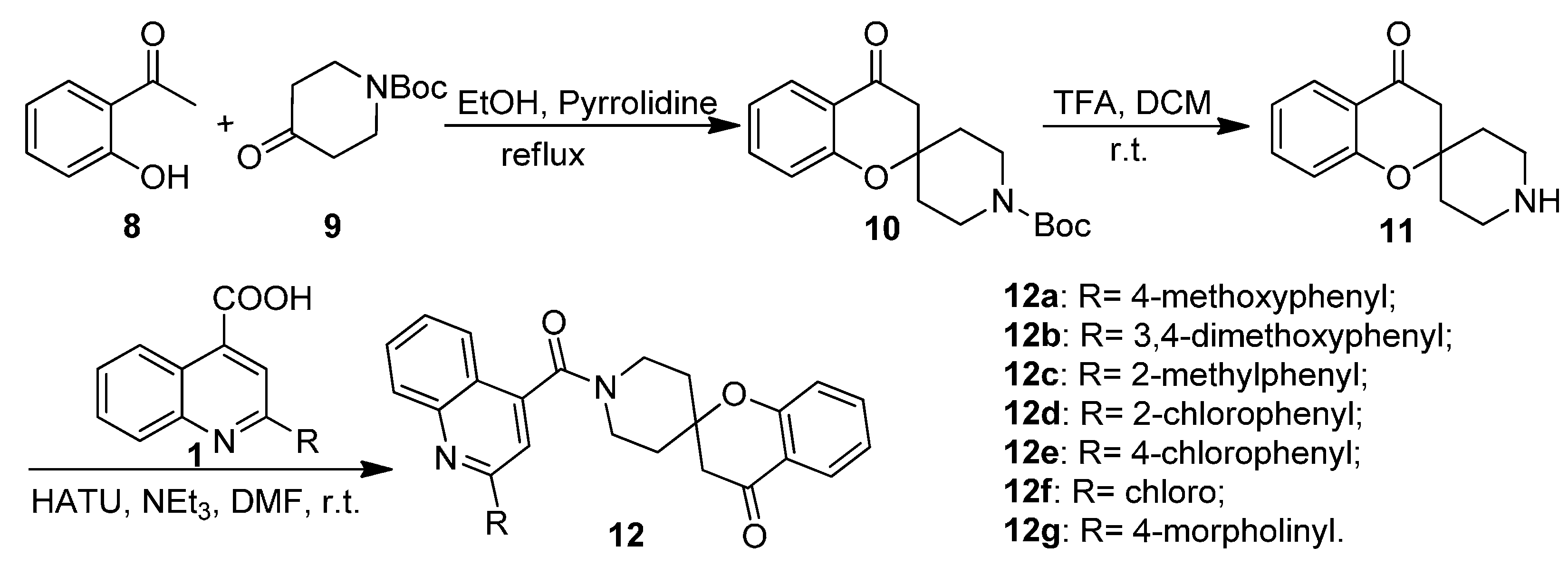
2.1. Chemistry


2.2. Biological Activities

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | ACC1 IC50 (nM) a | ACC2 IC50 (nM) a | TC50 (μM) a,b | LogP d | Drug-Likeness Model Score e |
|---|---|---|---|---|---|
| 7a | 189 (±3) | 172 (±7) | >100 | 3.48 | 1.14 |
| 7b | 750 (±9) | 360 (±10) | >100 | 5.03 | 1.61 |
| 7c | 940 (±5) | 205 (±9) | >100 | 4.38 | 1.49 |
| 7d | 620 (±12) | 294 (±3) | >100 | 3.48 | 1.38 |
| 7e | 750 (±11) | 382 (±15) | >100 | 4.03 | 1.4 |
| 7f | >1000 | 920 (±14) | N.T. c | 4.54 | 1.37 |
| 7g | 860 (±3) | 810 (±11) | >100 | 3.42 | 0.85 |
| 7h | >1000 | >1000 | N.T. c | 4.10 | 1.13 |
| 7i | >1000 | >1000 | N.T. c | 4.05 | 0.92 |
| 12a | 600 (±7) | 650 (±12) | >100 | 4.84 | 1.07 |
| 12b | 760 (±11) | 820 (±14) | >100 | 4.43 | 1.05 |
| 12c | >1000 | >1000 | N.T. c | 5.18 | 0.73 |
| 12d | >1000 | 940 | N.T. c | 5.41 | 0.81 |
| 12e | >1000 | >1000 | N.T. c | 5.46 | 1.04 |
| 12f | >1000 | >1000 | N.T. c | 3.91 | 0.65 |
| 12g | >1000 | >1000 | N.T. c | 3.18 | 0.41 |
| CP-640186 | 173 (±4) | 185 (±5) | >100 | 3.59 | 0.27 |
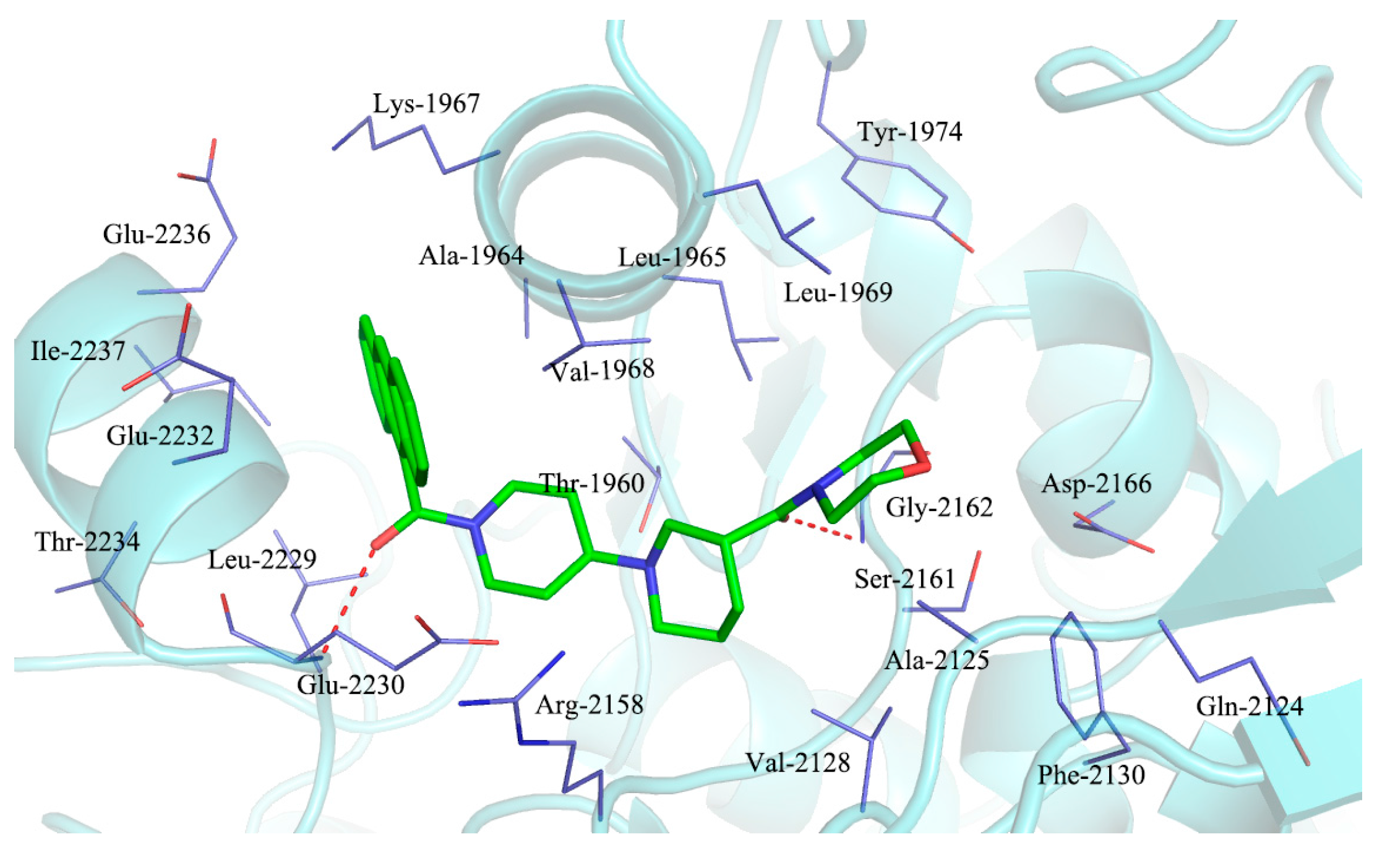
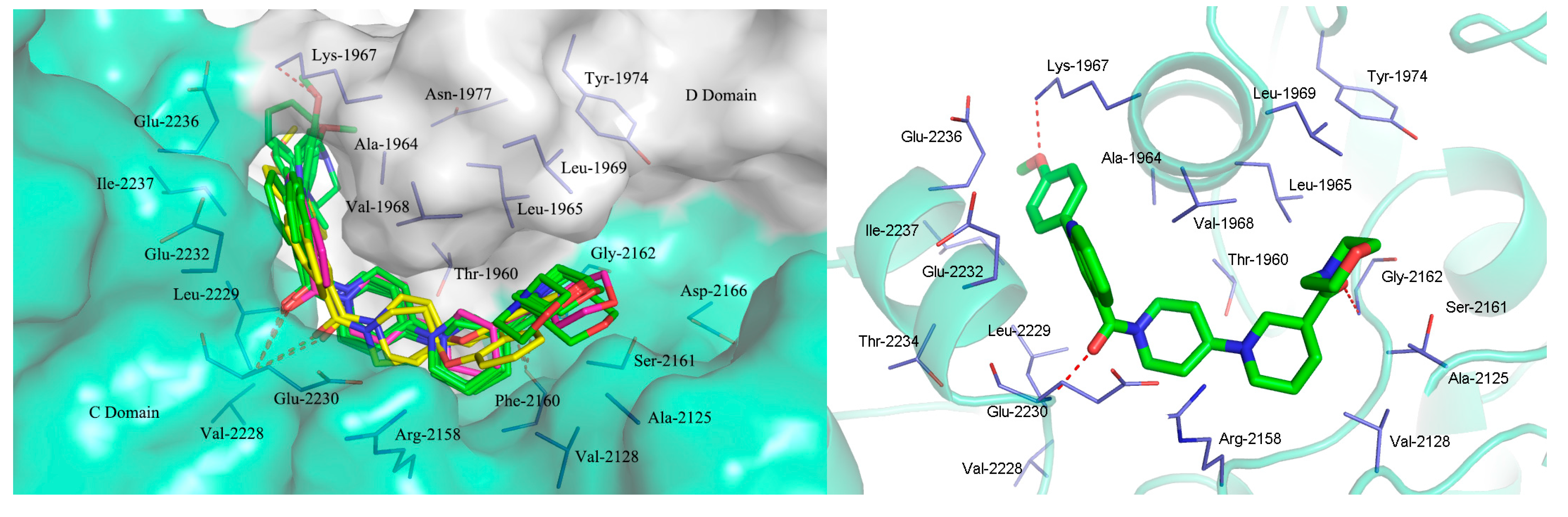
2.3. Molecular Docking Study

3. Experimental Section
3.1. General Information
