Abstract
Multicomponent reactions (MCRs) are extremely popular owing to their facile execution, high atom-efficiency and the high diversity of products. MCRs can be used to access various heterocycles and highly functionalized scaffolds, and thus have been invaluable tools in total synthesis, drug discovery and bioconjugation. Traditional isocyanide-based MCRs utilize an external nucleophile attacking the reactive nitrilium ion, the key intermediate formed in the reaction of the imine and the isocyanide. However, when reactants with multiple nucleophilic groups (bisfunctional reactants) are used in the MCR, the nitrilium intermediate can be trapped by an intramolecular nucleophilic attack to form various heterocycles. The implications of nitrilium trapping along with widely applied conventional isocyanide-based MCRs in drug design are discussed in this review.
1. Introduction
Multicomponent reactions are powerful tools for the rapid generation of molecular diversity and complexity. In a typical multicomponent process, more than two components are combined in a single reaction, thereby providing an operationally effective and highly modular approach towards the synthesis of structurally diverse molecular entities. Multicomponent reactions (MCRs) represent an excellent tool for the generation of libraries of small-molecule compounds and are indispensable for structure–activity relationship (SAR) studies. Some MCRs generate uncommon, if not unique, scaffolds so the ability to further functionalize or modify them is key to exploring the utility of the scaffold in the biological realm. The unusual structure of many of these scaffolds makes them suited for exploring biological targets that traditional scaffolds do not target. Novel scaffolds are becoming more and more sought after as pathogens mutate to become resistant to current medications, and anti-aging agents are needed to tackle diseases such as Alzheimer’s, Parkinson’s, diabetes, and cancer. While MCRs may not be directly responsible for the drugs that treat these diseases in the future, they will certainly be involved as scientists search for tomorrow’s remedies. Some of the classic MCRs include the Passerini, Ugi, Mannich, Kabachnik-Fields, Biginelli, Hantzsch, Bucherer-Bergs, van Leusen and Strecker reactions. Isocyanide-based reactions form the backbone of today’s MCR chemistry, and the most common examples are: the Passerini reaction and the Ugi reaction. This review will cover some aspects of isocyanide chemistry with particular emphasis on nitrilium trapping.
1.1. Passerini Reaction
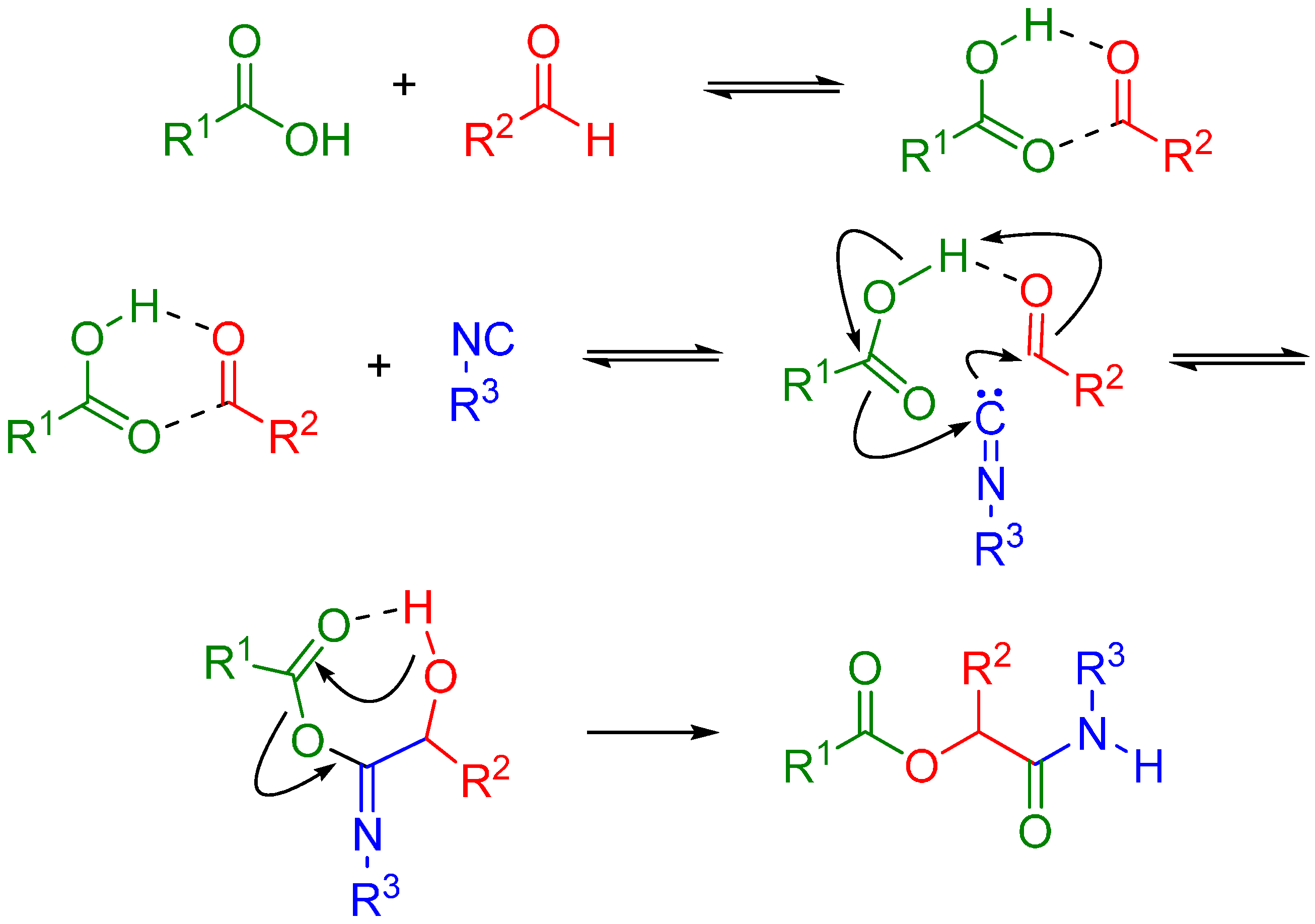
The Passerini reaction is named after Mario Passerini, who discovered this reaction in 1921. It is the first isocyanide based MCR and plays a central role in combinatorial chemistry today. It involves an aldehyde or ketone, an isocyanide, and a carboxylic acid and offers direct access to α-hydroxy carboxamides (Scheme 1). The exact mechanism is a subject of some uncertainty; the mechanism postulated by Ugi suggests a non-ionic pathway, since the reaction is accelerated in aprotic solvents (Scheme 2) [1]. The electrophilic activation of the carbonyl group is followed by a nucleophilic attack by the isocyanide. This creates a nitrilium intermediate, which is then attacked by the carboxylate.
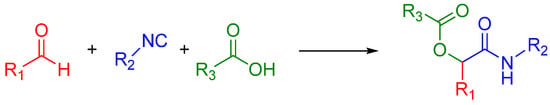
Scheme 1.
A general Passerini reaction yielding an α-acyloxy amide.
Scheme 1.
A general Passerini reaction yielding an α-acyloxy amide.
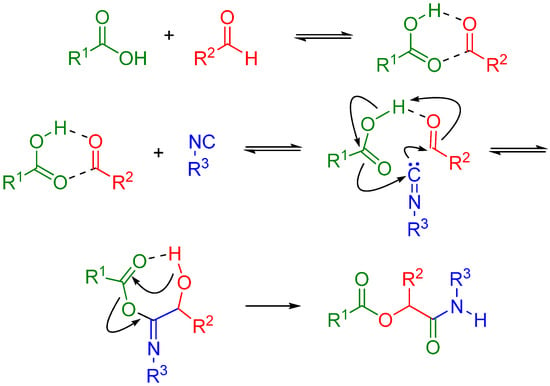
Scheme 2.
Possible mechanism of the Passerini reaction.
Scheme 2.
Possible mechanism of the Passerini reaction.
Asymmetric Passerini reactions are now known using a tridentate indan (pybox) Cu(II) Lewis acid complex developed by Schreiber [2] and other enantioselective catalysts (a combination of silicon tetrachloride and a chiral bisphosphoramide) developed by Denmark [3]. Recently, Wang et al. used commercially available chiral Lewis acids to achieve asymmetric reactions [4]. Passerini reactions with alcohols, isocyanides and carboxylic acids have been reported by the Zhu lab, thereby extending the potential utility of this reaction beyond carbonyl containing compounds. The methodology utilizes the conversion of an alcohol to an aldehyde using catalytic TEMPO, CuCl2 and NaNO2 [5]. In the presence of In(III), Passerini-type reactions have been reported between free alcohols (isopropanol), aldehydes (unsaturated and aryl) and isocyanides such as t-butyl isocyanide [6]. Pioneering work by El Kaïm and Grimaud led to the discovery of what is now known as the Passerini–Smiles reaction. In this case, an electron deficient phenol such as 2-nitrophenol (or other nitrogen heteroaromatic but electron deficient phenols) replaces the carboxylic acid. The mechanism is believed to involve activation of the aldehyde by the weakly acidic phenol (pKa ~ 4.2) which makes the carbonyl electrophilic vulnerable to attack by the isocyanide. The incipient nitrilium ion formed is attacked by the phenol followed by an SNAr leading to an α-aryloxy amide. The key step is believed to be the irreversible Smiles rearrangement of the intermediate phenoxyimidate adduct (Scheme 3) [7].
Some other variations of the Passerini reaction with surrogates of carboxylic acid involved in the intramolecular trapping of the nitrilium will be discussed later in the review. The Passerini reaction has recently been used to synthesize a library of monomers for the synthesis of polymers. By varying the R groups in the Passerini reaction, the group was able to tune the properties of the resulting polymer [8]. It also found use in the synthesis of depsipeptides on a dihydropyridinone core [9], dendrimers [10], photocaged compounds [11], cytotoxic/anticancer agents [12,13], imaging agents [14], and HCV NS3 protease inhibitors [15].
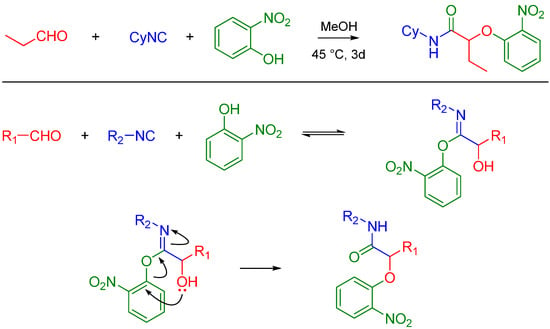
Scheme 3.
Passerini-Smiles reaction and its possible mechanism.
Scheme 3.
Passerini-Smiles reaction and its possible mechanism.
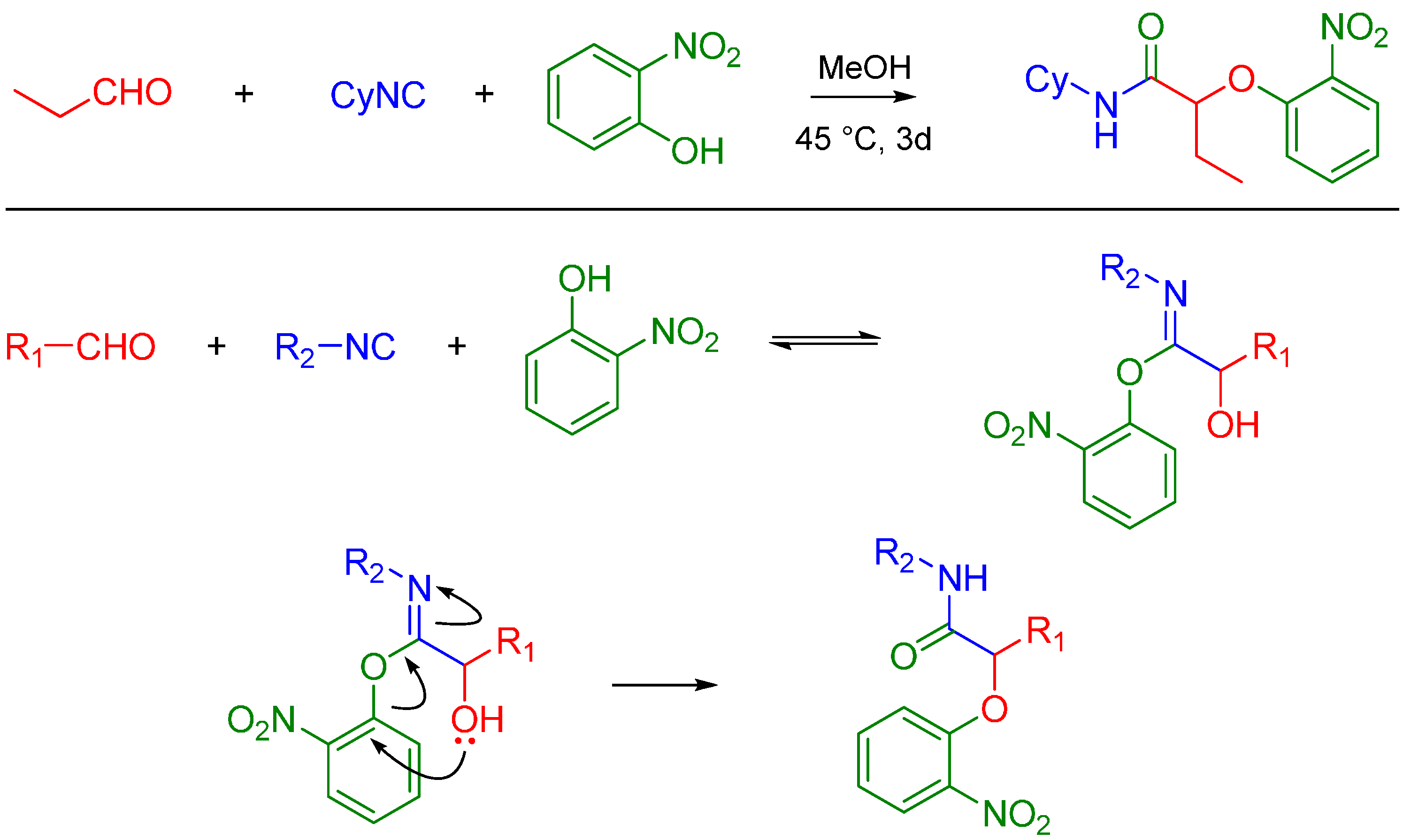
A more recent but relevant addition by Soeta et al. is use of silanols instead of carboxylic acids in the Passerini Reaction, which allows the synthesis of α-siloxyamides. The mechanism entails coordination of the silyl group to the oxygen of the carbonyl. This renders it susceptible to nucleophilic attack by an isocyanide, followed by intramolecular trapping of the nitrilium ion by the alcoholic functional group of the silanol (Scheme 4) [16].
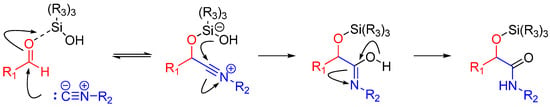
Scheme 4.
Mechanism of α-siloxyamide-forming Passerini reaction.
Scheme 4.
Mechanism of α-siloxyamide-forming Passerini reaction.
Soeta et al. also reported the one-pot synthesis of α-(sulfonyloxy)amide derivatives using an oxidative Passerini reaction. An aldehyde, an isocyanide and sulfinic acid were reacted, followed by the addition of meta-chloroperoxybenzoic acid giving the products in high yields (Scheme 5). The reaction has a wide scope on aldehydes and isocyanides [17].
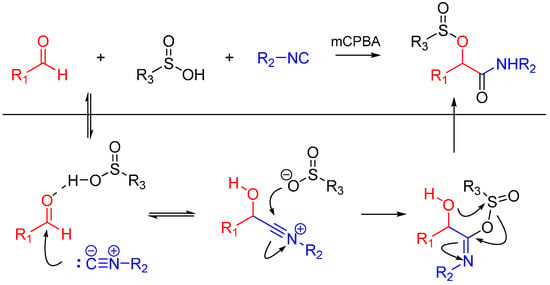
Scheme 5.
Passerini reaction yielding α-(sulfonyloxy)amides.
Scheme 5.
Passerini reaction yielding α-(sulfonyloxy)amides.
The same group reported a Passerini/Pudovik type reaction between aldehydes, isocyanides and phosphinic acids leading to α-(phosphinyloxy)amide derivatives. This report is the first instance of an isocyanide-based MCR using phosphinic acid instead of a carboxylic acid as a reactant. The nucleophilic phosphinate group undergoes a subsequent catalytic Pudovik-type reaction, affording the products in good yields (Scheme 6) [18].
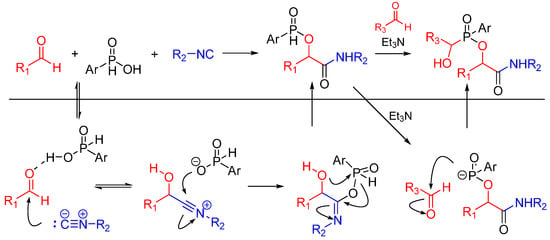
Scheme 6.
Passerini/Pudovik type reaction leading to highly functionalized α-(phosphinyloxy)amides.
Scheme 6.
Passerini/Pudovik type reaction leading to highly functionalized α-(phosphinyloxy)amides.
