
Phylogenetic and Diversity Analysis of Dactylis glomerata Subspecies Using SSR and IT-ISJ Markers



Abstract
:1. Introduction
2. Results
2.1. Polymorphism and Marker Efficiency Analysis of SSR and IT-ISJ Markers
2.2. Genetic Diversity and AMOVA Analysis of Dactylis Subspecies
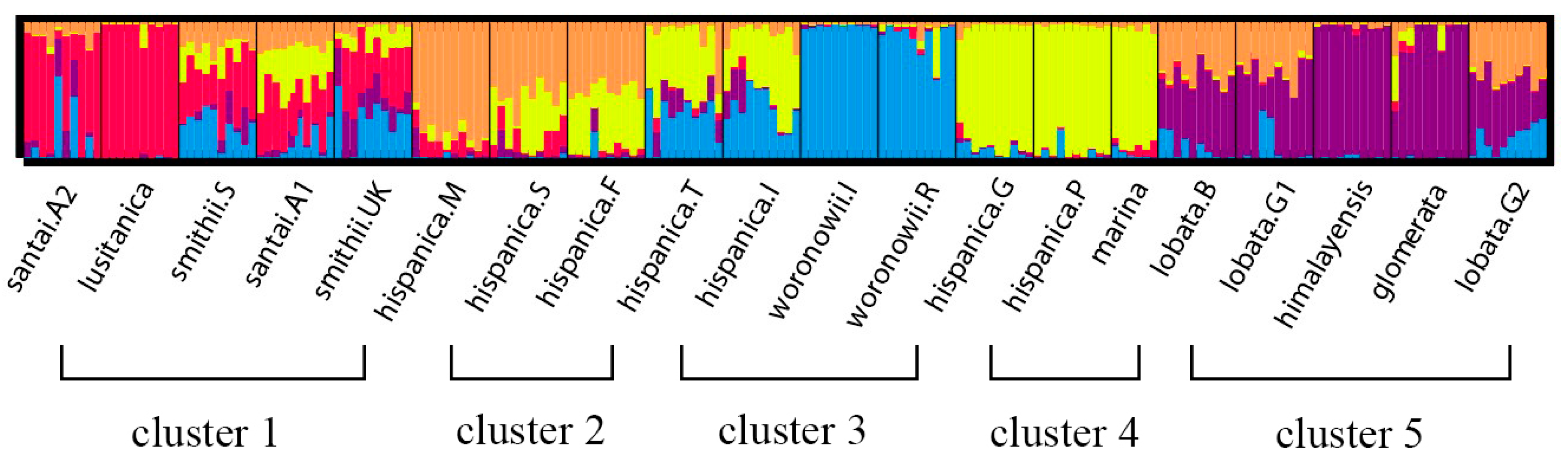
2.3. Phylogenetic Analysis
3. Discussion
3.1. Marker Efficiency Analysis
3.2. Genetic Diversity Analysis
3.3. Phylogenetic Analysis
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Extraction
4.3. Primer Selection and PCR Amplification
4.4. Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Watson, L.; Clifford, H.T.; Dallwitz, M.J. The classification of Poaceae: Subfamilies and supertribes. Aust. J. Bot. 1985, 33, 433–484. [Google Scholar] [CrossRef]
- Watson, L.; Dallwitz, M.J.; Johnston, C. Grass genera of the world: 728 detailed descriptions from an automated database. Aust. J. Bot. 1986, 34, 223–230. [Google Scholar] [CrossRef]
- Sanada, Y.; Gras M, C.; van Santen, E. Cocksfoot. In Fodder Crops and Amenity Grasses, 1st ed.; Boller, B., Posselt, U.K., Veronesi, F., Eds.; Springer: New York, NY, USA, 2010; pp. 317–328. [Google Scholar]
- Stewart, A.V.; Ellison, N.W. A molecular phylogenetic framework for cocksfoot (Dactylis glomerata L.) improvement. Crop Pasture Sci. 2014, 65, 780. [Google Scholar] [CrossRef]
- Stebbins, G.L.; Zohary, D. Cytogenetic and Evolutionary Studies in the Genus Dactylis: I: Morphology, Distribution, and Interrelationships of the Diploid Subspecies; University of California Press: Oakland, CA, USA, 1959. [Google Scholar]
- Beddows, A.R. Dactylis glomerata L. J. Ecol. 1959, 47, 223–239. [Google Scholar] [CrossRef]
- Xie, W.-G.; Zhang, X.-Q.; Cai, H.-W.; Liu, W.; Peng, Y. Genetic diversity analysis and transferability of cereal EST-SSR markers to orchardgrass (Dactylis glomerata L.). Biochem. Syst. Ecol. 2010, 38, 740–749. [Google Scholar] [CrossRef]
- Stewart, A.V.; Ellison, N. Origins of Diploid Dactylis from China as Determined by ITS Sequences. In Molecular Breeding of Forage and Turf, 1st ed.; Budak, H., Spangenberg, G., Eds.; Springer: Cham, Switzerland, 2015; pp. 165–171. [Google Scholar]
- Borrill, M.; Carroll, C. A chromosome atlas of the genus Dactylis (Part two). Cytologia 1969, 34, 6–17. [Google Scholar] [CrossRef]
- Lumaret, R.; Borrill, M. sCytology, genetics, and evolution in the genus Dactylis. Crit. Rev. Plant Sci. 1988, 7, 55–91. [Google Scholar] [CrossRef]
- Borrill, M. The pattern of morphological variation in diploid and tetraploid Dactylis. J. Linn. Soc. Lond. Bot. 1961, 56, 441–452. [Google Scholar] [CrossRef]
- Jones, K.; Borrill, M. Chromosomal status, gene exchange and evolution in Dactylis. Genetica 1962, 32, 296–322. [Google Scholar] [CrossRef]
- Jones, K.; Carroll, C.P.; Borrill, M. A chromosome atlas of the genus Dactylis L. Cytologia 1961, 26, 333–343. [Google Scholar] [CrossRef]
- Borrill, M.; Jones, K. Hexaploid Dactylis. Nature 1961, 190, 469–470. [Google Scholar] [CrossRef]