3.1.1. General Procedure for the Preparation of Substituted 1-(Quinoline-4-carbonyl)piperidin-4-ones 3
3.1.2. General Procedure for the Preparation of Substituted (R)-Ethyl 1′-(Quinoline-4-carbonyl)-[1,4′-bipiperidine]-3-carboxylates 5
3.1.3. General Procedure for the Preparation of Substituted (R)-1′-(Quinoline-4-carbonyl)-[1,4′-bipiperidine]-3-carboxamides 7a–7i
3.1.4. General Procedure for the Preparation of Tert-butyl 4-oxospiro[chroman-2,4' piperidine]-1'-carboxylate (10)
3.1.5. General Procedure for the Preparation of Substituted 1′-(Quinoline-4-carbonyl)spiro[chroman-2,4′-piperidin]-4-ones 12a–12g
3.2. Molecular Docking
3.3. ACC1 and ACC2 in Vitro Assay
3.4. In Vitro Cytotoxic Activities
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tsai, S.; Clemente-Casares, X.; Revelo, X.S.; Winer, S.; Winer, D.A. Are obesity-related insulin resistance and type 2 diabetes autoimmune Diseases? Diabetes 2015, 64, 1886–1897. [Google Scholar] [CrossRef] [PubMed]
- Abu-Elheiga, L.; Wu, H.; Gu, Z.; Bressler, R.; Wakil, S.J. Acetyl-CoA carboxylase 2−/− mutant mice are protected against fatty liver under high-fat, high-carbohydrate dietary and de novo lipogenic conditions. J. Biol. Chem. 2012, 287, 12578–12588. [Google Scholar] [CrossRef] [PubMed]
