
Stereoselective Formation of Substituted 1,3-Dioxolanes through a Three-Component Assembly during the Oxidation of Alkenes with Hypervalent Iodine(III)

Abstract
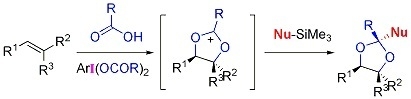
:
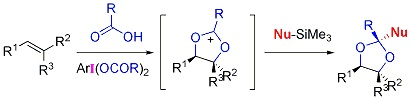
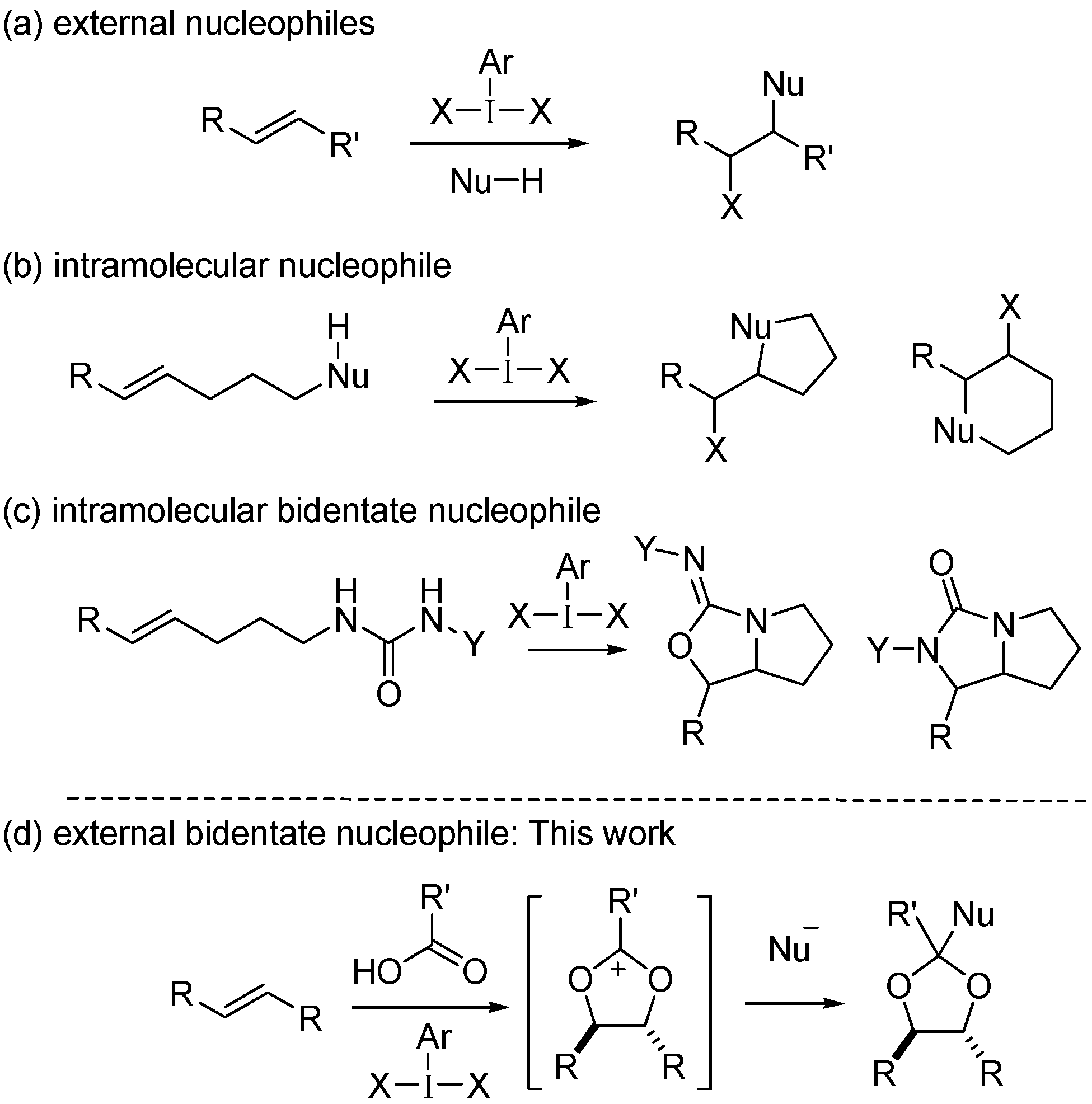
1. Introduction

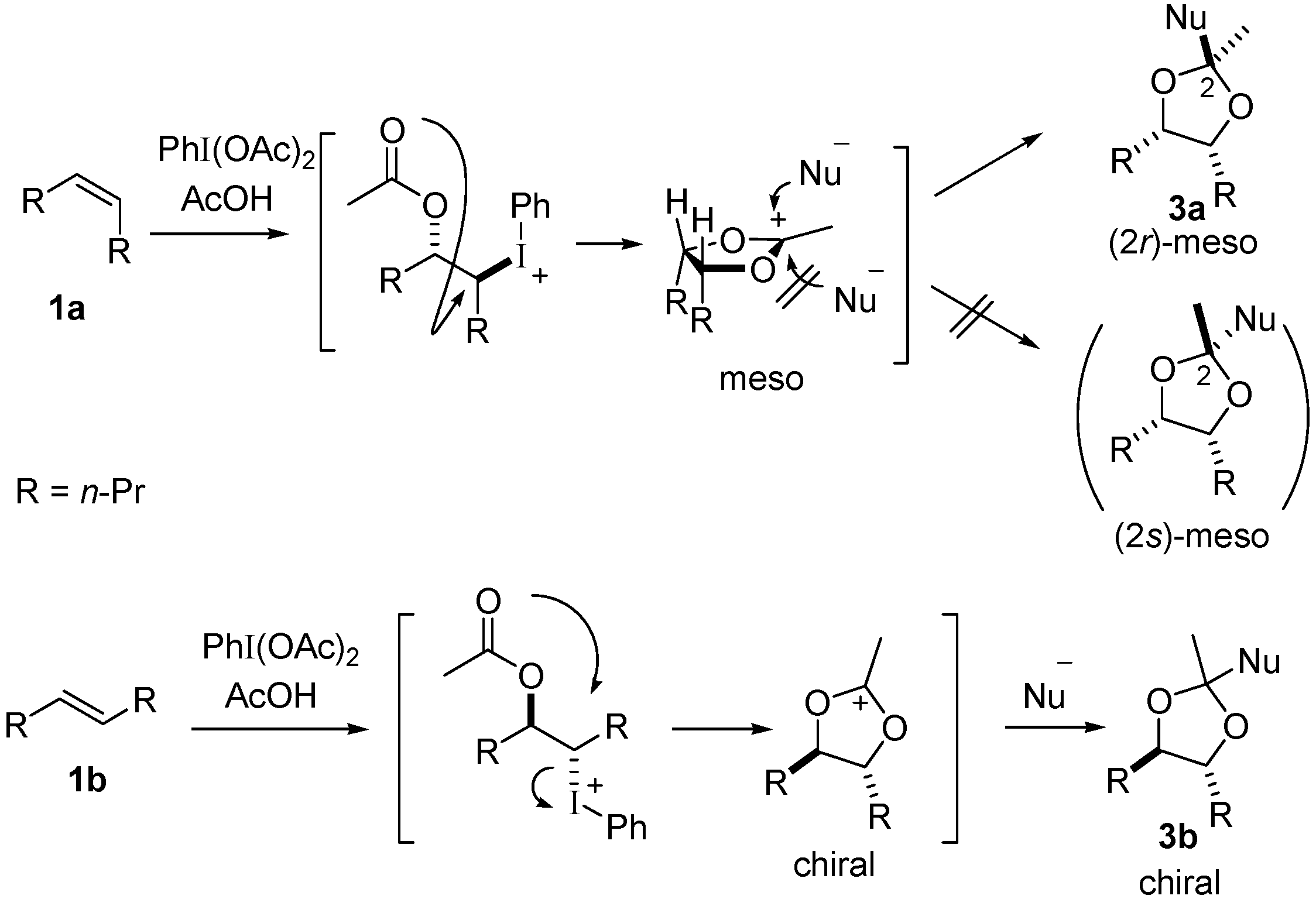
2. Results and Discussion


| Entry | 1 | AcOH (mmol) | 2a (mmol) | Quench Temp. | Yield (%) |
|---|---|---|---|---|---|
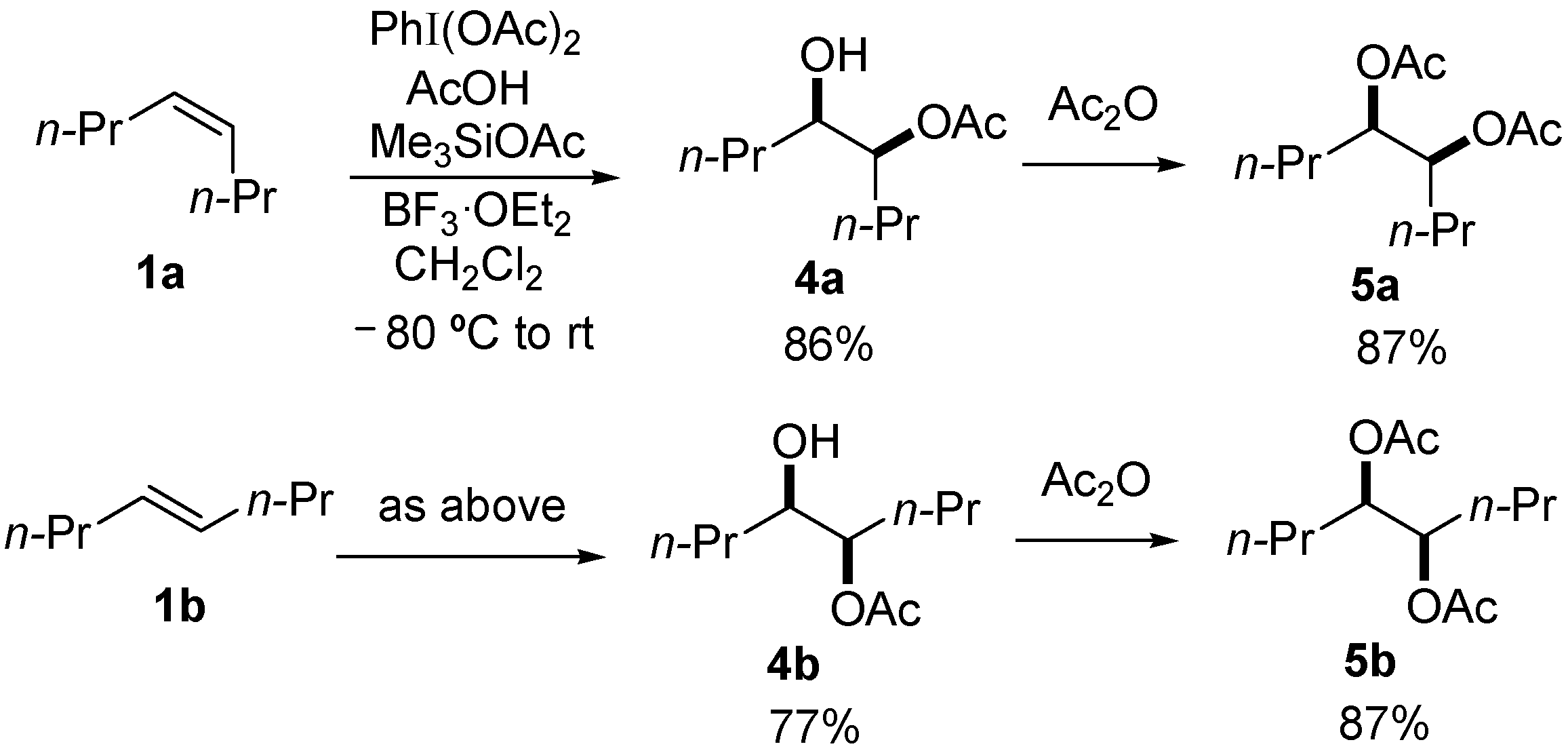
| 1 | 1a | 1.7 | 0.5 | rt | 3a, 2; 4a, 58 |
| 2 | 1a | 0.5 | 1.5 | rt | 3a, 46 |
| 3 | 1a | 0.5 | 1.5 | −30 °C | 3a, 62 |
| 4 | 1b | 0.5 | 1.5 | −30 °C | 3b, 48 |



| Entry | 1 | Nu-SiMe3 | Product | Yield (%) |
|---|---|---|---|---|
| 1 b |  |  |  | 15 (1.2:1) c |
| 2 | 56 d | |||
| 3 b |  | 2a |  | 25 (1:1.2) c |
| 4 | 40 d | |||
| 5 |  | 2a |  | 52 d |
| 6 |  | 2a |  | 46 d |
| 7 |  |  |  | 39 d |
| 8 |  | 2b |  | 37 |
| 9 | 1c | 2b |  | 29 d |
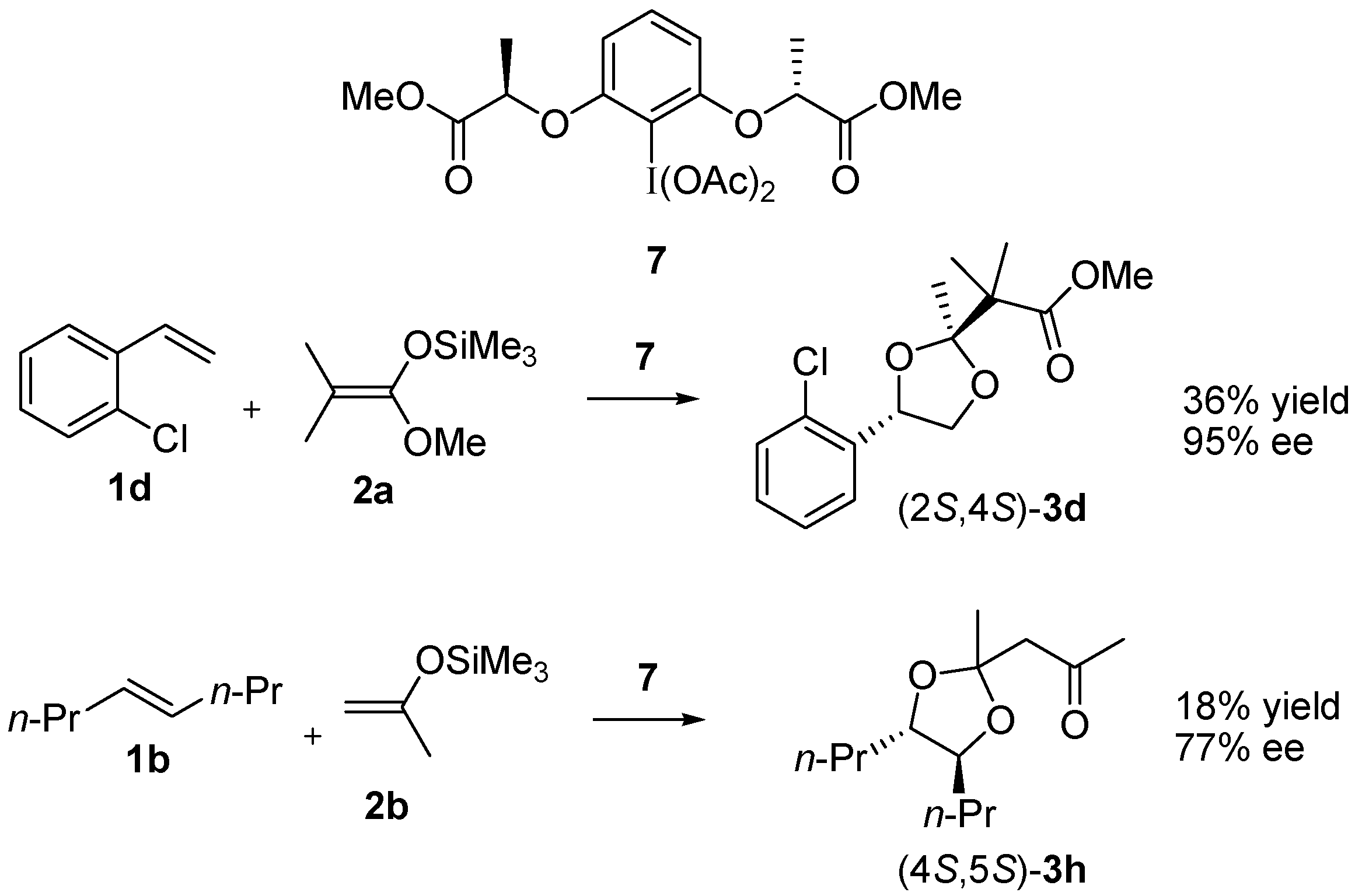
| 10 | 1d | 2b |  | 25 (8:1) c |
| 11 | 1a |  |  | 34 d |
| 12 | 1b | 2c |  | 21 |
| 13 | 1d | 2c |  | 29 (7:1) c |
| 14 | 1b |  |  | 27 |
 ); d Diastereomer was not detected by 1H-NMR.
); d Diastereomer was not detected by 1H-NMR.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R | Yield (%) |
|---|---|---|
| 1 | Et | 3o, 41 |
| 2 | i-Pr | 3p, 35 |
| 3 | t-Bu | b |
| 4 | Ph | 3q, 26 |


3. Experimental Section
3.1. General Information
3.2. Typical Procedure for the Three-Component Assembly Reaction
3.3. Isomerization of 3d Mediated by a Lewis Acid
3.4. Enantioselective Reaction of 1d with 7 and 2a
3.5. Enantioselective Reaction of 1b with 7 and 2b
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Varvoglis, A. The Organic Chemistry of Polycoordinated Iodine; VCH: New York, NY, USA, 1992. [Google Scholar]
- Varvoglis, A. Hypervalent Iodine in Organic Synthesis; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Wirth, T. Hypervalent Iodine Chemistry; Springer: Berlin, Germany, 2003. [Google Scholar]