- Levan, A.V. Beitrag zur Kenntnis der Chromosomen in der Gattung Dactylis L. Bot. Not. 1930, 1930, 95–104. [Google Scholar]
- Kattermann, G. Chromosomenuntersuchungen bei Gramineen. Planta 1930, 12, 19–37. [Google Scholar] [CrossRef]
- Müntzing, A. Quadrivalent formation and aneuploidy in Dactylis glomerata. Bot Not. 1933, 84, 198–205. [Google Scholar]
- Müntzing, A. The effects of chromosomal variation in Dactylis. Hereditas 1937, 23, 113–235. [Google Scholar] [CrossRef]
- Borrill, M. Dactylis marina, Borrill, sp. nov. a natural group of related tetraploid forms. J. Linn. Soc. Lond. Bot. 1961, 56, 431–439. [Google Scholar] [CrossRef]
- Borrill, M. Chromosomal status, gene exchange and evolution in Dactylis. Genetica 1962, 32, 94–117. [Google Scholar] [CrossRef]
- Fiasson, J.; Ardouin, P.; Jay, M. A Phylogenetic Groundplan the Specific Complex Dactylis glomerara. Biochem. Syst. Ecol. 1987, 15, 225–229. [Google Scholar] [CrossRef]
- Stewart, A.V.; Ellison, N.W. Dactylis. In Wild Crop Relatives: Genomic and Breeding Resources, 1st ed.; Kole, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 73–87. [Google Scholar]
- Hamada, H.; Petrino, M.G.; Kakunaga, T. A novel repeated element with Z-DNA-forming potential is widely found in evolutionarily diverse eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1982, 79, 6465–6469. [Google Scholar] [CrossRef] [PubMed]
- Bowcock, A.; Ruiz-Linares, A.; Tomfohrde, J.; Minch, E.; Kidd, J.; Cavalli-Sforza, L.L. High resolution of human evolutionary trees with polymorphic microsatellites. Nature 1994, 368, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-S.; Tanksley, S.D. Abundance, polymorphism and genetic mapping of microsatellites in rice. Mol. Gen. Genet. 1993, 241, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Panaud, O.; Chen, X.; McCouch, S. Development of microsatellite markers and characterization of simple sequence length polymorphism (SSLP) in rice (Oryza sativa L.). Mol. Gen. Genet. 1996, 252, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Beckmann, J.; Soller, M. Toward a unified approach to genetic mapping of eukaryotes based on sequence tagged microsatellite sites. Biotechnology 1990, 8, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.M.; Hopkins, M.S.; Mitchell, S.E.; Senior, M.L.; Wang, T.Y.; Duncan, R.R.; Gonzalez-Candelas, F.; Kresovich, S. Multiple methods for the identification of polymorphic simple sequence repeats (SSRs) in sorghum [Sorghum bicolor (L.) Moench]. Theor. Appl. Genet. 1996, 93, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.; Yang, Y.; Yang, X.; Li, X.; Li, L. Phylogenetic relationship and diversity among Agropyron Gaertn. germplasm using SSRs markers. Plant Syst. Evol. 2015, 301, 163–170. [Google Scholar] [CrossRef]