1.2. Ugi Reaction
A traditional Ugi four component reaction (U4CR, Scheme 7) employs a ketone or aldehyde, a carboxylic acid, an isocyanide, and an amine. The reaction is typically carried out in methanol or 2,2,2-trifluoroethanol in high reactant concentrations. The initial step is the formation of an imine from the amine and the carbonyl compound. This is followed by the nucleophilic attack of the isocyanide, resulting in the formation of the highly reactive nitrilium intermediate. The nitrilium is then attacked by the carboxylic acid, and as a result of intramolecular Mumm rearrangement, the reaction yields a central bis-amide (Scheme 8).
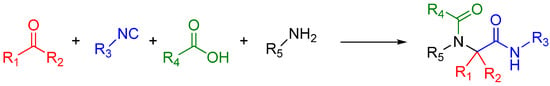
Scheme 7.
A standard U4CR. The reaction can tolerate a wide variety of R groups.
Scheme 7.
A standard U4CR. The reaction can tolerate a wide variety of R groups.
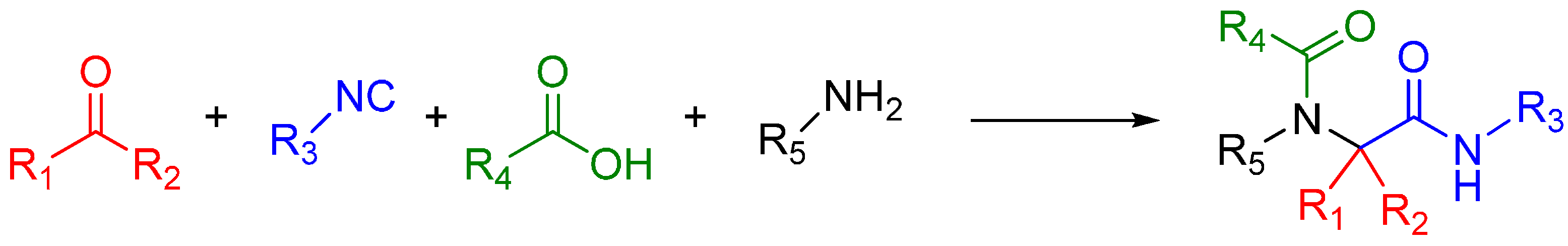
Scheme 8.
Mechanism of the Ugi 4-component reaction.
Scheme 8.
Mechanism of the Ugi 4-component reaction.
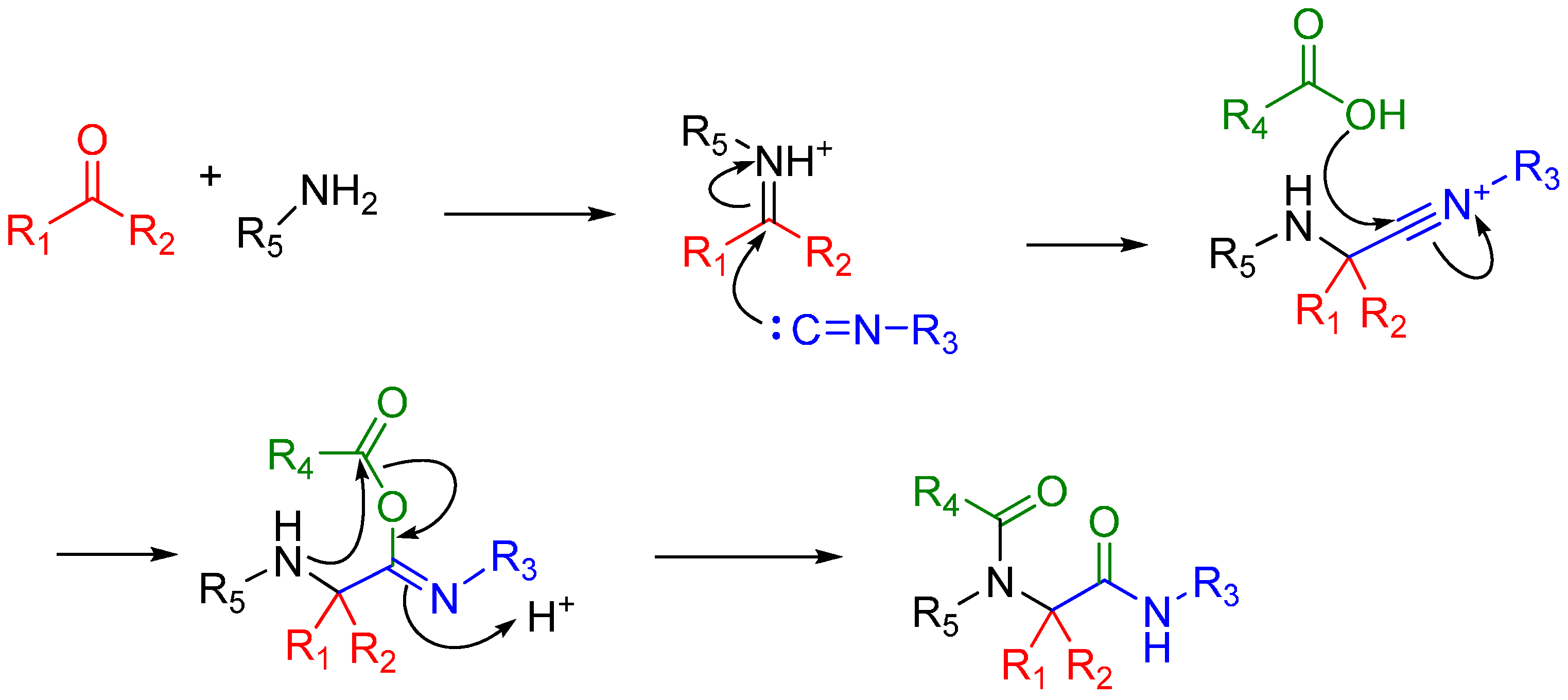
Depending upon the R groups, post Ugi reactions have been reported. Most notable are the Ugi-Heck [19], Ugi-Diels-Alder [20], Ugi-click [21] and Ugi-Buchwald-Hartwig [22] reactions, whereby the Ugi bis-amide with reactive functional groups undergoes secondary reactions to form a ring. Linear bis-amides on the other hand are useful in the synthesis of peptides (linear and cyclic) and peptidomimetics [23].
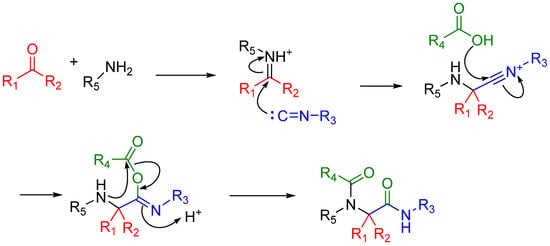
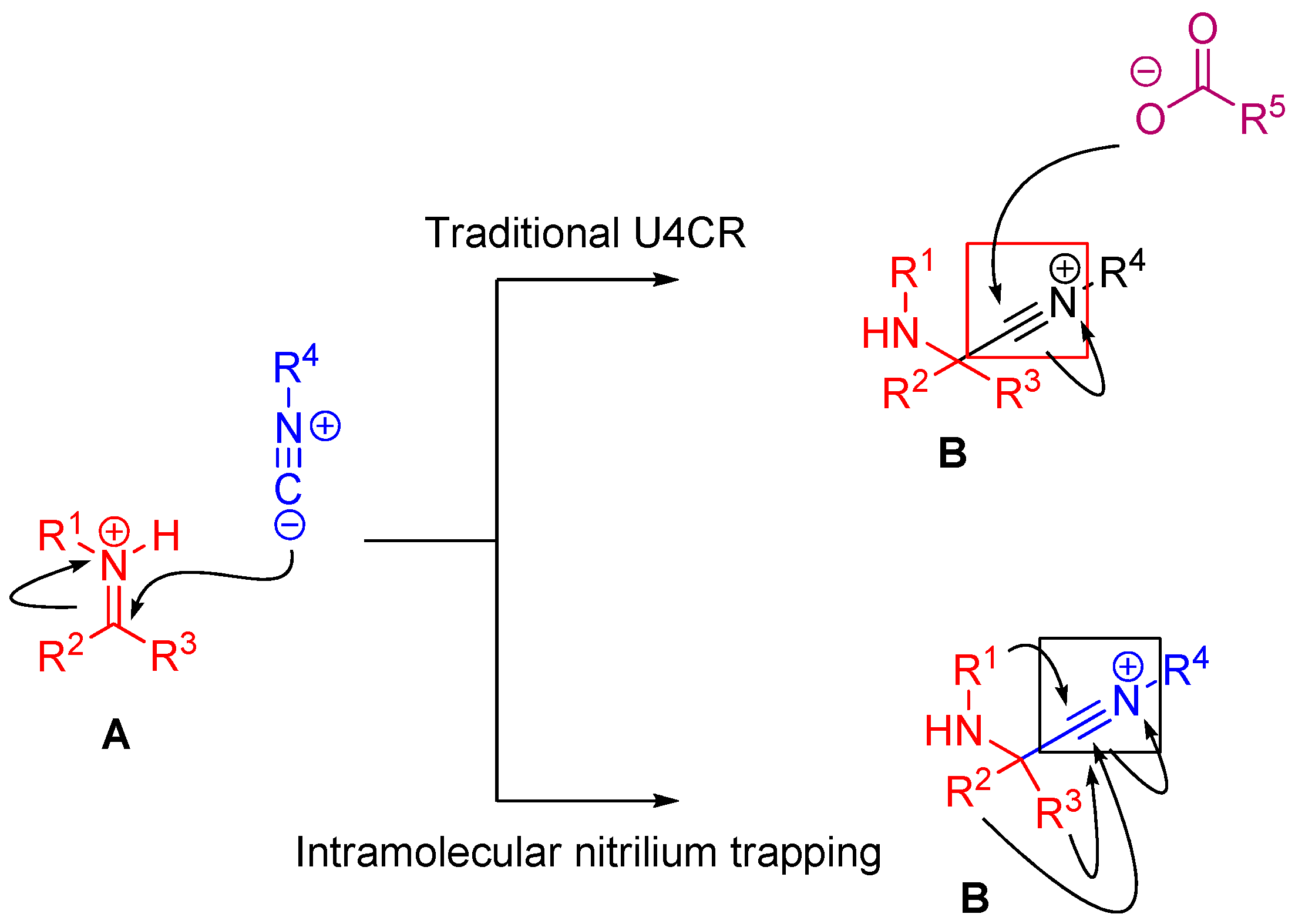
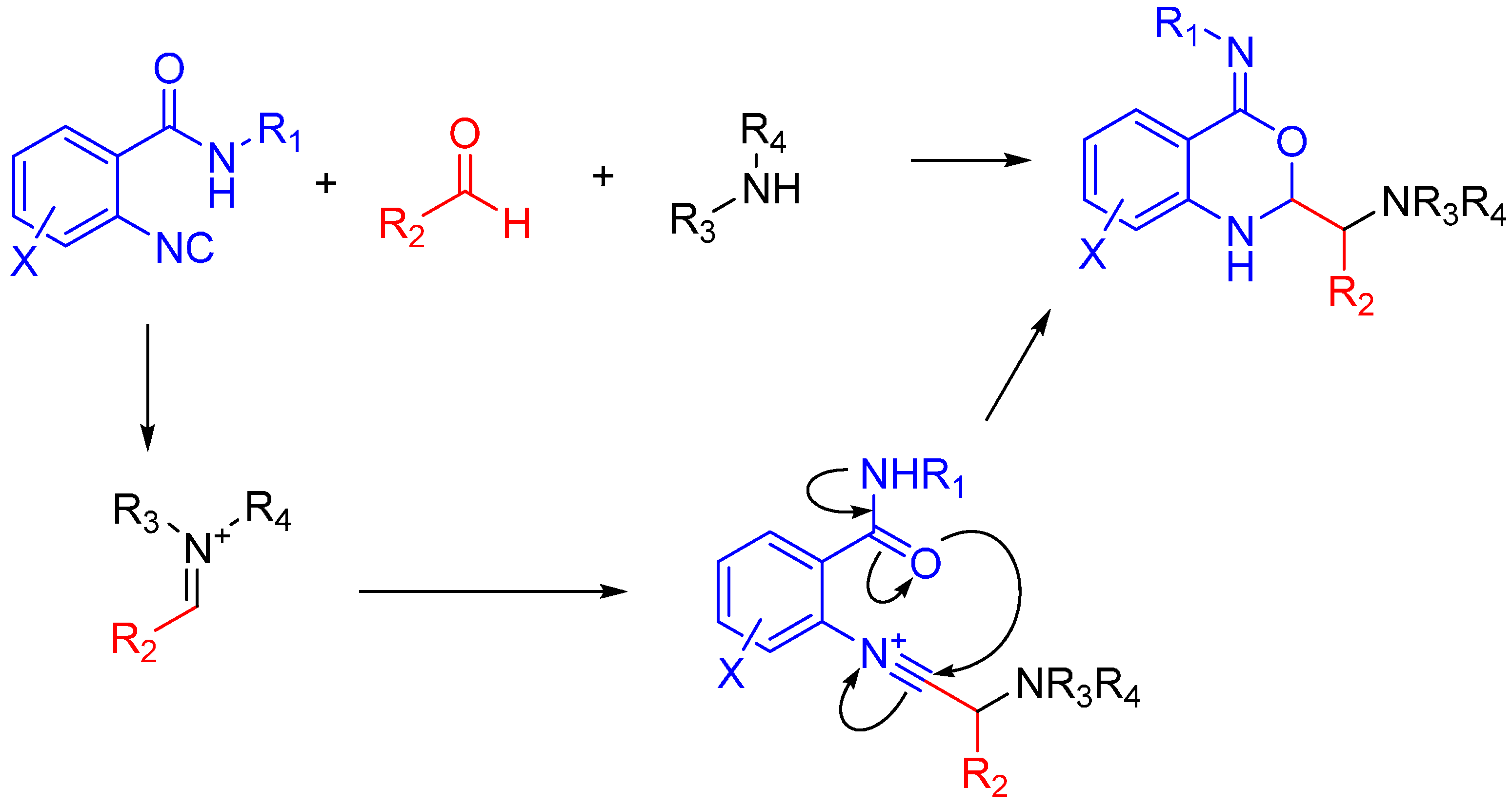
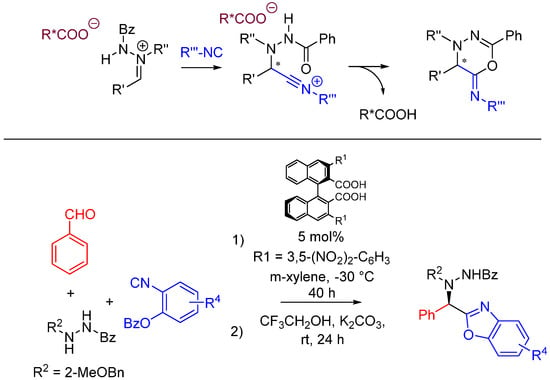
In the case of Ugi and Passerini reactions, an important intermediate in the mechanism is the reactive nitrilium. The mechanism of the Ugi reaction involves the nucleophilic attack of the isocyanide on the imine species (A) formed by the condensation of the amine and the ketone or aldehyde (Scheme 9). Lewis acids (such as TiCl4) are effective in aiding the formation of the imine by activating the aldehyde or ketone, thereby facilitating a nucleophilic attack by an amine [24]. The resulting nitrilium intermediate (B) itself is highly reactive as well and readily reacts with nucleophiles. In a traditional U4CR, the reaction partner of the nitrilium (B) is the carboxylic acid. Besides being a nucleophilic partner in the Ugi reaction, the carboxylic acid also plays a part in activating the nitrilium ion. Metal triflates have been shown to activate this nitrilium intermediate making them more susceptible to nucleophilic attack by carboxylic acids and thereby increasing rates of U4CR product formation by about two- to seven-fold [25]. However, when reactants with multiple nucleophilic groups (bisfunctional components) are used in the Ugi reaction, the nitrilium intermediate can be trapped by an intramolecular nucleophilic attack (Scheme 9).
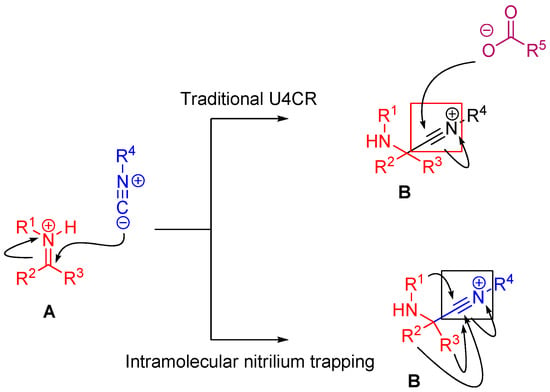
Scheme 9.
In nitrilium trapping reactions, an external or intramolecular nucleophile attacks the nitrilium species (B).
Scheme 9.
In nitrilium trapping reactions, an external or intramolecular nucleophile attacks the nitrilium species (B).
1.3. Nitrilium Trapping by an External Nucleophile
The traditional Ugi reaction utilizes a carboxylic acid as the fourth component in trapping the nitrilium ion. In the absence of a carboxylic acid, water can act as the nucleophilic partner too. One well-known example of utilizing the Ugi reaction in bioactive molecule synthesis is the synthesis of the local anesthetic lidocaine (Scheme 10) [26]. Trapping of nitrilium ion by water is usually carried out under acidic conditions [27,28].
Scheme 10.