- Zhdankin, V.V. Hypervalent Iodine Chemistry; John Wiley & Sons: Chichester, UK, 2014. [Google Scholar]
- Moriarty, R.M. Organohypervalent iodine: Development, applications, and future directions. J. Org. Chem. 2005, 70, 2893–2903. [Google Scholar] [CrossRef] [PubMed]
- Wirth, T. Hypervalent iodine chemistry in synthesis: Scope and new directions. Angew. Chem. Int. Ed. 2005, 44, 3656–3665. [Google Scholar] [CrossRef] [PubMed]
- Zhdankin, V.V.; Stang, P.J. Chemistry of polyvalent iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, M.; Miyamoto, K. Catalytic version of and reuse in hypervalent organo-λ3- and -λ5-iodane oxidation. Eur. J. Org. Chem. 2008, 4229–4239. [Google Scholar] [CrossRef]
- Ngatimin, M.; Lupton, D.W. The discovery of catalytic enantioselective polyvalent iodine mediated reactions. Aust. J. Chem. 2010, 63, 653–658. [Google Scholar] [CrossRef]
- Liang, H.; Ciufolini, M.A. Chiral hypervalent iodine reagents in asymmetric reactions. Angew. Chem. Int. Ed. 2011, 50, 11849–11851. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M.; Ishihara, K. Conformationally-flexible chiral hypervalent organoiodine catalysts for enantioselective oxidative transformations. J. Synth. Org. Chem. Jpn. 2012, 70, 1116–1122. [Google Scholar] [CrossRef]
- Rawling, M.J.; Tomkinson, N.C.O. Metal-free syn-dioxygenation of alkenes. Org. Biomol. Chem. 2013, 11, 1434–1440. [Google Scholar] [CrossRef] [PubMed]
- Parra, A.; Reboredo, S. Chiral hypervalent iodine reagents: Synthesis and reactivity. Chem. Eur. J. 2013, 19, 17244–17260. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Farid, U.; Wirth, T. Hypervalent iodine reagents as powerful electrophiles. Synlett 2013, 24, 424–431. [Google Scholar]
- Dohi, T.; Kita, Y. New site-selective organoradical based on hypervalent iodine reagent for controlled alkane sp3 C–H oxidations. ChemCatChem 2014, 6, 76–78. [Google Scholar] [CrossRef]