- Zu, X.Y.; Zhong, J.; Luo, D.X.; Tan, J.J.; Zhang, Q.H.; Wu, Y.; Liu, J.H.; Cao, R.X.; Wen, G.B.; Cao, D.L. Chemical genetics of acetyl-CoA carboxylases. Molecules 2013, 18, 1704–1719. [Google Scholar]
- Lee, J.; Walsh, M.C.; Hoehn, K.L.; James, D.E.; Wherry, E.J.; Choi, Y. Regulator of fatty acid metabolism, acetyl Coenzyme A carboxylase 1, controls T cell. J. Immunol. 2014, 192, 3190–3199. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Hudson, R.; Sintim, H.O. Inhibitors of fatty acid synthesis in prokaryotes and eukaryotes as anti-infective, anticancer and anti-obesity drugs. Future Med. Chem. 2012, 4, 1113–1151. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Oda, Z.; Hara, Y.; Fujinama, K.; Okawa, M.; Ohbuchi, K.; Yonemoto, M.; Ikeda, Y.; Ohwaki, K.; Aragane, K.; et al. Hepatic de novo lipogenesis is present in liver-specific ACC1-deficient mice. Mol. Cell. Biol. 2007, 27, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Freeman-Cook, K.D.; Amor, P.; Bader, S.; Buzon, L.M.; Coffey, S.B.; Corbett, J.W.; Dirico, K.J.; Doran, S.D.; Elliott, R.L.; Esler, W.P.; et al. Maximizing lipophilic efficiency: The use of free-wilson analysis in the design of inhibitors of Acetyl-CoA carboxylase. J. Med. Chem. 2012, 55, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Harwood, H.J.; Petras, S.F.; Shelly, L.D.; Zaccaro, L.M.; Perry, D.A.; Makowski, M.R.; Hargrove, D.M.; Martin, K.A.; Tracey, W.R.; Chapman, J.G.; et al. Isozyme-nonselective N-substituted bipiperidylcarboxamide acetyl-CoA carboxylase inhibitors reduce tissue malonyl-CoA concentrations, inhibit fatty acid synthesis and increase fatty acid oxidation in cultured cells and in experimental animals. J. Biol. Chem. 2003, 278, 37099–37111. [Google Scholar] [CrossRef] [PubMed]
- Kamata, M.; Yamashita, T.; Kina, A.; Funata, M.; Mizukami, A.; Sasaki, M.; Tani, A.; Funami, M.; Amano, N.; Fukatsu, K. Design, synthesis, and structure-activity relationships of novel spiro-piperidines as acetyl-CoA carboxylase inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 3643–3647. [Google Scholar] [CrossRef] [PubMed]
- Bourbeau, M.P.; Bartberger, M.D. Recent advances in the development of acetyl-CoA carboxylase (ACC) inhibitors for the treatment of metabolic disease. J. Med. Chem. 2015, 58, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Pfizer Pipeline. As of 8 May 2014. Available online: http://www.pfizer.com/sites/default/files/product-pipeline/May%208%2C%202014%20Pipeline%20Update_Final_to%20BT.pdf (accessed on 10 August 2015).