Lidocaine is a local anesthetic on the WHO Model List of Essential Medicines, which can be synthesized in one step via an U3CR.
Scheme 10.
Lidocaine is a local anesthetic on the WHO Model List of Essential Medicines, which can be synthesized in one step via an U3CR.
Besides carboxylic acids, hydrazoic acid, HOCN, HSCN, and H2Se are some other external nucleophiles which participate as the fourth component in a U4CR (Scheme 11) [29].
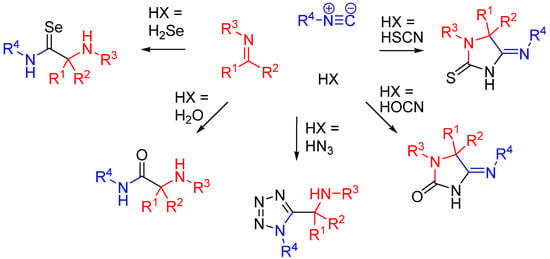
Scheme 11.
Replacement of the carboxylic acid in an U4CR.
Scheme 11.
Replacement of the carboxylic acid in an U4CR.
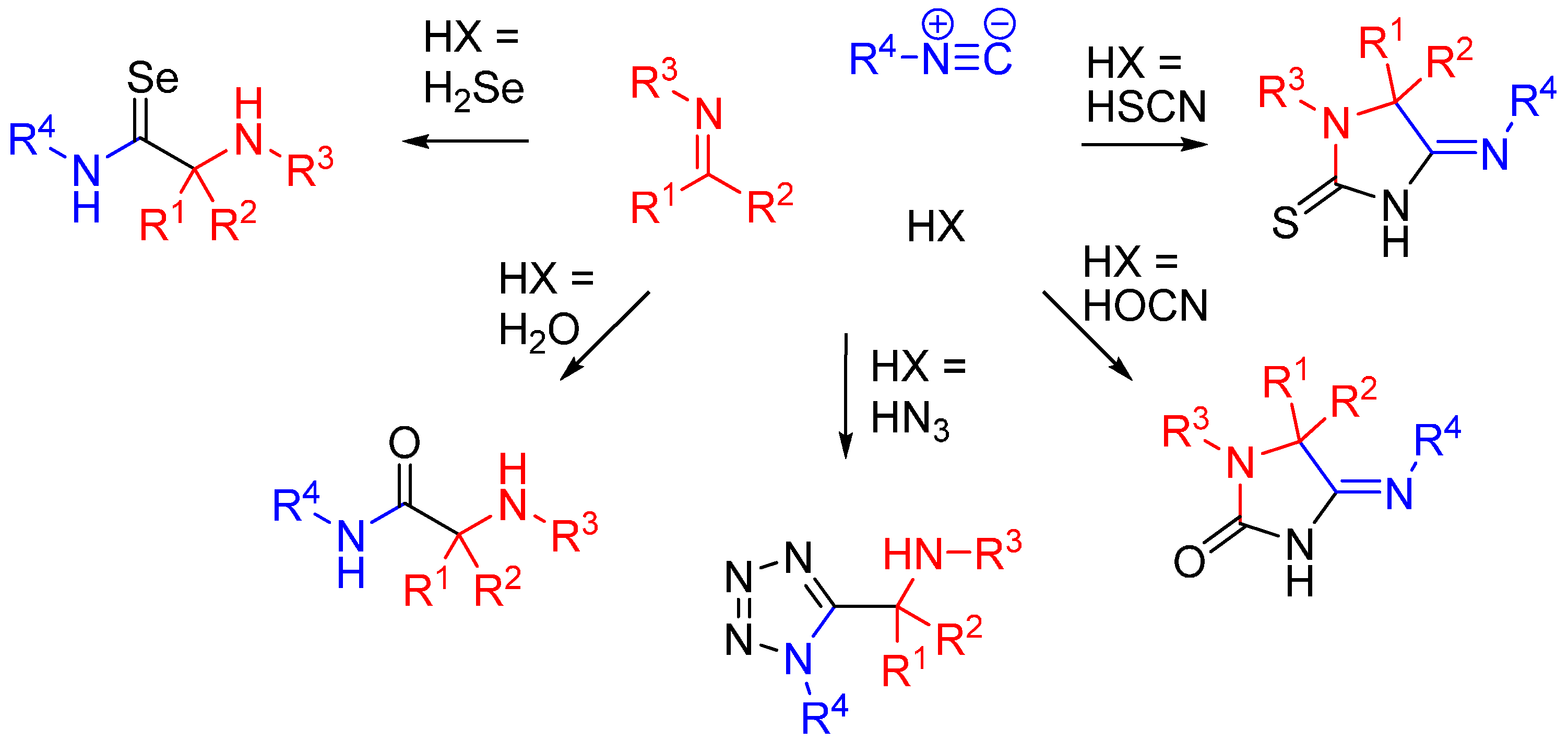
Nitrilium trapping by primary amines leads to the synthesis of amidines [30] (Scheme 12A), while with electron deficient phenols leads to N-aryl amines [31,32] (Ugi-Smiles coupling, Scheme 12B).
Scheme 12.
(A) Reaction of amines with the nitrilium yields amidines; (B) trapping with phenols leads to N-aryl amines (Ugi-Smiles reaction).
Scheme 12.
(A) Reaction of amines with the nitrilium yields amidines; (B) trapping with phenols leads to N-aryl amines (Ugi-Smiles reaction).
2. Intramolecular Trapping of Nitrilium Ion
This review will focus on the chemistry of nitrilium trapping, and its possible application in the synthesis of biologically active heterocyclic compounds. For excellent reviews on the usefulness of MCRs in drug development, see the papers by Dömling and Hulme [26,33,34]. In the absence of an external nucleophile, the reactive nitrilium species can be trapped intramolecularly by a bisfunctional nucleophilic component. This component could be an amine, carboxylic acid, phenol, amide, imine, hydrazide, activated carbon or oxime.
2.1. Trapping by a Carboxylic Acid
In the case where both an aldehyde and a carboxylic acid are present on the same molecule, as in 2-formylbenzoic acid, it has been shown that the carboxylic acid will trap the nitrilium ion [35]. Whereas an U4CR yields a linear scaffold, this trapping generates a cyclic product. This has been utilized for the rapid and high-yielding synthesis of functionalized isocoumarins, such as the anticoagulant warfarin (Scheme 13). Treatment of the product with a catalytic amount of acid led to an isomerization to the expected product.
Scheme 13.
Intramolecular trapping with a carboxylic acid.
Scheme 13.
Intramolecular trapping with a carboxylic acid.
2.2. Trapping by Amide
Nitrilium ions can also be trapped by isocyanides containing a secondary amide. The nitrilium is trapped by the amide after attacking the imine, but contrary to what one would expect the nitrogen does not directly attack the isocyanide carbon. Instead, the electrons flow from the nitrogen through the oxygen, and only then to the target atom (Scheme 14). The reaction proceeds even when R1−4 are bulky substituents, showing that the scope of the reaction is not limited by the trapping when compared with a traditional Ugi [36].
Scheme 14.
The isocyanide attacks the nitrilium ion but is then trapped by the amide substituent before hydrolysis can occur.
Scheme 14.
The isocyanide attacks the nitrilium ion but is then trapped by the amide substituent before hydrolysis can occur.
2.3. Trapping by an Imine
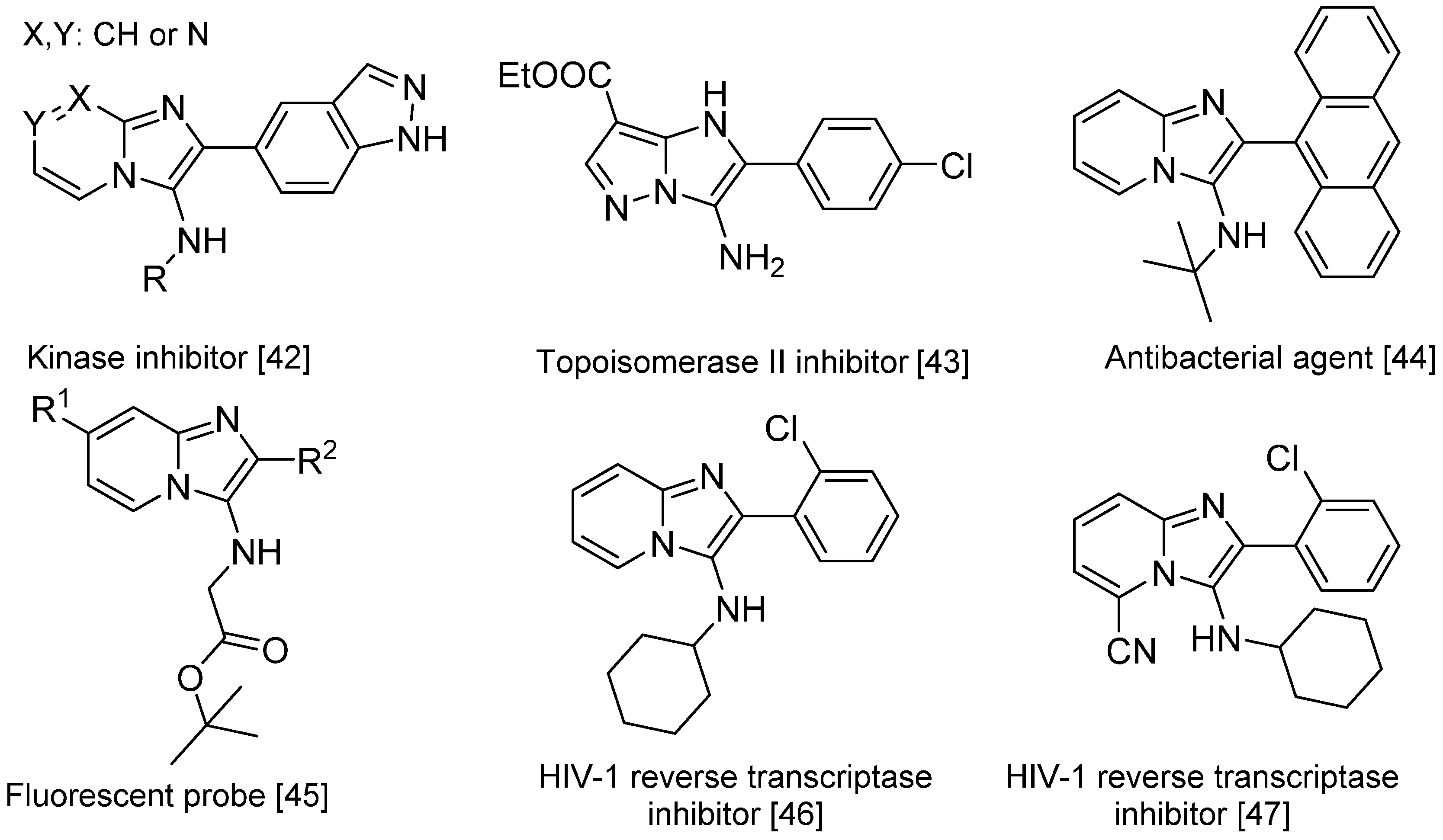
The Groebke-Blackburn-Bienaymé reaction [37,38,39] yields 3-aminoimidizoles in a non-concerted [4 + 1] reaction between an imine formed by the reaction between an aldehyde and an amine, and an isocyanide. The heterocyclic nitrogen traps the nitrilium ion, which leads to the formation of an imidazole ring following a rearrangement (Scheme 15). This reaction is widely used for the generation of highly diverse small molecule libraries [40,41]; the bioactive compounds synthesized this way include kinase inhibitors [42], topoisomerase II inhibitors [43], antibacterials effective against methicillin-resistant Staphylococcus aureus [44], fluorescent probes [45] and HIV-1 reverse transcriptase inhibitors (Scheme 16) [46,47].
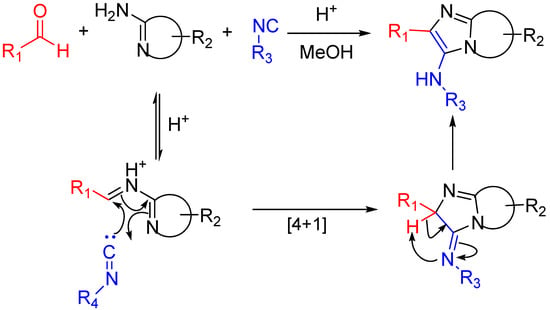
Scheme 15.
The isocyanide in the Groebke–Blackburn–Bienaymé reaction is trapped by the presence of an imine-like moiety [39].
Scheme 15.
The isocyanide in the Groebke–Blackburn–Bienaymé reaction is trapped by the presence of an imine-like moiety [39].
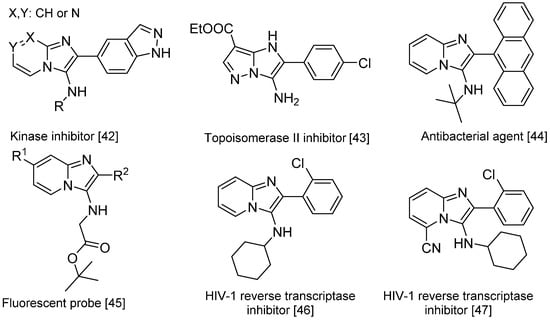
Scheme 16.
Bioactive compounds synthesized via the Groebke–Blackburn–Bienaymé reaction.
Scheme 16.
Bioactive compounds synthesized via the Groebke–Blackburn–Bienaymé reaction.
2.4. Trapping by a Secondary Amine
When an aldehyde and 4-morpholinepropionitrile are combined in the presence of trimethylsilyltriflate, the aldehyde is trapped by the TMS group and the morpholino nitrogen stabilizes the nitrilium ion (Scheme 17). The trimethylsilyltriflate coordinates to and activates the aldehyde to attack by an isocyanide, similar to how the carboxylic acid normally activates the aldehyde through hydrogen bonding. If a ketone is present on the α-carbon of the isocyanide, the reaction can yield a cyclized product as well [48].
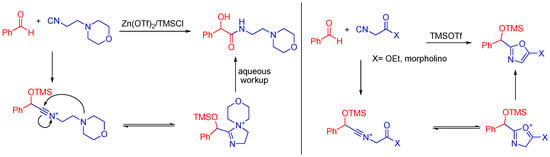
Scheme 17.
A morpholino isocyanide yields a linear product, while an α-keto morpholino isocyanide cyclizes.
Scheme 17.
A morpholino isocyanide yields a linear product, while an α-keto morpholino isocyanide cyclizes.
2.5. Trapping by Amine
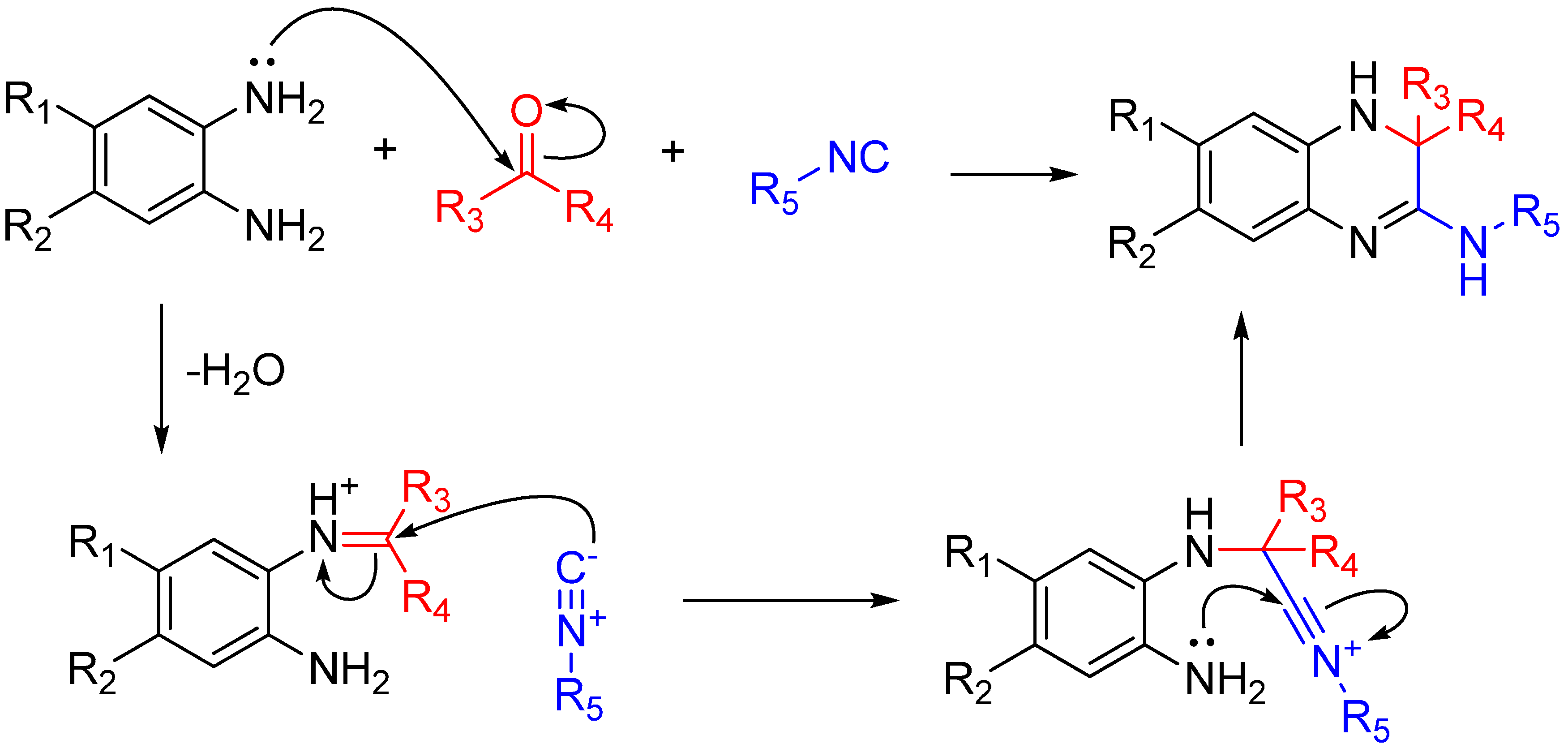
If the amine reagent is a diamine, one amino group will form an imine and the other will react with the nitrilium after it attacks that imine. This trapping, like many others shown here, forms a highly substituted heterocycle. By using 1,2-diaminobenzenes, the reaction yields quinoxalinones in a single step (Scheme 18). As noted by the authors, several quinoxalinone compounds have affinity for various central nervous system (CNS) receptors, and also may be useful as antidiabetic agents and antiretrovirals [49].