- Singh, F.V.; Wirth, T. Hypervalent iodine-catalyzed oxidative functionalizations including stereoselective reactions. Chem. Asian J. 2014, 9, 950–971. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.M.; Wöste, T.H.; Muñiz, K. Vicinal difunctionalization of alkenes with iodine(III) reagents and catalysts. Chem. Asian J. 2014, 9, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Harned, A.M. Asymmetric oxidative dearomatizations promoted by hypervalent iodine(III) reagents: An opportunity for rational catalyst design? Tetrahedron Lett. 2014, 55, 4681–4689. [Google Scholar] [CrossRef] [PubMed]
- Berthiol, F. Reagent and catalyst design for asymmetric hypervalent iodine oxidations. Synthesis 2015, 47, 587–603. [Google Scholar] [CrossRef]
- Kang, Y.-B.; Gade, L.H. The nature of the catalytically active species in olefin dioxygenation with PhI(OAc)2: Metal or proton? J. Am. Chem. Soc. 2011, 133, 3658–3667. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Yang, J.; Meng, X.; Li, Z. BF3·OEt2-Promoted diastereoselective diacetoxylation of alkenes by PhI(OAc)2. J. Org. Chem. 2011, 76, 9997–10004. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Studer, A. Transition-metal-free trifluoromethylaminoxylation of alkenes. Angew. Chem. Int. Ed. 2012, 51, 8221–8224. [Google Scholar] [CrossRef] [PubMed]
- Nocquet-Thibault, S.; Retailleau, P.; Cariou, K.; Dodd, R.H. Iodine(III)-mediated umpolung of bromide salts for the ethoxybromination of enamides. Org. Lett. 2013, 15, 1842–1845. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Yang, Y.; Zhang, P.; Du, Z.; Wang, C. Alkene oxyalkylation enabled by merging rhenium catalysis with hypervalent iodine(III) reagents via decarboxylation. J. Am. Chem. Soc. 2013, 135, 18048–18051. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-T.; Li, L.-H.; Yang, Y.-F.; Zhou, Z.-Z.; Hua, H.-L.; Liu, X.-Y.; Liang, Y.-M. Copper-catalyzed intermolecular cyanotrifluoromethylation of alkenes. Org. Lett. 2014, 16, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Danneman, M.W.; Hong, K.B.; Johnston, J.N. Oxidative inter-/intermolecular alkene diamination of hydroxy styrenes with electron-rich amines. Org. Lett. 2015, 17, 2558–2561. [Google Scholar] [CrossRef] [PubMed]