- Zhang, H.; Tweel, B.; Li, J.; Tong, L. Crystal structure of the carboxyltransferase domain of acetyl-coenzyme A carboxylase in complex with CP-640186. Structure 2004, 12, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.W.; Freeman-Cook, K.D.; Elliott, R.; Vajdos, F.; Rajamohan, F.; Kohls, D.; Marr, E.; Zhang, H.L.; Tong, L.; Tu, M.H.; et al. Discovery of small molecule isozyme non-specific inhibitors of mammalian acetyl-CoA carboxylase 1 and 2. Bioorg. Med. Chem. Lett. 2010, 20, 2383–2388. [Google Scholar] [CrossRef] [PubMed]
- Madauss, K.P.; Burkhart, W.A.; Consler, T.G.; Cowan, D.J.; Gottschalk, W.K.; Miller, A.B.; Short, S.A.; Tran, T.B.; Williams, S.P. The human ACC2 CT-domain C-terminus is required for full functionality and has a novel twist. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Park, E.H.; Kim, H.S.; Eom, S.J.; Kim, K.T.; Paik, H.D. Antioxidative and Anticanceric Activities of Magnolia (Magnolia denudata) Flower Petal Extract Fermented by Pediococcus acidilactici KCCM 11614. Molecules 2015, 20, 12154–12165. [Google Scholar] [CrossRef] [PubMed]
- Brylinski, M.; Waldrop, G.L. Computational redesign of bacterial biotin carboxylase inhibitors using structure-based virtual screening of combinatorial libraries. Molecules 2014, 19, 4021–4045. [Google Scholar] [CrossRef] [PubMed]
- Molinspiration Cheminformatics. Available online: http://www.molinspiration.com (accessed on 10 August 2015).
- Drug-Likeness and Molecular Property Prediction. Available online: http://www.molsoft.com/mprop/ (accessed on 10 August 2015).
- Zhang, H.L.; Yang, Z.; Shen, Y. Crystal structure of the carboxyltransferase domain of acetyl-coenzyme A carboxylase. Science 2003, 299, 2064–2067. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.P.; Kim, Y.S.; Tong, L. Mechanism for the inhibition of the carboxyltransferase domain of acetyl-coenzyme A carboxylase by pinoxaden. Proc. Natl. Acad. Sci. USA 2010, 107, 22072–22077. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.T.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. Dock 6: Combining technique to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.K.; Lin, J.; Chen, Y.; Zhong, W.; Zhao, G.M.; Liu, H.Y.; Li, S.; Wang, L.L.; Li, S. Novel fatty acid synthase (FAS) inhibitors: Design, synthesis, biological evaluation, and molecular docking studies. Bioorg. Med. Chem. 2009, 17, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 7a–7i and 12a–12g are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, T.; Sun, J.; Wang, Q.; Gao, J.; Liu, Y. Synthesis, Biological Evaluation and Molecular Docking Studies of Piperidinylpiperidines and Spirochromanones Possessing Quinoline Moieties as Acetyl-CoA Carboxylase Inhibitors. Molecules 2015, 20, 16221-16234. https://doi.org/10.3390/molecules200916221
Huang T, Sun J, Wang Q, Gao J, Liu Y. Synthesis, Biological Evaluation and Molecular Docking Studies of Piperidinylpiperidines and Spirochromanones Possessing Quinoline Moieties as Acetyl-CoA Carboxylase Inhibitors. Molecules. 2015; 20(9):16221-16234. https://doi.org/10.3390/molecules200916221
Chicago/Turabian StyleHuang, Tonghui, Jie Sun, Qianqian Wang, Jian Gao, and Yi Liu. 2015. "Synthesis, Biological Evaluation and Molecular Docking Studies of Piperidinylpiperidines and Spirochromanones Possessing Quinoline Moieties as Acetyl-CoA Carboxylase Inhibitors" Molecules 20, no. 9: 16221-16234. https://doi.org/10.3390/molecules200916221