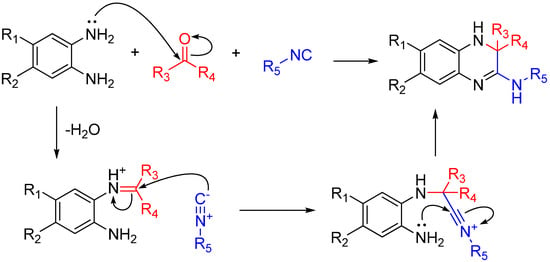
Scheme 18.
Synthesis of 3,4-dihydroquinoxalin-2-amine and a library of substituted analogs using 1,2-diaminobenzenes.
Scheme 18.
Synthesis of 3,4-dihydroquinoxalin-2-amine and a library of substituted analogs using 1,2-diaminobenzenes.
An intriguing way to ensure the generation of a heterocycle from a trapped MCR is to use a reactant that contains both the isocyanophile, which is an imine in a typical Ugi, and a nucleophile to trap the nitrilium after its attack (Scheme 19). While similar to using a diamino reactant, this approach goes one step further and includes the “imine” with the nucleophile rather than forming it in situ [50].
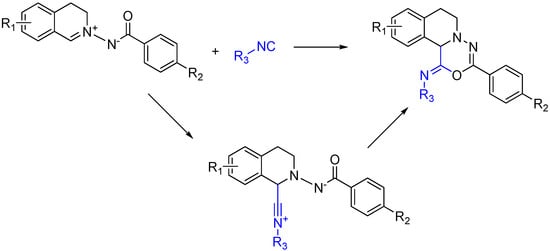
Scheme 19.
[5 + 1] Isocyanide cycloaddition yields six membered heterocycles.
Scheme 19.
[5 + 1] Isocyanide cycloaddition yields six membered heterocycles.
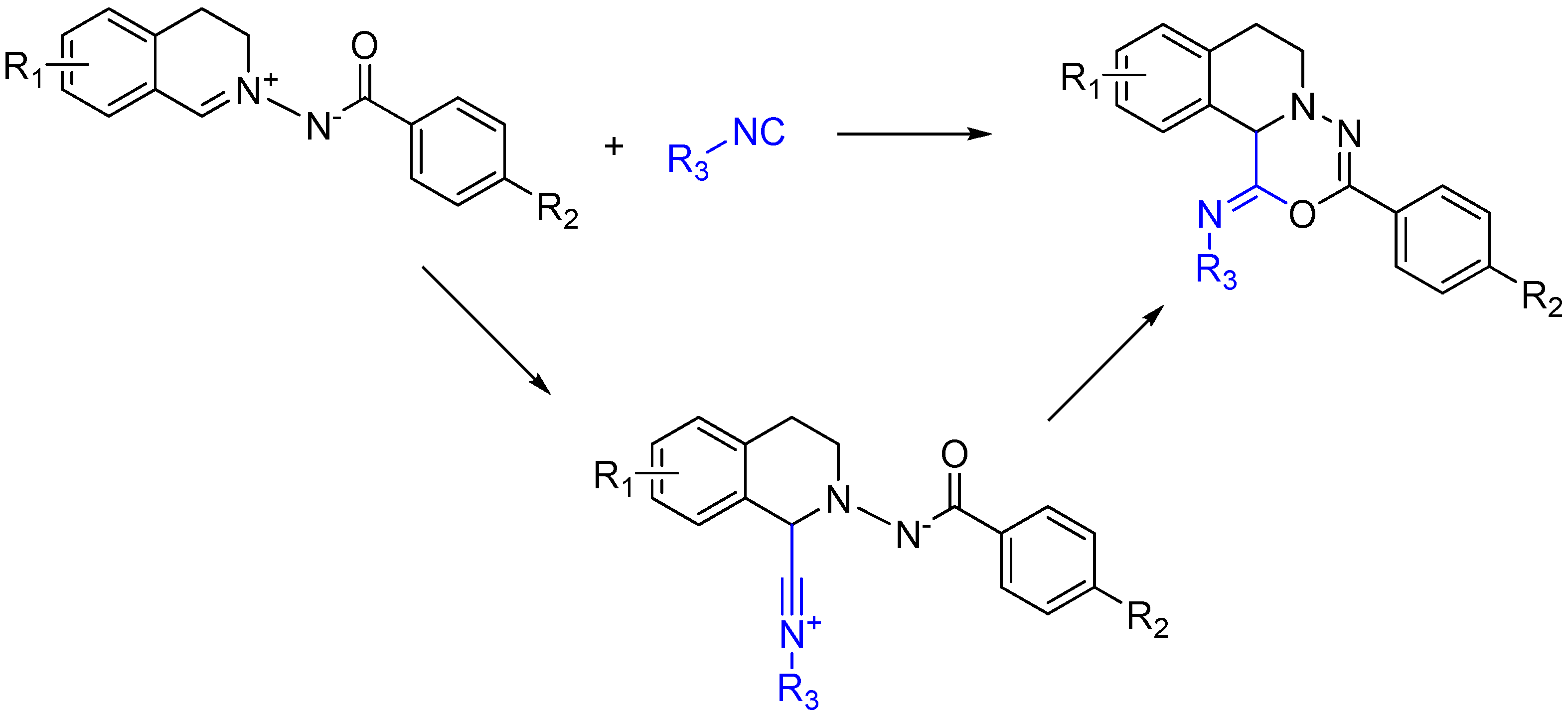
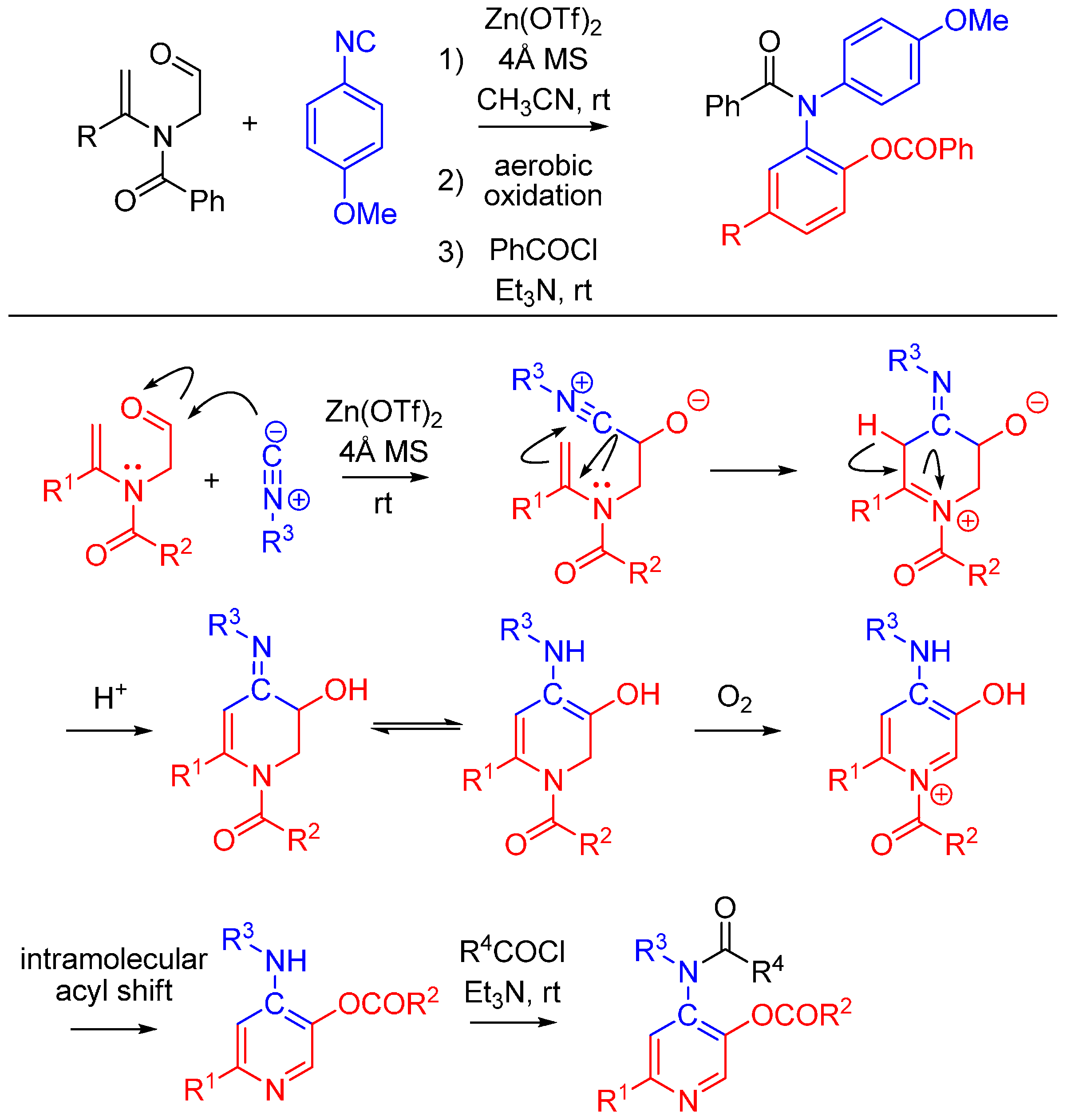
2.6. Trapping by Enamide
Lei and co-workers report a novel approach to synthesize pyridines [51]. The method features an unprecedented α-addition of aldehyde and enamide to an isocyanide. While not an Ugi reaction, this cycloaddition is an intriguing example of nitrilium trapping outside the realm of isocyanide-based MCRs. The cascade reaction involves a Zn(OTf)2-promoted [1 + 5] cycloaddition of the isocyanide with a substituted enamide, followed by the aerobic oxidative aromatization and intramolecular acyl transfer and finally an acylation by an external acyl chloride to yield 2-substituted 4-acylamino-5-acyloxypyridines (Scheme 20). The method provides a straightforward route to diversely substituted pyridines that are analogues of acetylcholinesterase inhibitors [52].
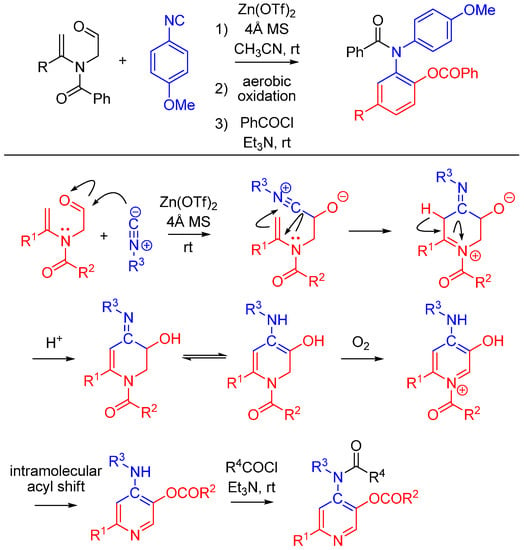
Scheme 20.
[1 + 5] cycloaddition with isocyanides and enamides yields substituted pyridines.
Scheme 20.
[1 + 5] cycloaddition with isocyanides and enamides yields substituted pyridines.
2.7. Trapping by Hydrazide
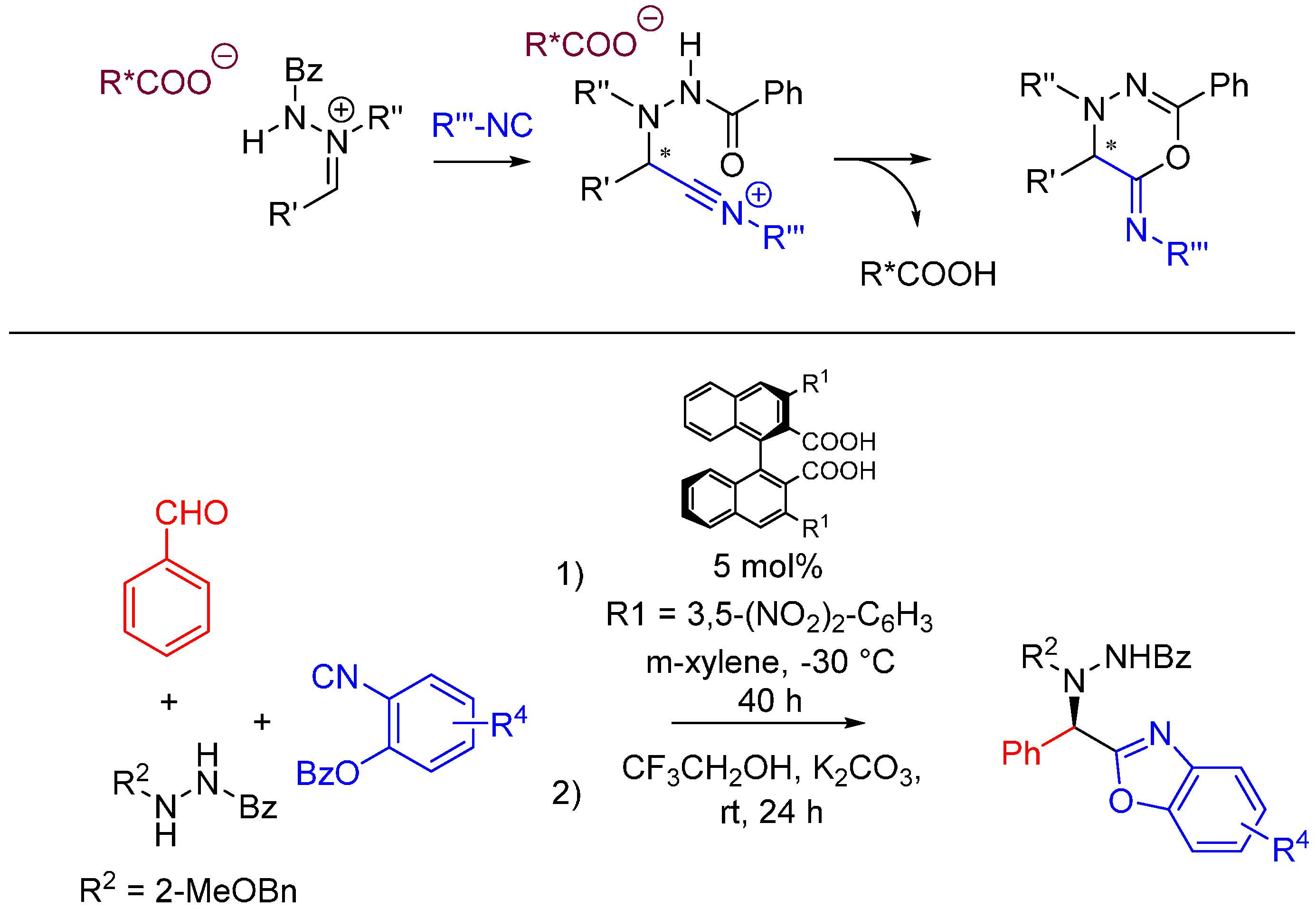
Achieving stereocontrol is among the current challenges of MCR chemistry. A recent example by Hashimoto et al. uses acyclic azomethine imines as prochiral electrophiles generated with catalytic amounts of axially chiral dicarboxylic acids [53]. The nitrilium intermediate following the attack of the isocyanide is trapped intramolecularly by the oxygen of the hydrazide. The imine is generated from benzaldehyde and N’-benzylbenzohydrazide in the presence of a chiral carboxylic acid. 2-benzoyloxyphenyl isocyanides [54] are used for this reaction, and a one-pot basic hydrolysis is performed on the Ugi products to generate asymmetric benzoxazoles in high yield and ee (Scheme 21).
Scheme 21.
Chiral, acid-catalyzed, asymmetric Ugi reaction.
Scheme 21.
Chiral, acid-catalyzed, asymmetric Ugi reaction.