- Lovick, H.M.; Michael, F.E. Metal-free highly regioselective aminotrifluoroacetoxylation of alkenes. J. Am. Chem. Soc. 2010, 132, 1249–1251. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, D.J.; Bowen, E.G.; Forslund, R.E.; Sussman, A.D.; Weerasekera, S.L. Intramolecular oxamidation of unsaturated O-alkyl hydroxamates: A remarkably versatile entry to hydroxy lactams. J. Am. Chem. Soc. 2010, 132, 1188–1189. [Google Scholar] [CrossRef] [PubMed]
- Karila, D.; Leman, L.; Dodd, R.H. Copper-catalyzed iminoiodane-mediated aminolactonization of olefins: Application to the synthesis of 5,5-disubstituted butyrolactones. Org. Lett. 2011, 13, 5830–5833. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.-L.; Piou, T.; Dufour, J.; Neuville, L.; Zhu, J. Iodo-carbocyclization of electron-deficient alkenes: Synthesis of oxindoles and spirooxindoles. Org. Lett. 2011, 13, 2244–2247. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Buchwald, S.L. Copper-catalyzed oxytrifluoromethylation of unactivated alkenes. J. Am. Chem. Soc. 2012, 134, 12462–12465. [Google Scholar] [CrossRef] [PubMed]
- Tu, D.; Ma, L.; Tong, X.; Deng, X.; Xia, C. Synthesis of pyrrolo[2,3-b]indole via iodine(III)-mediated intramolecular annulation. Org. Lett. 2012, 14, 4830–4833. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, X.; Ren, C.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Synthesis of oxazoles from enamides via phenyliodine diacetate-mediated intramolecular oxidative cyclization. J. Org. Chem. 2012, 77, 10353–10361. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Mori, K.; Shimogaki, M.; Sugimura, T. Asymmetric synthesis of 4,8-dihydroxyisochroman-1-one polyketide metabolites using chiral hypervalent iodine(III). Org. Lett. 2012, 14, 1294–1297. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Buchwald, S.L. Enantioselective functionalization of radical intermediates in redox catalysis: Copper-catalyzed asymmetric oxytrifluoromethylation of alkenes. Angew. Chem. Int. Ed. 2013, 52, 12655–12658. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Mori, K.; Shimogaki, M.; Sugimura, T. Total synthesis of (12R)- and (12S)-12-hydroxymonocerins: Stereoselective oxylactonization using a chiral hypervalent iodine(III) species. RSC Adv. 2013, 3, 17717–17725. [Google Scholar] [CrossRef]
- Li, L.; Deng, M.; Zheng, S.-C.; Xiong, Y.-P.; Tan, B.; Liu, X.-Y. Metal-free direct intramolecular carbotrifluoromethylation of alkenes to functionalized trifluoromethyl azaheterocycles. Org. Lett. 2014, 16, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Kaga, A.; Chiba, S. Diastereoselective aminooxygenation and diamination of alkenes with amidines by hypervalent iodine(III) reagents. Org. Lett. 2014, 16, 6136–6139. [Google Scholar] [CrossRef] [PubMed]
- Takesue, T.; Fujita, M.; Sugimura, T.; Akutsu, H. A series of two oxidation reactions of ortho-alkenylbenzamide with hypervalent iodine(III): A concise entry into (3R,4R)-4-hydroxymellein and (3R,4R)-4-hydroxy-6-methoxymellein. Org. Lett. 2014, 16, 4634–4637. [Google Scholar] [CrossRef] [PubMed]
- Alhalib, A.; Kamouka, S.; Moran, W.J. Iodoarene-catalyzed cyclizations of unsaturated amides. Org. Lett. 2015, 17, 1453–1456. [Google Scholar] [CrossRef] [PubMed]
- Steuff, J.; Hövelmann, C.H.; Nieger, M.; Muñiz, K. Palladium(II)-catalyzed intramolecular diamination of unfunctionalized alkenes. J. Am. Chem. Soc. 2005, 127, 14586–14587. [Google Scholar] [CrossRef] [PubMed]
- Muñiz, K.; Hövelmann, C.H.; Steuff, J. Oxidative diamination of alkenes with ureas as nitrogen sources: Mechanistic pathways in the presence of a high oxidation state palladium catalyst. J. Am. Chem. Soc. 2008, 130, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Cochran, B.M.; Michael, F.E. Metal-free oxidative cyclization of urea-tethered alkenes with hypervalent iodine. Org. Lett. 2008, 10, 5039–5042. [Google Scholar] [CrossRef] [PubMed]
- Mizar, P.; Laverny, A.; El-Sherbini, M.; Farid, U.; Brown, M.; Malmedy, F.; Wirth, T. Enantioselective diamination with novel chiral hypervalent iodine catalysts. Chem. Eur. J. 2014, 20, 9910–9913. [Google Scholar] [CrossRef] [PubMed]
- Anumandla, D.; Littlefield, R.; Jeffrey, C.S. Oxidative 1,4-diamination of dienes using simple urea derivatives. Org. Lett. 2014, 16, 5112–5115. [Google Scholar] [CrossRef] [PubMed]
- Karade, N.N.; Shirodkar, S.G.; Patil, M.N.; Potrelar, R.A.; Larade, H.N. Diacetoxyiodobenzene-mediated oxidative addition of 1,3-dicarbonyl compounds to olefins: An efficient one-pot synthesis of 2,3-dihydrofuran derivatives. Tetrahedron Lett. 2003, 44, 6729–6731. [Google Scholar] [CrossRef]
- Yoshimura, A.; Middleton, K.R.; Todora, A.D.; Kastern, B.J.; Koski, S.R.; Maskaev, A.V.; Zhdankin, V.V. Hypervalent iodine catalyzed generation of nitrile oxides from oximes and their cycloaddition with alkenes or alkynes. Org. Lett. 2013, 15, 4010–4013. [Google Scholar] [CrossRef] [PubMed]
- Pittman, C.U., Jr.; McManus, S.P.; Larsen, J.W. 1,3-Dioxolan-2-ylium and related heterocyclic cations. Chem. Rev. 1972, 72, 357–438. [Google Scholar] [CrossRef]
- Lorenz, W.; Maas, G. O-Acylation of α-diazo ketones. A novel route to alkenediazonium and 1,3-dioxolium salts. J. Org. Chem. 1987, 52, 375–381. [Google Scholar]
- Prévost, C. Sur un complexe iodo-argento-benzoïque et son application à l’oxydation des combinaisons éthyléniques en α-glycols. Comptes Rendus 1933, 196, 1129–1131. [Google Scholar]
- Woodward, R.B.; Brutcher, F.V., Jr. cis-Hydroxylation of a synthetic steroid intermediate with iodine, silver acetate and wet acetic acid. J. Am. Chem. Soc. 1958, 80, 209–211. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Tykwinski, R.; Berglund, B.; Mullikin, M.; Caple, R.; Zefirov, N.S.; Koz’min, A.S. Iodosobenzene tetrafluoroborate, hexafluoroantimonate, and hexafluorophosphate: Stable electrophilic hypervalent iodine reagents without nucleophilic ligands. J. Org. Chem. 1989, 54, 2609–2612. [Google Scholar] [CrossRef]