2.8. Trapping by Activated Aryl Carbon
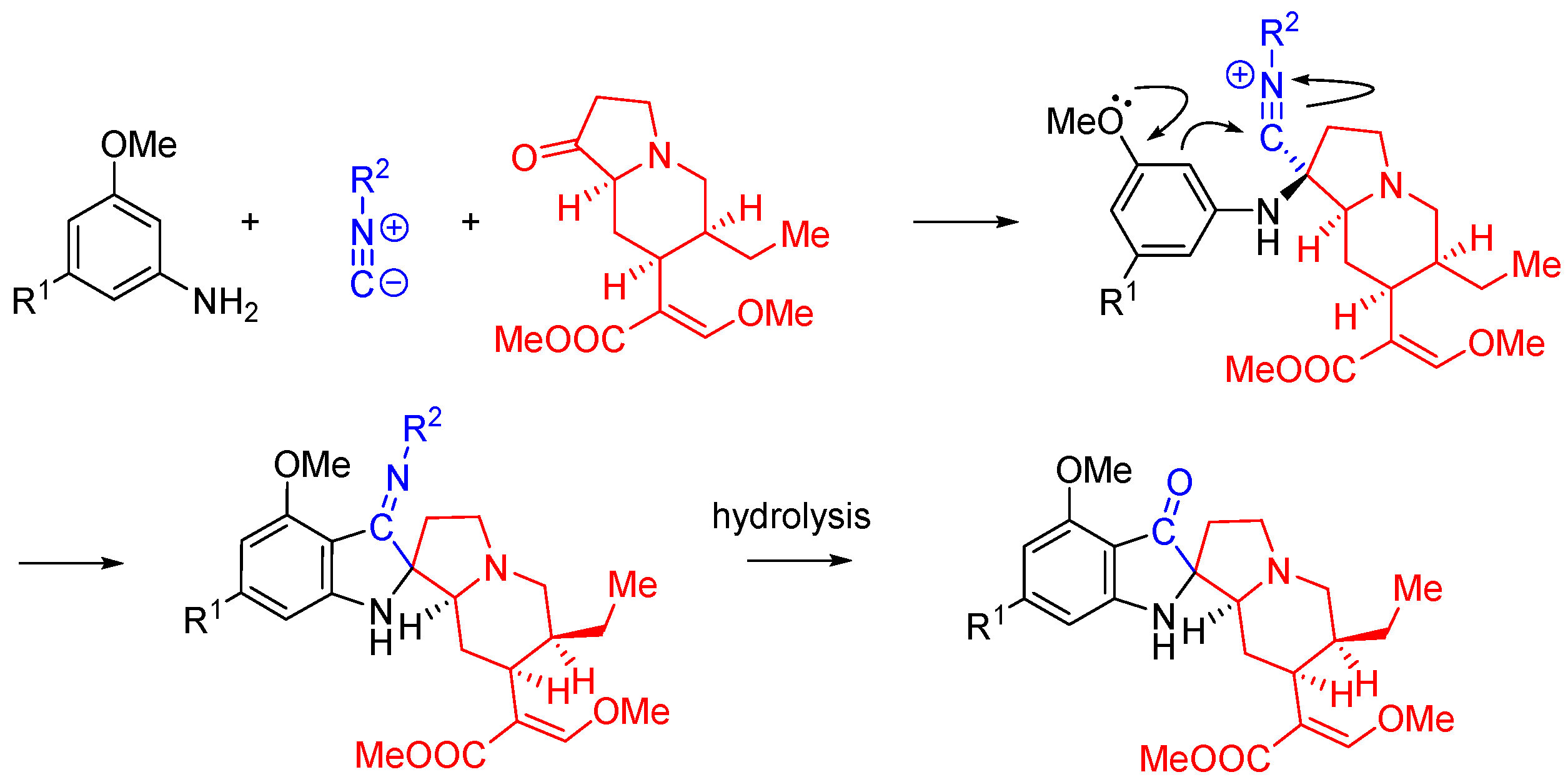
Nitrilium trapping can be utilized in the synthesis of natural product analogs as demonstrated by the work of Kim et al [55]. In this work, the synthesis of 11-methoxymitragynine pseudoindoxyl, a derivative of the opioid natural product mitragynine, is reported. The electron donating properties of the C-9 phenolic group are utilized to direct the annulation reaction through the intramolecular trapping of the nitrilium species, forming the spirocyclic indoxyl ring system. The Ugi three-component reaction is interrupted by a Houben-Hoesch type cyclization (Scheme 22) [56].
Scheme 22.
The interrupted Ugi approach to synthesize mitragynine pseudoindoxyl analogs.
Scheme 22.
The interrupted Ugi approach to synthesize mitragynine pseudoindoxyl analogs.
Kysil et al. report a multicomponent reaction between ethylenediamines, isocyanides, ketones and aldehydes that affords highly substituted 3,4,5,6-tetrahydropyrazin-2-amines, including spirocyclic compounds (Scheme 23) [57]. The tetrahydropyrazine ring is formed as a result of the intramolecular trapping of the nitrilium intermediate by the second ethylenediamine amino group and the subsequent tautomerization. The reaction is promoted by Lewis acids, particularly trimethylchlorosilane.
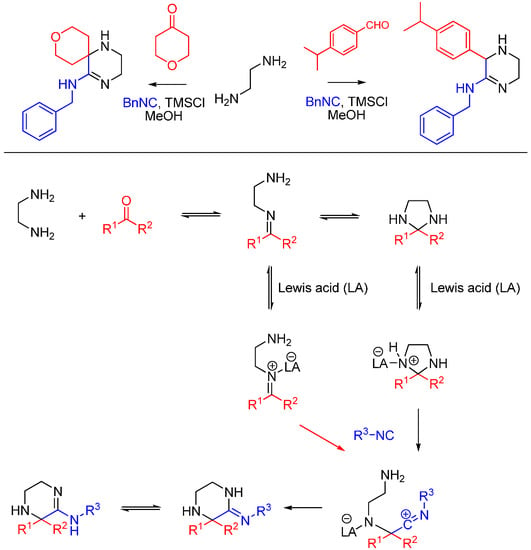
Scheme 23.
Synthesis of tetrahydropyrazines using ethylenediamine and Lewis acid catalysis.
Scheme 23.
Synthesis of tetrahydropyrazines using ethylenediamine and Lewis acid catalysis.
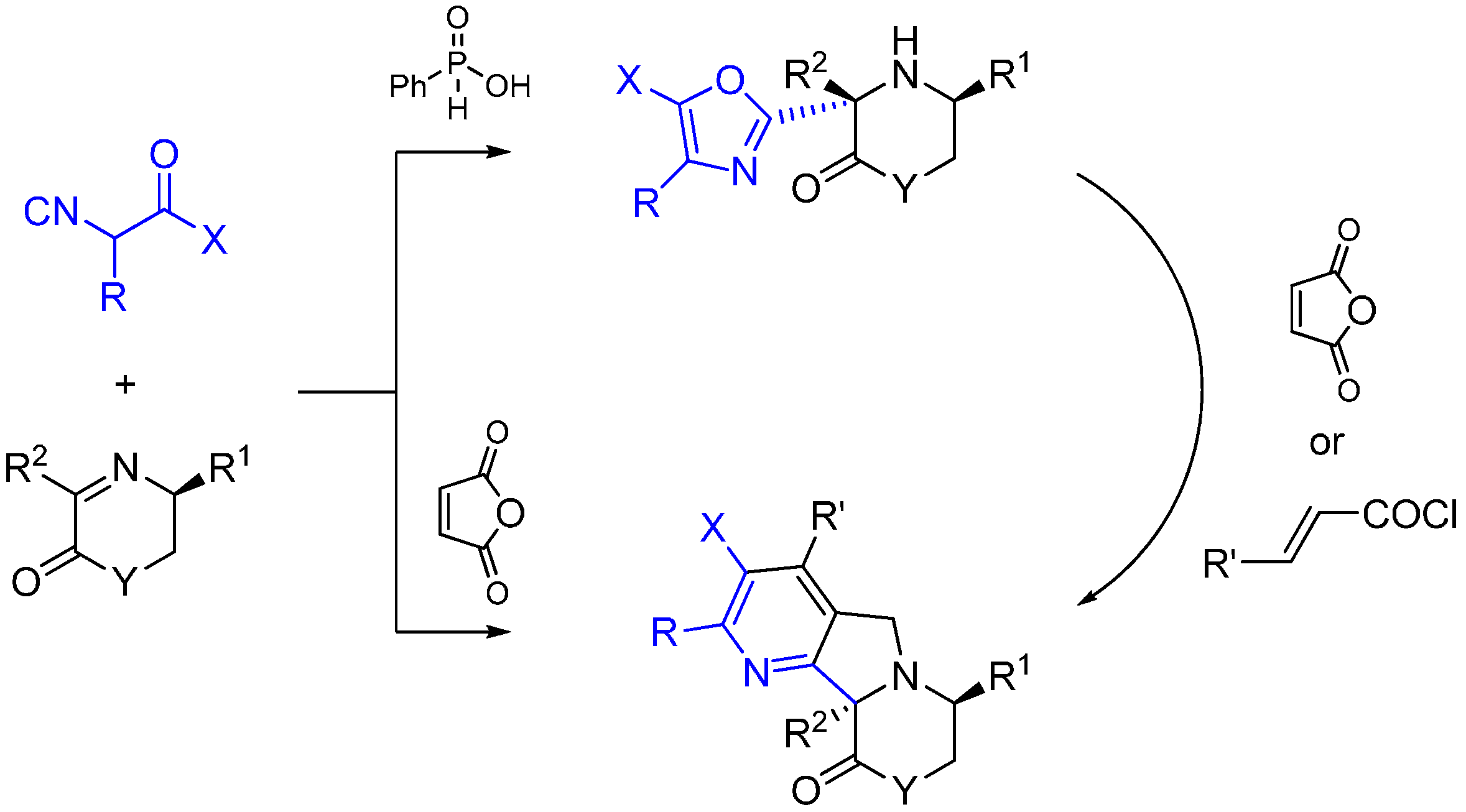
Xia and co-workers report an asymmetric Ugi-type reaction between α-isocyanoacetamides and chiral imines catalyzed by phenylphosphilic acid leading to 3-oxazolyl morpholin-2-one or piperazin-2-one derivatives [58]. The oxazol ring is formed following the intramolecular attack of the carbonyl oxygen of the isocyanoacetmide amide group. The subsequent reaction of aminooxazoles with maleic anhydride led to fused heterocycles through a multiple domino reaction sequence (Scheme 24).
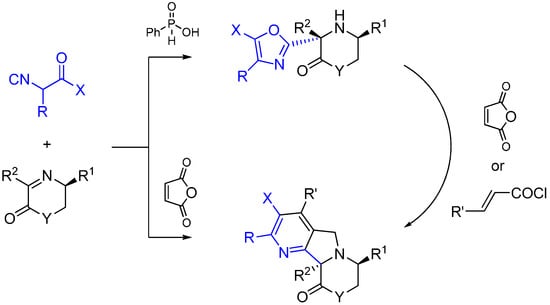
Scheme 24.
Synthesis of tetrahydropyrazines using ethylenediamine and Lewis acid catalysis.
Scheme 24.
Synthesis of tetrahydropyrazines using ethylenediamine and Lewis acid catalysis.
3. Intramolecular Nitrilium Trapping by Aminophenols
Heterocycles play an important role in the design and synthesis of bioactive small molecules: the vast majority of marketed drugs contain at least one heterocyclic ring. Numerous nitrogen-containing heterocycles including dihydrothiazoles, benzoxazoles, benzothiazoles and benzimidazoles possess biological activity [59,60,61,62,63].
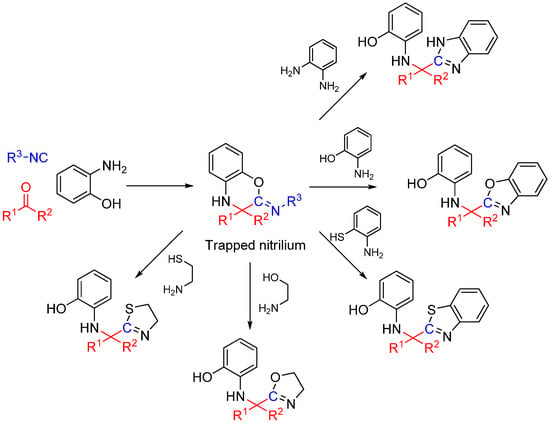
Scheme 25.
Synthesis of heterocyclic scaffolds using a three-component reaction.
Scheme 25.
Synthesis of heterocyclic scaffolds using a three-component reaction.
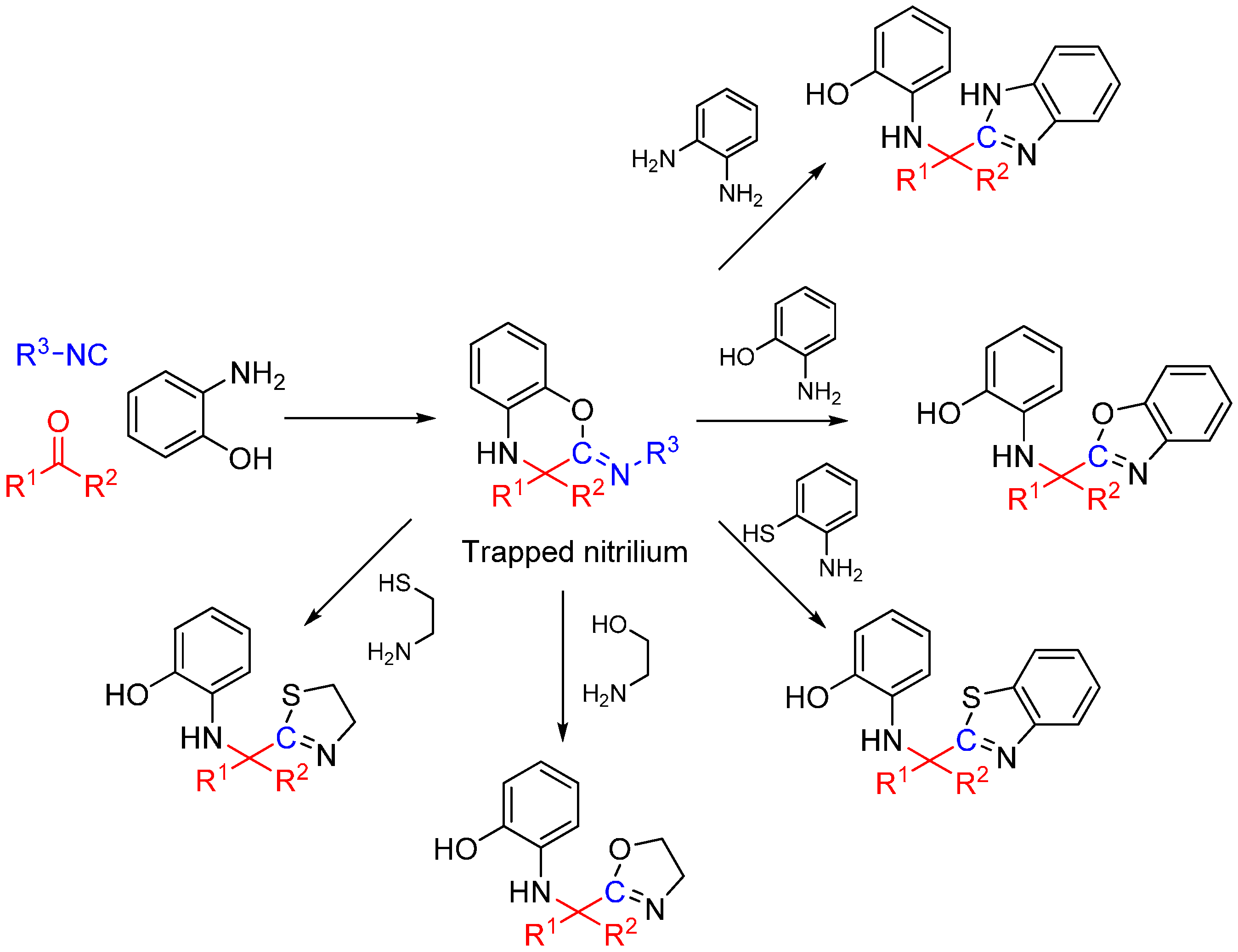
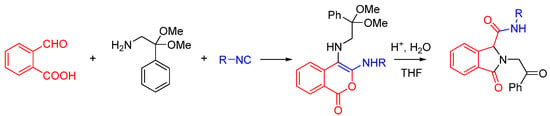
We recently reported a novel MCR between α-ketones, 2-aminophenol, and isocyanide that takes advantage of intramolecular nitrilium trapping yielding benzoxazoles and other heterocycles (Scheme 25) [64]. The reaction proceeds via a benzo[b][1,4]oxazine intermediate. Owing to the reactive nature of the trapped intermediate, the ring can be opened by a second molecule of aminophenol or other bis-nucleophiles yielding benzoxazoles and other diverse heterocyclic scaffolds. Furthermore, the reaction generates two additional diversity handles that were utilized to synthesize various substituted bis-heterocyclic derivatives. Synthesis of heterocycles is traditionally accomplished through multistep routes and/or harsh reaction conditions, particularly of benzoxazoles [65], and while MCR approaches can also be taken [29,66], there is a need for a more resource-efficient, preferably one-pot method for their synthesis [67,68]. Metal catalyzed insertion of isocyanides leading to heterocycles are known and usually utilize harsh conditions and have somewhat limited scope in terms of diversity [69,70].