- Ochiai, M.; Miyamoto, K.; Shiro, M.; Ozawa, T.; Yamaguchi, K. Isolation, characterization, and reaction of activated iodosylbenzene monomer hydroxy(phenyl)iodonium ion with hypervalent bonding: Supramolecular complex PhI+OH·18-crown-6 with secondary I···O interactions. J. Am. Chem. Soc. 2003, 125, 13006–13007. [Google Scholar] [CrossRef] [PubMed]
- Emmanuvel, L.; Shaikh, T.M.A.; Sudalai, A. NaIO4/LiBr-mediated diastereoselective dihydroxylation of olefins: A catalytic approach to the Prevost−Woodward reaction. Org. Lett. 2005, 7, 5071–5074. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, D.; Dong, V.M. Palladium-catalyzed olefin dioxygenation. J. Am. Chem. Soc. 2008, 130, 2962–2964. [Google Scholar] [CrossRef] [PubMed]
- Seayad, J.; Seayad, A.M.; Chai, C.L.L. Copper-catalyzed diacetoxylation of olefins using PhI(OAc)2 as oxidant. Org. Lett. 2010, 12, 1412–1415. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Wakita, M.; Sugimura, T. Enantioselective Prévost and Woodward reactions using chiral hypervalent iodine(III): Switchover of stereochemical course of an optically active 1,3-dioxolan-2-yl cation. Chem. Commun. 2011, 47, 3983–3985. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Liu, S.; Yang, J.; Meng, X.; Li, Z. Metal-free, organocatalytic syn diacetoxylation of alkenes. Org. Lett. 2012, 14, 3336–3339. [Google Scholar] [CrossRef] [PubMed]
- Mukaiyama, T.; Hayashi, Y.; Hashimoto, Y. Regioselective alkylation of 1,3-dioxolan-2-ylium cation derived from α,β-unsaturated aldehyde ethylene acetal with lithium organo compounds. Chem. Lett. 1986, 1627–1630. [Google Scholar] [CrossRef]
- Hayashi, Y.; Wariishi, K.; Mukaiyama, T. Oxidative carbon-carbon bond forming reaction via a 1,3-dioxolan-2-ylium cation. Chem. Lett. 1987, 1243–1246. [Google Scholar] [CrossRef]
- An enamine nucleophile, 4-(1-phenylvinyl)morpholine was used instead of 2d in the reaction of 1b. However, no 3n was observed.