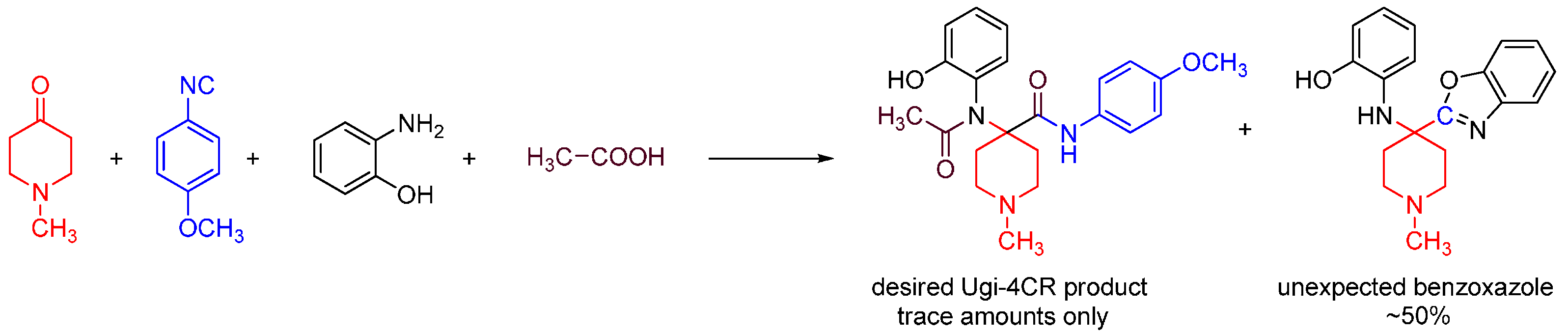
We discovered this reaction while performing a routine U4CR in 2,2,2-trifluoroethanol (TFE) between 2-aminophenol, N-methyl-4-piperidone, 4-methoxyphenyl isocyanide and acetic acid at 55 °C, we observed an unusual heterocyclic (benzo[d]oxazol-2-yl)-1-methylpiperidine product (benzoxazole, 1) along with the expected U4CR product seen only in trace amounts (Scheme 26).
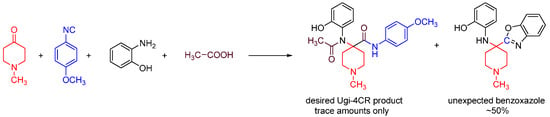
Scheme 26.
Our MCR yielded an unexpected benzoxazole instead of the U4CR product.
Scheme 26.
Our MCR yielded an unexpected benzoxazole instead of the U4CR product.
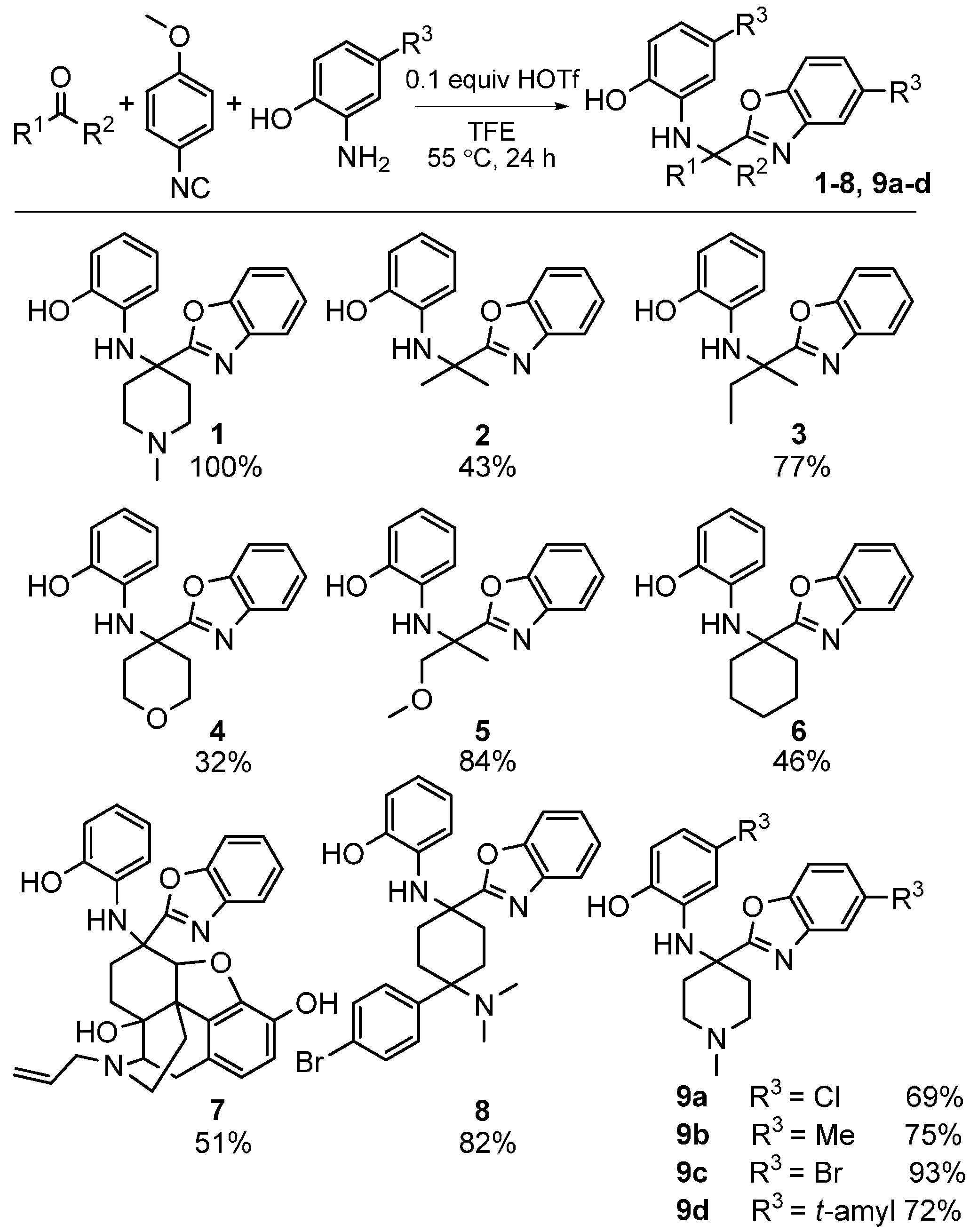
We investigated the scope of this heterocycle-forming MCR using various isocyanides. While 4-methoxyphenylisocyanide provided the best yields, other isocyanides were also effective in the reaction. It should be pointed out that the isocyanide contributes only one carbon atom towards benzoxazole; therefore, the reaction outcome is independent of the nature of isocyanide used. The scope of this reaction was evaluated by using different ketones and substituted 2-aminophenols (Scheme 27). The reaction was effective with a variety of ketones including the complex, multifunctional semi-synthetic natural product-like naloxone (product 7), a clinically used opioid antagonist, highlighting the good functional group tolerance of this reaction.
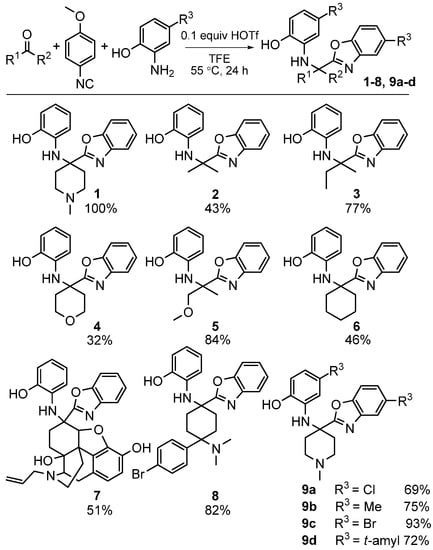
Scheme 27.
Scope of our MCR yielding benzoxazoles.
Scheme 27.
Scope of our MCR yielding benzoxazoles.
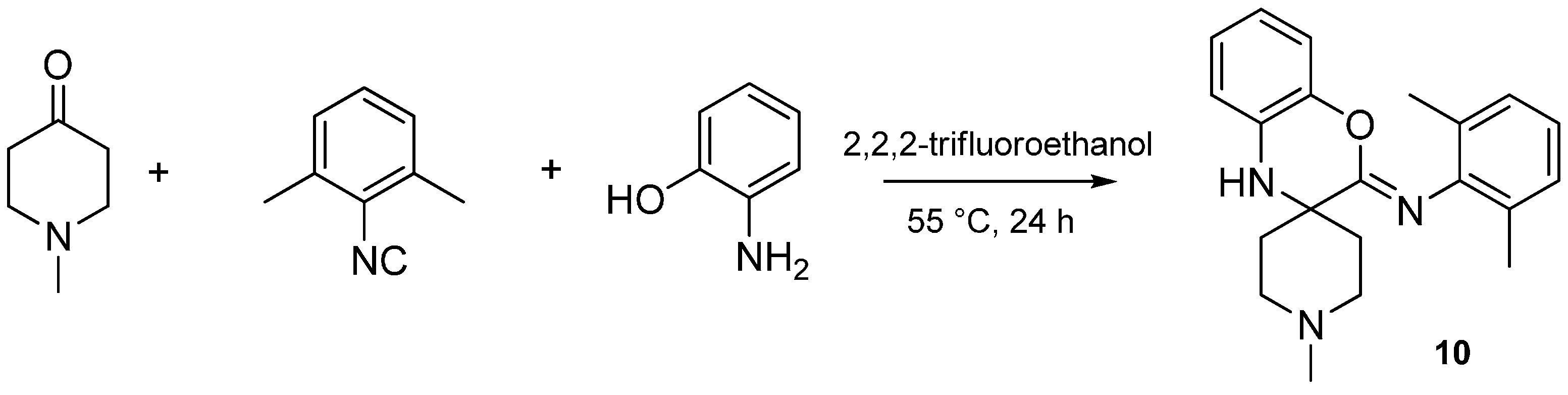
Isocyanides play an unusual role in this MCR, contributing only one carbon atom to the benzoxazole. The aryl or alkyl amine moiety acts as the leaving group, leading to an insertion of only the isocyanide carbon into the benzoxazole moiety. While investigating the effect of various isocyanides, we found that 2,6-dimethylphenyl isocyanide, instead of the expected benzoxazole, yielded a spiro[benzo[b][1,4]oxazine]-imine (benzoxazine, 10) scaffold when reacted with NMP and 2-aminophenol (Scheme 28). The isolation and structure elucidation of this benzoxazine derivative provided us the first clues about the mechanism of this reaction.
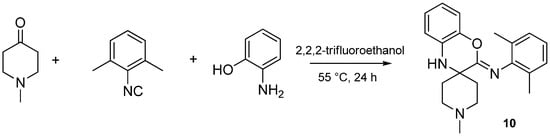
Scheme 28.
Sterically hindered isocyanide yields a [benzo[b][1,4]oxazine]-imine (10).
Scheme 28.
Sterically hindered isocyanide yields a [benzo[b][1,4]oxazine]-imine (10).
We hypothesized that the benzoxazine 10 was an intermediate in the reaction route leading to the final product benzoxazole 1. We envisioned the reaction mechanism for the formation of benzoxazoles shown in Scheme 29.
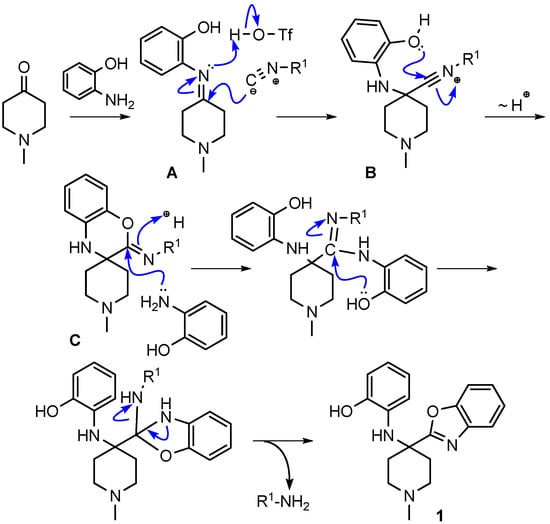
Scheme 29.
Proposed mechanism for the formation of benzoxazole 1.
Scheme 29.
Proposed mechanism for the formation of benzoxazole 1.
To test our proposed mechanism that the benzoxazine C is an intermediate in the synthesis of benzoxazoles, we isolated the benzoxazine obtained using 4-methoxyphenylisocyanide. We enhanced the scope of our MCR by reacting the isolated benzoxazine intermediate with a series of bis-nucleophiles including 1,2-diaminobenzene, 2-aminothiophenol, and cysteamine to synthesize benzimidazole, benzothiazole and dihydrothiazole derivatives, respectively.
To enhance the diversity and, thus, the possible biological relevance of the benzoxazole scaffold, we decided to exploit the free phenolic OH and secondary aromatic amine of the benzoxazole products to make second-generation derivatives (Figure 1).
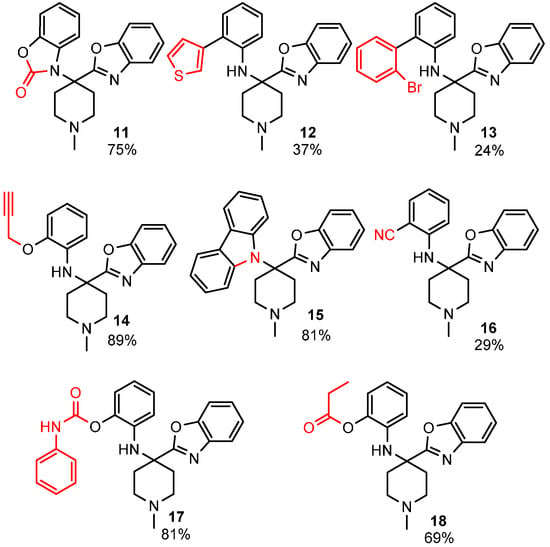
Figure 1.
Diversification of the benzoxazole scaffold.
Figure 1.
Diversification of the benzoxazole scaffold.
In summary, we discovered a convenient, Brønsted acid catalyzed, isocyanide based heterocycle-forming three-component reaction between 2-aminophenols, isocyanides, and ketones that leads to benzoxazoles and other heterocyclic products. The reaction progresses via a benzoxazine intermediate formed by intramolecular nucleophilic trapping of the reactive nitrilium intermediate by an adjacent phenolic group. The reactivity of benzoxazine to bis-nucleophiles was leveraged to synthesize an array of heterocyclic scaffolds including benzimidazoles, dihydrothiazoles and benzothiazoles. The reaction showed good functional group tolerance, and is compatible with different ketones and aminophenols, ideal for the generation of molecule libraries under mild reaction conditions, without the use of transition metal catalysts. We have further exploited the reactivity of the phenolic hydroxyl and the aromatic secondary amine to diversify the scaffolds.
4. Synthesis of Opioids by the Ugi Reaction
Morphine and its semi-synthetic congeners are the most important agents used for the treatment of moderate to severe pain. The desired analgesic effect is accompanied by various dangerous adverse effects, which are predominantly mediated by mu opioid receptors (MOR). One possible way to overcome MOR-mediated adverse effects is to synthesize mixed partial agonists or compounds with mixed MOR agonist/delta opioid receptor (DOR) antagonist properties [71,72]. The ability of morphine to induce tolerance and dependence may be countered by co-administering DOR antagonists without sacrificing antinociception [73,74]. Similarly, partial activation of multiple opioid receptors, for example by a dual MOR-DOR partial agonist ligand, can alleviate these side effects. However, the previously reported MOR/DOR mixed agonists are mostly peptides, therefore their clinical applicability is low [75,76,77].
Synthetic aryl anilido piperidines including fentanyl and carfentanil (19) are potent analgesics acting primarily on MOR, and their scaffold can be accessed by the Ugi reaction. Lofentanyl, structurally closely related to carfentanil, has been shown to possess high affinity for DOR as well [78]. The synthesis of carfentanil was recently reported using an U4CR followed by methanolysis of the carfentanil amide Ugi product (Scheme 30) [79]. The same approach has been used by Portoghese and co-workers to prepare carfentanil-based bivalent ligands for the treatment of opioid withdrawal [80,81].
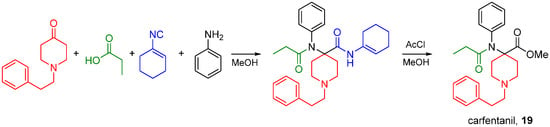
Scheme 30.
Synthesis of carfentanil by the U4CR.
Scheme 30.
Synthesis of carfentanil by the U4CR.
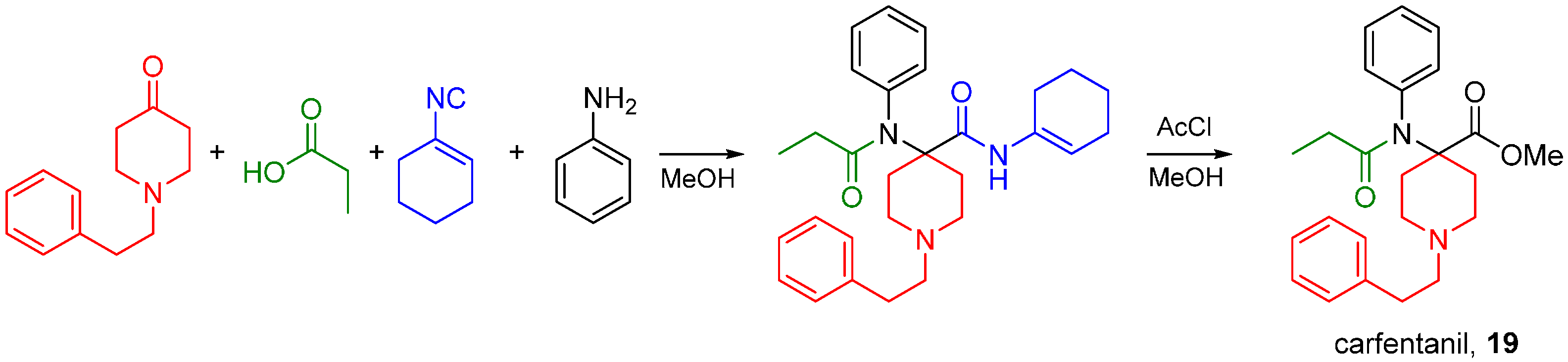
Interested in creating highly potent synthetic opioids acting as MOR and DOR dual agonists, our research group recently reported the synthesis of carfentanil amides using the U4CR approach [82]. We substituted the ester moiety with an amide using various isocyanides and determined if the amide analogs were analgesics with improved side effect profiles compared to morphine. Previously, only the primary amide was reported [83,84]. Four-component Ugi reactions were carried out between N-alkylpiperidones, aniline, propionic acid and an array of aliphatic isocyanides (Scheme 31).
Scheme 31.
Synthesis of carfentanil amides by the Ugi reaction.
Scheme 31.
Synthesis of carfentanil amides by the Ugi reaction.
Piperidine-4-ones with N-phenylethyl and N-cyclopropylmethyl substituents were employed in the reactions. The carfentanil amides were isolated in moderate to good yields. The use of Ugi reactions to access the carfentanil scaffold makes diversification library-friendly because of the commercial availability of the simple building blocks used in this reaction.
The synthesized derivatives (Figure 2, 20–29) were characterized using in vitro receptor binding and in vivo analgesia assays in a mouse model. Our lead compound, the N-cycloheptyl analog (30), was subjected to detailed pharmacological characterization.
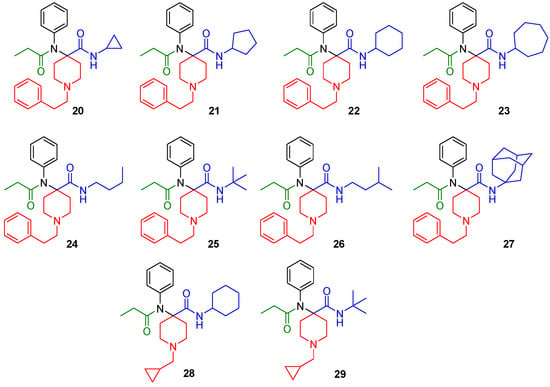
Figure 2.
Structures of the synthesized carfentanil amide analogs 20–29.
Figure 2.
Structures of the synthesized carfentanil amide analogs 20–29.
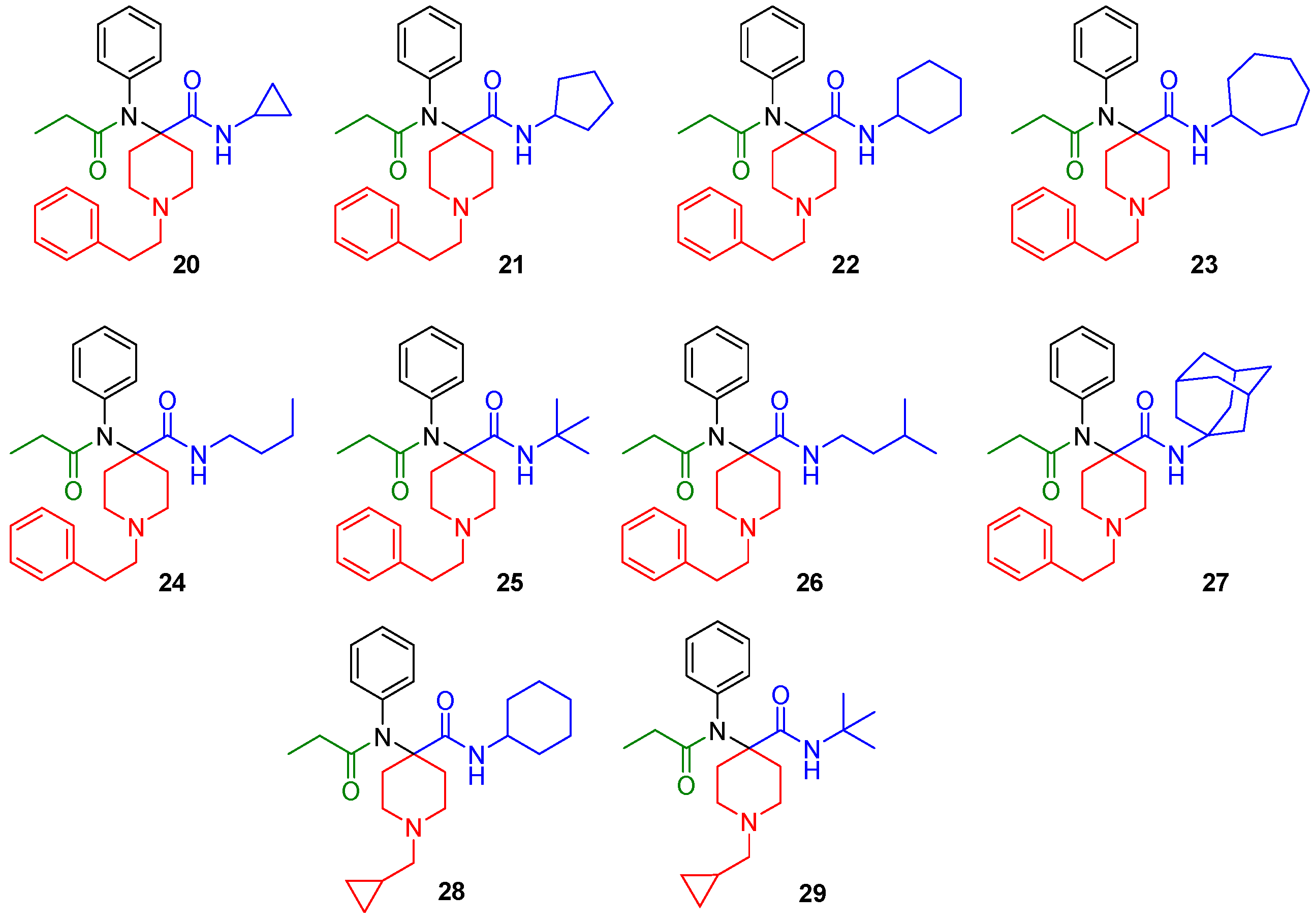
The synthesized compounds were characterized in in vitro radioligand binding assays in CHO cell lines stably transfected with murine MOR, DOR, and KOR (Table 1). Analogs with N-cyclopropylmethyl (28, 29) displayed low affinity for opioid receptors (Ki > 100 nM), whereas analogs with N-phenylethyl substituents (20–27) showed moderate to high affinity, particularly for MOR. Most analogs had low affinity (Ki > 100 nM) for KOR. Three compounds, 22 (cyclohexyl), 23 (cycloheptyl) and 27 (adamantyl) had DOR affinity of less than 10 nM. Given our hypothesis that MOR-DOR compounds may be useful for overcoming side effects, we studied the pharmacology of compounds with affinity at MOR and DOR. We selected 23 for detailed pharmacological evaluation because its affinity for DOR was the highest in the series.

Table 1.
Summary of in vitro receptor binding and in vivo tail-flick analgesia.
| Cmpd. | MOR-CHO | DOR-CHO | KOR-CHO | ED50 (mg/kg) |
|---|---|---|---|---|
| 20 | 10.3 ± 5.1 | >100 | 87.6 ± 29 | 0.78 ± 0.26 |
| 21 | 29.4 ± 15 | 90.7 ± 23 | >100 | 9.92 ± 0.08 |
| 22 | 0.84 ± 0.34 | 2.65 ± 0.32 | 0.44 ± 0.05 | 3.10 ± 0.19 |
| 23 | 2.66 ± 1.3 | 8.90 ± 7.7 | >100 | 10.0 ± 0.00 |
| 24 | 21.1 ± 11 | 87.9 ± 4.8 | >100 | >10 |
| 25 | 2.73 ± 2.2 | 71.2 ± 8.7 | >100 | 1.09 ± 0.05 |
| 26 | 27.0 ± 20 | 27.0 ± 3.6 | >100 | >10 |
| 27 | 25.0 ± 9.8 | 8.83 ± 0.63 | >100 | >10 |
| 28 | >100 | >100 | >100 | >10 |
| 29 | >100 | >100 | >100 | >10 |
The compounds were evaluated in in vivo antinociception assays in mice. Analogs 20–23 and 25 showed analgesia at the highest given dose of 10 mg/kg. Three compounds in the series (20, 22, 25) were more potent than morphine (ED50 ~5 mg/kg, sc) [85]. The analgesic ED50 values of 21 and our lead compound 23 (ED50 = 10 mg/kg, sc) was about two-fold lower than that of morphine (Table 1). Compound 23 was found to be a full agonist at MOR and a partial agonist at DOR in [35S]GTPγS binding assays.
We looked at the side-effect profile of 23 in mouse models of respiratory depression (RD) and physical dependence. At doses 4 × ED50 (40 mg/kg, sc), 23 did show some signs of RD. Repeated administration of traditional opioids leads to both tolerance and physical dependence, reducing the effectiveness of the drug over time and making it liable to cause addiction. Chronic dosing of 23 produced tolerance, however, 23-tolerant mice challenged with naloxone (a common opioid antagonist) demonstrated fewer withdrawal symptoms than morphine. Another serious side effect of MOR opioids is constipation. At doses 2× ED50 (20 mg/kg) and 5× ED50 (50 mg/kg), 23 showed no signs of inhibition of gastrointestinal motility, while morphine caused constipation at its ED50 dose. In summary, the cycloheptyl amide analog (23) of carfentanil may be useful in negating multiple major side effects seen with classic MOR analgesics. We hope to optimize the structure of this carfentanil amide scaffold to maintain receptor affinities and MOR agonism, while reducing the DOR efficacy to attain a MOR agonist/DOR antagonist based pharmacophore to address respiratory depression, which was observed with the current lead molecule. The utilization of Ugi chemistry to diversify the amine and carboxylic acid ends with commercially available reagents makes further derivatives readily accessible.
The presented examples underline the greatest advantage of multicomponent reactions: the possibility to create diverse bioactive compounds from simple building blocks in just a few or even only one step.
MCRs are an extremely useful and quickly evolving field of synthetic organic chemistry. In recent years, countless varieties of isocyanide-based MCRs have emerged, among which numerous intriguing reactions can be found that take advantage of nitrilium trapping, the nucleophilic attack on the extremely reactive nitrilium intermediate. These reactions have not only introduced us to novel chemical methodologies and reaction mechanisms, but also made new areas of chemical space easily accessible. Based on the diversity of structures that can be accessed by MCRs that involve nitrilium trapping, these methodologies will have a bright future in drug design and synthesis.
Acknowledgments
This work was supported by research grants from the National Institute on Drug Abuse (DA034106) to S.M. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748.
Author Contributions
SM conceived and designed the review article. AV, TP and RD did literature research on relevant articles for this review. AV, TP and SM wrote the review.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ugi, I.; Meyr, R.; Isonitrile, V. Erweiterter anwendungsbereich der passerini-reaktion. Chem. Ber. 1961, 94, 2229–2233. [Google Scholar] [CrossRef]
- Andreana, P.R.; Liu, C.C.; Schreiber, S.L. Stereochemical control of the passerini reaction. Org. Lett. 2004, 6, 4231–4233. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Fan, Y. Catalytic, enantioselective α-additions of isocyanides: Lewis base catalyzed passerini-type reactions. J. Org. Chem. 2005, 70, 9667–9676. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-X.; Wang, M.-X.; Wang, D.-X.; Zhu, J. Catalytic enantioselective passerini three-component reaction. Angew. Chem. Int. Ed. 2008, 47, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Brioche, J.; Masson, G.; Zhu, J. Passerini three-component reaction of alcohols under catalytic aerobic oxidative conditions. Org. Lett. 2010, 12, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Yanai, H.; Oguchi, T.; Taguchi, T. Direct alkylative passerini reaction of aldehydes, isocyanides, and free aliphatic alcohols catalyzed by indium(III) triflate. J. Org. Chem. 2009, 74, 3927–3929. [Google Scholar] [CrossRef] [PubMed]
- El Kaim, L.; Gizolme, M.; Grimaud, L. O-Arylative Passerini reactions. Org. Lett. 2006, 8, 5021–5023. [Google Scholar] [CrossRef] [PubMed]
- Sehlinger, A.; Kreye, O.; Meier, M.A.R. Tunable polymers obtained from Passerini multicomponent reaction derived acrylate monomers. Macromolecules 2013, 46, 6031–6037. [Google Scholar] [CrossRef]
- Paravidino, M.; Scheffelaar, R.; Schmitz, R.F.; de Kanter, F.J.J.; Groen, M.B.; Ruijter, E.; Orru, R.V.A. A flexible six-component reaction to access constrained depsipeptides based on a dihydropyridinone core. J. Org. Chem. 2007, 72, 10239–10242. [Google Scholar] [CrossRef] [PubMed]
- Jee, J.-A.; Song, S.; Rudick, J.G. Enhanced reactivity of dendrons in the Passerini three-component reaction. Chem. Commun. 2015, 51, 5456–5459. [Google Scholar] [CrossRef] [PubMed]
- Szymański, W.; Velema, W.A.; Feringa, B.L. Photocaging of carboxylic acids: A modular approach. Angew. Chem. Int. Ed. 2014, 53, 8682–8686. [Google Scholar] [CrossRef] [PubMed]
- Shaaban, S.; Diestel, R.; Hinkelmann, B.; Muthukumar, Y.; Verma, R.P.; Sasse, F.; Jacob, C. Novel peptidomimetic compounds containing redox active chalcogens and quinones as potential anticancer agents. Eur. J. Med. Chem. 2012, 58, 192–205. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, W.; Zwolinska, M.; Klossowski, S.; Młynarczuk-Biały, I.; Biały, Ł.; Issat, T.; Malejczyk, J.; Ostaszewski, R. Synthesis of novel, peptidic kinase inhibitors with cytostatic/cytotoxic activity. Bioorg. Med. Chem. 2014, 22, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Zha, Z.; Ploessl, K.; Lieberman, B.P.; Zhu, L.; Wise, D.R.; Thompson, C.B.; Kung, H.F. Synthesis of optically pure 4-fluoro-glutamines as potential metabolic imaging agents for tumors. J. Am. Chem. Soc. 2011, 133, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Velázquez, F.; Venkatraman, S.; Wu, W.; Blackman, M.; Prongay, A.; Girijavallabhan, V.; Shih, N.-Y.; Njoroge, F.G. Application of ring-closing metathesis for the synthesis of macrocyclic peptidomimetics as inhibitors of hcv ns3 protease. Org. Lett. 2007, 9, 3061–3064. [Google Scholar] [CrossRef] [PubMed]
- Soeta, T.; Kojima, Y.; Ukaji, Y.; Inomata, K. O-Silylative Passerini reaction: A new one-pot synthesis of α-siloxyamides. Org. Lett. 2010, 12, 4341–4343. [Google Scholar] [CrossRef] [PubMed]
- Soeta, T.; Matsuzaki, S.; Ukaji, Y. A one-pot o-sulfinative passerini/oxidation reaction: Synthesis of α-(sulfonyloxy)amide derivatives. J. Org. Chem. 2015, 80, 3688–3694. [Google Scholar] [CrossRef] [PubMed]
- Soeta, T.; Matsuzaki, S.; Ukaji, Y. A one-pot o-phosphinative passerini/pudovik reaction: Efficient synthesis of highly functionalized α-(phosphinyloxy)amide derivatives. Chem. Eur. J. 2014, 20, 5007–5012. [Google Scholar] [CrossRef] [PubMed]
- Sunderhaus, J.D.; Martin, S.F. Applications of multicomponent reactions to the synthesis of diverse heterocyclic scaffolds. Chem.-Eur. J. 2009, 15, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Paulvannan, K. Preparation of tricyclic nitrogen heterocycles via tandem four-component condensation/intramolecular Diels-Alder reaction. Tetrahedron Lett. 1999, 40, 1851–1854. [Google Scholar] [CrossRef]
- Koopmanschap, G.; Ruijter, E.; Orru, R.V.A. Isocyanide-based multicomponent reactions towards cyclic constrained peptidomimetics. Beilstein J. Org. Chem. 2014, 10, 544–598. [Google Scholar] [CrossRef] [PubMed]
- Bonnaterre, F.; Bois-Choussy, M.; Zhu, J. Rapid access to oxindoles by the combined use of an Ugi four-component reaction and a microwave-assisted intramolecular buchwald−hartwig amidation reaction. Org. Lett. 2006, 8, 4351–4354. [Google Scholar] [CrossRef] [PubMed]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Godet, T.; Bonvin, Y.; Vincent, G.; Merle, D.; Thozet, A.; Ciufolini, M.A. Titanium catalysis in the Ugi reaction of α-amino acids with aromatic aldehydes. Org. Lett. 2004, 6, 3281–3284. [Google Scholar] [CrossRef] [PubMed]
- Okandeji, B.O.; Gordon, J.R.; Sello, J.K. Catalysis of Ugi four component coupling reactions by rare earth metal triflates. J. Org. Chem. 2008, 73, 5595–5597. [Google Scholar] [CrossRef] [PubMed]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef] [PubMed]
- Shaabani, A.; Keshipour, S.; Shaabani, S.; Mahyari, M. Zinc chloride catalyzed three-component Ugi reaction: Synthesis of n-cyclohexyl-2-(2-hydroxyphenylamino)acetamide derivatives. Tetrahedron Lett. 2012, 53, 1641–1644. [Google Scholar] [CrossRef]
- Pan, S.C.; List, B. Catalytic three-component Ugi reaction. Angew. Chem. Int. Ed. 2008, 47, 3622–3625. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, E.; Scheffelaar, R.; Orru, R.V.A. Multicomponent reaction design in the quest for molecular complexity and diversity. Angew. Chem. Int. Ed. 2011, 50, 6234–6246. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.T.; R, S.B.; Lal, M.; Mir, M.H. Formation of unexpected [small alpha]-amino amidine through three-component “Ugi condensation reaction”. RSC Adv. 2012, 2, 5506–5509. [Google Scholar] [CrossRef]
- El Kaïm, L.; Grimaud, L.; Oble, J. Phenol Ugi–smiles systems: Strategies for the multicomponent n-arylation of primary amines with isocyanides, aldehydes, and phenols. Angew. Chem. Int. Ed. 2005, 44, 7961–7964. [Google Scholar] [CrossRef] [PubMed]
- El Kaim, L.; Gizolme, M.; Grimaud, L.; Oble, J. Direct access to heterocyclic scaffolds by new multicomponent Ugi−Smiles couplings. Org. Lett. 2006, 8, 4019–4021. [Google Scholar] [CrossRef] [PubMed]