- Fujita, M.; Okuno, S.; Lee, H.J.; Sugimura, T.; Okuyama, T. Enantiodifferentiating tetrahydrofuranylation of but-3-enyl carboxylates using optically active hypervalent iodine(III) reagents via a 1,3-dioxan-2-yl cation intermediate. Tetrahedron Lett. 2007, 48, 8691–8694. [Google Scholar] [CrossRef]
- Uyanik, M.; Yasui, T.; Ishihara, K. Enantioselective Kita oxidative spirolactonization catalyzed by in situ generated chiral hypervalent iodine(III) species. Angew. Chem. Int. Ed. 2010, 49, 2175–2177. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Yoshida, Y.; Miyata, K.; Wakisaka, A.; Sugimura, T. Enantiodifferentiating endo-selective oxylactonization of ortho-alk-1-enylbenzoate with a lactate-derived aryl-λ3-iodane. Angew. Chem. Int. Ed. 2010, 49, 7068–7071. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M.; Yasui, T.; Ishihara, K. Chiral hypervalent iodine-catalyzed enantioselective oxidative Kita spirolactonization of 1-naphthol derivatives and one-pot diastereo-selective oxidation to epoxyspirolactones. Tetrahedron 2010, 66, 5841–5851. [Google Scholar] [CrossRef]
- Röben, C.; Souto, J.A.; González, Y.; Lishchynskyi, A.; Muñiz, K. Enantioselective metal-free diamination of styrenes. Angew. Chem. Int. Ed. 2011, 50, 9478–9482. [Google Scholar] [CrossRef] [PubMed]
- Farid, U.; Wirth, T. Highly stereoselective metal-free oxyaminations using chiral hypervalent iodine reagents. Angew. Chem. Int. Ed. 2012, 51, 3462–3465. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Feige, P.; de Haro, T.; Nevado, C. Regio- and enantioselective aminofluorination of alkenes. Angew. Chem. Int. Ed. 2013, 52, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Farid, U.; Malmedy, F.; Claveau, R.; Albers, L.; Wirth, T. Stereoselective rearrangements with chiral hypervalent iodine reagents. Angew. Chem. Int. Ed. 2013, 52, 7018–7022. [Google Scholar] [CrossRef] [PubMed]
- Shimogaki, M.; Fujita, M.; Sugimura, T. Enantioselective oxidation of alkenylbenzoates catalyzed by chiral hypervalent iodine(III) to yield 4-hydroxyisochroman-1-ones. Eur. J. Org. Chem. 2013, 2013, 7128–7138. [Google Scholar] [CrossRef]
- Wu, H.; He, Y.-P.; Xu, L.; Zhang, D.-Y.; Gong, L.-Z. Asymmetric organocatalytic direct C(sp2)–H/C(sp3)–H oxidative cross-coupling by chiral iodine reagents. Angew. Chem. Int. Ed. 2014, 53, 3466–3469. [Google Scholar] [CrossRef] [PubMed]
- Lethbridge, A.; Norman, R.O.C.; Thomas, C.B.; Parr, W.J.E. Oxidation of oct-1-ene and trans-oct-4-ene by lead(IV), thallium(III), and mercury(II) acetates. J. Chem. Soc. Perkin Trans. 1975, 1, 231–241. [Google Scholar] [CrossRef]
- Uemura, S.; Ohe, K.; Fukuzawa, S.-I.; Patil, S.R.; Sugita, N. Dominant cis-diacetoxylation of alkenes with tellurium(IV) oxide and lithium bromide in acetic acid. J. Organomet. Chem. 1986, 316, 67–78. [Google Scholar] [CrossRef]
- Fujioka, H.; Matsunaga, N.; Kitagawa, H.; Nagatomi, Y.; Kondo, M.; Kita, Y. Asymmetric synthesis using C2-symmetric diols: Use of (5R,6R)-3-acetoxy-5,6-diphenyl-1,4-dioxan-2-one as a chiral synthetic equivalent of 1,2-ethanediol 1,2-dicarbocation. Tetrahedron Asymmetry 1995, 6, 2117–2020. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimogaki, M.; Fujita, M.; Sugimura, T. Stereoselective Formation of Substituted 1,3-Dioxolanes through a Three-Component Assembly during the Oxidation of Alkenes with Hypervalent Iodine(III). Molecules 2015, 20, 17041-17057. https://doi.org/10.3390/molecules200917041
Shimogaki M, Fujita M, Sugimura T. Stereoselective Formation of Substituted 1,3-Dioxolanes through a Three-Component Assembly during the Oxidation of Alkenes with Hypervalent Iodine(III). Molecules. 2015; 20(9):17041-17057. https://doi.org/10.3390/molecules200917041
Chicago/Turabian StyleShimogaki, Mio, Morifumi Fujita, and Takashi Sugimura. 2015. "Stereoselective Formation of Substituted 1,3-Dioxolanes through a Three-Component Assembly during the Oxidation of Alkenes with Hypervalent Iodine(III)" Molecules 20, no. 9: 17041-17057. https://doi.org/10.3390/molecules200917041
APA StyleShimogaki, M., Fujita, M., & Sugimura, T. (2015). Stereoselective Formation of Substituted 1,3-Dioxolanes through a Three-Component Assembly during the Oxidation of Alkenes with Hypervalent Iodine(III). Molecules, 20(9), 17041-17057. https://doi.org/10.3390/molecules200917041