- Dömling, A.; Ugi, I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Hulme, C.; Gore, V. “Multi-component reactions: Emerging chemistry in drug discovery” “from xylocain to crixivan”. Curr. Med. Chem. 2003, 10, 51–80. [Google Scholar] [CrossRef] [PubMed]
- Faggi, C.; García-Valverde, M.A.; Marcaccini, S.; Menchi, G. Isolation of Ugi four-component condensation primary adducts: A straightforward route to isocoumarins. Org. Lett. 2010, 12, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Bonne, D.; Dekhane, M.; Zhu, J. Exploiting the dual reactivity of o-isocyanobenzamide: Three-component synthesis of 4-imino-4H-3,1-benzoxazines. Org. Lett. 2005, 7, 5285–5288. [Google Scholar] [CrossRef] [PubMed]
- Groebke, K.; Weber, L.; Mehlin, F. Synthesis of imidazo[1,2-a] annulated pyridines, pyrazines and pyrimidines by a novel three-component condensation. Synlett 1998, 6, 661–663. [Google Scholar] [CrossRef]
- Blackburn, C.; Guan, B.; Fleming, P.; Shiosaki, K.; Tsai, S. Parallel synthesis of 3-aminoimidazo[1,2-a]pyridines and pyrazines by a new three-component condensation. Tetrahedron Lett. 1998, 39, 3635–3638. [Google Scholar] [CrossRef]
- Bienaymé, H.; Bouzid, K. A new heterocyclic multicomponent reaction for the combinatorial synthesis of fused 3-aminoimidazoles. Angew. Chem. Int. Ed. 1998, 37, 2234–2237. [Google Scholar] [CrossRef]
- Hulme, C.; Lee, Y.-S. Emerging approaches for the syntheses of bicyclic imidazo[1,2-x]-heterocycles. Mol. Divers. 2008, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Devi, N.; Rawal, R.K.; Singh, V. Diversity-oriented synthesis of fused-imidazole derivatives via Groebke–Blackburn–Bienayme reaction: A review. Tetrahedron 2015, 71, 183–232. [Google Scholar] [CrossRef]
- Akritopoulou-Zanze, I.; Wakefield, B.D.; Gasiecki, A.; Kalvin, D.; Johnson, E.F.; Kovar, P.; Djuric, S.W. Scaffold oriented synthesis. Part 4: Design, synthesis and biological evaluation of novel 5-substituted indazoles as potent and selective kinase inhibitors employing heterocycle forming and multicomponent reactions. Bioorg. Med. Chem. Lett. 2011, 21, 1480–1483. [Google Scholar] [CrossRef] [PubMed]
- Baviskar, A.T.; Madaan, C.; Preet, R.; Mohapatra, P.; Jain, V.; Agarwal, A.; Guchhait, S.K.; Kundu, C.N.; Banerjee, U.C.; Bharatam, P.V. N-Fused imidazoles as novel anticancer agents that inhibit catalytic activity of topoisomerase IIα and induce apoptosis in g1/s phase. J. Med. Chem. 2011, 54, 5013–5030. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.M.; Salunke, D.B.; Yoo, E.; Mutz, C.A.; Balakrishna, R.; David, S.A. Antibacterial activities of groebke-blackburn-bienayme-derived imidazo[1,2-a]pyridin-3-amines. Bioorg. Med. Chem. Lett. 2012, 20, 5850–5863. [Google Scholar] [CrossRef] [PubMed]
- Burchak, O.N.; Mugherli, L.; Ostuni, M.; Lacapère, J.J.; Balakirev, M.Y. Combinatorial discovery of fluorescent pharmacophores by multicomponent reactions in droplet arrays. J. Am. Chem. Soc. 2011, 133, 10058–10061. [Google Scholar] [CrossRef] [PubMed]
- Elleder, D.; Baiga, T.; Russell, R.; Naughton, J.; Hughes, S.; Noel, J.; Young, J. Identification of a 3-aminoimidazo[1,2-a]pyridine inhibitor of hiv-1 reverse transcriptase. Virol. J. 2012, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Bode, M.L.; Gravestock, D.; Moleele, S.S.; van der Westhuyzen, C.W.; Pelly, S.C.; Steenkamp, P.A.; Hoppe, H.C.; Khan, T.; Nkabinde, L.A. Imidazo[1,2-a]pyridin-3-amines as potential HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2011, 19, 4227–4237. [Google Scholar] [CrossRef] [PubMed]
- Ganem, B. Strategies for innovation in multicomponent reaction design. Acc. Chem. Res. 2009, 42, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Shaabani, A.; Maleki, A.; Mofakham, H.; Khavasi, H.R. Novel isocyanide-based three-component synthesis of 3,4-dihydroquinoxalin-2-amine derivatives. J. Comb. Chem. 2008, 10, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Soeta, T.; Tamura, K.; Ukaji, Y. [5 + 1] cycloaddition of c,n-cyclic n′-acyl azomethine imines with isocyanides. Org. Lett. 2012, 14, 1226–1229. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.-H.; Wang, D.-X.; Zhao, L.; Zhu, J.; Wang, M.-X. Synthesis of substituted pyridines from cascade [1 + 5] cycloaddition of isonitriles to n-formylmethyl-substituted enamides, aerobic oxidative aromatization, and acyl transfer reaction. J. Amer. Chem. Soc. 2013, 135, 4708–4711. [Google Scholar] [CrossRef] [PubMed]
- Shutske, G.M.; Kapples, K.J.; Tomer, J.D.; Hrib, N.J.; Jurcak, J.J. Substituted-4-amino-3-pyridinols, a Process for Their Preparation and Their Use as Medicaments. EP0477903 A2, 1 April 1991. [Google Scholar]
- Hashimoto, T.; Kimura, H.; Kawamata, Y.; Maruoka, K. A catalytic asymmetric Ugi-type reaction with acyclic azomethine imines. Angew. Chem. Int. Ed. Engl. 2012, 51, 7279–7281. [Google Scholar] [CrossRef] [PubMed]
- Pirrung, M.C.; Ghorai, S. Versatile, fragrant, convertible isonitriles. J. Am. Chem. Soc. 2006, 128, 11772–11773. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Schneekloth, J.S.; Sorensen, E.J. A chemical synthesis of 11-methoxy mitragynine pseudoindoxyl featuring the interrupted ugi reaction. Chem. Sci. 2012, 3, 2849–2852. [Google Scholar] [CrossRef] [PubMed]
- Schneekloth, J.S.; Jimin, K., Jr.; Sorensen, E.J. An interrupted Ugi reaction enables the preparation of substituted indoxyls and aminoindoles. Tetrahedron 2009, 65, 3096–3101. [Google Scholar] [CrossRef] [PubMed]
- Kysil, V.; Tkachenko, S.; Khvat, A.; Williams, C.; Tsirulnikov, S.; Churakova, M.; Ivachtchenko, A. Tmscl-promoted isocyanide-based mcr of ethylenediamines: An efficient assembling of 2-aminopyrazine core. Tetrahedron Lett. 2007, 48, 6239–6244. [Google Scholar] [CrossRef]
- Xia, L.; Li, S.; Chen, R.; Liu, K.; Chen, X. Catalytic ugi-type condensation of α-isocyanoacetamide and chiral cyclic imine: Access to asymmetric construction of several heterocycles. J. Org. Chem. 2013, 78, 3120–3131. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharappa, A.P.; Badiger, S.E.; Dubey, P.K.; Panigrahi, S.K.; Manukonda, S.R. Design and synthesis of 2-substituted benzoxazoles as novel ptp1b inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2579–2584. [Google Scholar] [CrossRef] [PubMed]
- Ulhaq, S.; Chinje, E.C.; Naylor, M.A.; Jaffar, M.; Stratford, I.J.; Threadgill, M.D. Heterocyclic analogues of l-citrulline as inhibitors of the isoforms of nitric oxide synthase (nos) and identification of nδ-(4,5-dihydrothiazol-2-yl)ornithine as a potent inhibitor. Bioorg. Med. Chem. Lett. 1999, 7, 1787–1796. [Google Scholar] [CrossRef]
- Horton, D.A.; Bourne, G.T.; Smythe, M.L. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem. Rev. 2003, 103, 893–930. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Hirao, A.; Imai, A.; Nakamura, H.; Murata, Y.; Ohashi, K.; Nakata, E. Novel non-peptide nociceptin/orphanin fq receptor agonist, 1-[1-(1-methylcyclooctyl)-4-piperidinyl]-2-[(3R)-3-piperidinyl]-1H-benzimidazole: Design, synthesis, and structure−activity relationship of oral receptor occupancy in the brain for orally potent antianxiety drug(1, 2). J. Med. Chem. 2009, 52, 610–625. [Google Scholar] [PubMed]
- Siracusa, M.A.; Salerno, L.; Modica, M.N.; Pittalà, V.; Romeo, G.; Amato, M.E.; Nowak, M.; Bojarski, A.J.; Mereghetti, I.; Cagnotto, A.; et al. Synthesis of new arylpiperazinylalkylthiobenzimidazole, benzothiazole, or benzoxazole derivatives as potent and selective 5-ht1a serotonin receptor ligands†. J. Med. Chem. 2008, 51, 4529–4538. [Google Scholar] [CrossRef] [PubMed]
- Varadi, A.; Palmer, T.C.; Notis, P.R.; Redel-Traub, G.N.; Afonin, D.; Subrath, J.J.; Pasternak, G.W.; Hu, C.; Sharma, I.; Majumdar, S. Three-component coupling approach for the synthesis of diverse heterocycles utilizing reactive nitrilium trapping. Org. Lett. 2014, 16, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.V. Synthetic strategies towards benzoxazole ring systems: A review. Asian J. Chem. 2004, 16, 1241–1260. [Google Scholar] [CrossRef]
- Tempest, P.; Ma, V.; Thomas, S.; Hua, Z.; Kelly, M.G.; Hulme, C. Two-step solution-phase synthesis of novel benzimidazoles utilizing a udc (Ugi/de-Boc/cyclize) strategy. Tetrahedron Lett. 2001, 42, 4959–4962. [Google Scholar] [CrossRef]
- Boissarie, P.J.; Hamilton, Z.E.; Lang, S.; Murphy, J.A.; Suckling, C.J. A powerful palladium-catalyzed multicomponent process for the preparation of oxazolines and benzoxazoles. Org. Lett. 2011, 13, 6256–6259. [Google Scholar] [CrossRef] [PubMed]
- Spatz, J.H.; Bach, T.; Umkehrer, M.; Bardin, J.; Ross, G.; Burdack, C.; Kolb, J. Diversity oriented synthesis of benzoxazoles and benzothiazoles. Tetrahedron Lett. 2007, 48, 9030–9034. [Google Scholar] [CrossRef]
- El Kaim, L.; Grimaud, L. Beyond the ugi reaction: Less conventional interactions between isocyanides and iminium species. Tetrahedron 2009, 65, 2153–2171. [Google Scholar] [CrossRef]
- Vlaar, T.; Ruijter, E.; Maes, B.U.W.; Orru, R.V.A. Palladium-catalyzed migratory insertion of isocyanides: An emerging platform in cross-coupling chemistry. Angew. Chem. Int. Ed. 2013, 52, 7084–7097. [Google Scholar] [CrossRef] [PubMed]
- Mosberg, H.I.; Yeomans, L.; Anand, J.P.; Porter, V.; Sobczyk-Kojiro, K.; Traynor, J.R.; Jutkiewicz, E.M. Development of a bioavailable mu opioid receptor (mopr) agonist, delta opioid receptor (dopr) antagonist peptide that evokes antinociception without development of acute tolerance. J. Med. Chem. 2014, 57, 3148–3153. [Google Scholar] [CrossRef] [PubMed]
- Ananthan, S. Opioid ligands with mixed mu/delta opioid receptor interactions: An emerging approach to novel analgesics. AAPS J. 2006, 8, E118–E125. [Google Scholar] [CrossRef] [PubMed]
- Ananthan, S.; Kezar, H.S.; Carter, R.L.; Saini, S.K.; Rice, K.C.; Wells, J.L.; Davis, P.; Xu, H.; Dersch, C.M.; Bilsky, E.J.; et al. Synthesis, opioid receptor binding, and biological activities of naltrexone-derived pyrido- and pyrimidomorphinans. J. Med. Chem. 1999, 42, 3527–3538. [Google Scholar] [CrossRef] [PubMed]
- Schiller, P.W.; Fundytus, M.E.; Merovitz, L.; Weltrowska, G.; Nguyen, T.M.D.; Lemieux, C.; Chung, N.N.; Coderre, T.J. The opioid μ agonist/δ antagonist dipp-nh2[ψ] produces a potent analgesic effect, no physical dependence, and less tolerance than morphine in rats. J. Med. Chem. 1999, 42, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Purington, L.C.; Pogozheva, I.D.; Traynor, J.R.; Mosberg, H.I. Pentapeptides displaying mu opioid receptor agonist and delta opioid receptor partial agonist/antagonist properties. J. Med. Chem. 2009, 52, 7724–7731. [Google Scholar] [CrossRef] [PubMed]
- Lowery, J.J.; Raymond, T.J.; Giuvelis, D.; Bidlack, J.M.; Polt, R.; Bilsky, E.J. In vivo characterization of mmp-2200, a mixed delta/mu opioid agonist, in mice. J. Pharmacol. Exp. Ther. 2011, 336, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo, G.P.; Polt, R.; Bilsky, E.J.; Rice, K.C.; Negus, S.S. Behavioral pharmacology of the μ/δ opioid glycopeptide mmp2200 in rhesus monkeys. J. Pharmacol. Exp. Ther. 2008, 326, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Maguire, P.; Tsai, N.; Kamal, J.; Cometta-Morini, C.; Upton, C.; Loew, G. Pharmacological profiles of fentanyl analogs at mu, delta and kappa opiate receptors. Eur. J. Pharmacol. 1992, 213, 219–225. [Google Scholar] [CrossRef]
- Malaquin, S.; Jida, M.; Gesquiere, J.-C.; Deprez-Poulain, R.; Deprez, B.; Laconde, G. Ugi reaction for the synthesis of 4-aminopiperidine-4-carboxylic acid derivatives. Application to the synthesis of carfentanil and remifentanil. Tetrahedron Lett. 2010, 51, 2983–2985. [Google Scholar] [CrossRef]
- Pentel, P.R.; Portoghese, P.S.; Pravetoni, M.; Naour, M.C.P.L. Compositions and Methods of Treating Opioid Addiction. US20140093525 A1, 3 April 2014. [Google Scholar]
- Portoghese, P.; Eyup, A. Analgesic Conjugates. WO2014124317 A1, 14 August 2014. [Google Scholar]
- Váradi, A.; Palmer, T.C.; Haselton, N.; Afonin, D.; Subrath, J.J.; le Rouzic, V.; Hunkele, A.; Pasternak, G.W.; Marrone, G.F.; Borics, A.; et al. Synthesis of carfentanil amide opioids using the ugi multicomponent reaction. ACS Chem. Neurosci. 2015, 6, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Srulevitch, D.B.; Lien, E.J. Design, synthesis and sar of analgesics. Prog. Clin. Biol. Res. 1989, 291, 377–381. [Google Scholar] [PubMed]
- Srulevitch, D.B.; Lien, E.J. 4-phenylamidopiperidines: Synthesis, pharmacological testing and sar analysis. Acta Pharm. Jugosl. 1991, 41, 89–106. [Google Scholar]
- Kolesnikov, Y.A.; Pick, C.G.; Ciszewska, G.; Pasternak, G.W. Blockade of tolerance to morphine but not to kappa opioids by a nitric oxide synthase inhibitor. Proc. Nat. Acad. Sci. USA 1993, 90, 5162–5166. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).