- Nishikawa, T.; Vaughan, D.A.; Kadowaki, K.-I. Phylogenetic analysis of Oryza species, based on simple sequence repeats and their flanking nucleotide sequences from the mitochondrial and chloroplast genomes. Theor. Appl. Genet. 2005, 110, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Q.; Zhang, T.; Wang, B.; Li, J. Diversity comparison and phylogenetic relationships of S. bicolor and S. sundanense as revealed by SSR markers. Plant Sci. 2008, 174, 9–16. [Google Scholar] [CrossRef]
- Wu, X.; He, C.; Chen, S.; Zhuang, B.; Wang, K.; Wang, X. [Phylogenetic analysis of interspecies in genus Glycine through SSR markers]. Yi Chuan Xue Bao 2000, 28, 359–366. [Google Scholar]
- Sethy, N.K.; Choudhary, S.; Shokeen, B.; Bhatia, S. Identification of microsatellite markers from Cicer reticulatum: Molecular variation and phylogenetic analysis. Theor. Appl. Genet. 2006, 112, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, L.; Zhang, X.; Wang, J.; Yan, D.; Ji, L.; Tang, L.; Li, X.; Shi, T. Construction of high-density genetic linkage map and identification of flowering-time QTLs in orchardgrass using SSRs and SLAF-seq. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Lu, X.; Zhang, X.; Huang, L.; Cheng, L. Genetic variation and comparison of orchardgrass (Dactylis glomerata L.) cultivars and wild accessions as revealed by SSR markers. Genet. Mol. Res. 2012, 11, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.G.; Zhang, X.Q.; Cheng, Y.X. Identification and genetic variation analysis of orchardgrass hybrids (Dactylis glomerata) by SSR molecular markers. Acta Prataculturae Sin. 2010, 19, 212–217. [Google Scholar]
- Zheng, J.; Zhang, Z.; Chen, L.; Wan, Q.; Hu, M.; Wang, W.; Zhang, K.; Liu, D.; Chen, X.; Wei, X. Intron-targeted intron-exon splice conjunction (IT-ISJ) marker and its application in construction of upland cotton linkage map. Agric. Sci. Chin. 2008, 7, 1172–1180. [Google Scholar] [CrossRef]
- Weining, S.; Langridge, P. Identification and mapping of polymorphisms in cereals based on the polymerase chain reaction. Theor. Appl. Genet. 1991, 82, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Chabane, K.; Hendre, P.S.; Aggarwal, R.K.; Graner, A. Comparative assessment of EST-SSR, EST-SNP and AFLP markers for evaluation of genetic diversity and conservation of genetic resources using wild, cultivated and elite barleys. Plant Sci. 2007, 173, 638–649. [Google Scholar] [CrossRef]
- Powell, W.; Morgante, M.; Andre, C.; Hanafey, M.; Vogel, J.; Tingey, S.; Rafalki, A. The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol. Breed. 1996, 2, 225–238. [Google Scholar] [CrossRef]
- Gupta, P.; Balyan, H.; Edwards, K.; Isaac, P.; Korzun, V.; Röder, M.; Gautier, M.-F.; Joudrier, P.; Schlatter, A.; Dubcovsky, J.; et al. Genetic mapping of 66 new microsatellite (SSR) loci in bread wheat. Theor. Appl. Genet. 2002, 105, 413–422. [Google Scholar] [PubMed]
- Song, Q.; Marek, L.; Shoemaker, R.; Lark, K.; Concibido, V.; Delannay, X.; Specht, J.; Cregan, P. A new integrated genetic linkage map of the soybean. Theor. Appl. Genet. 2004, 109, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Agrama, H.; Tuinstra, M. Phylogenetic diversity and relationships among sorghum accessions using SSRs and RAPDs. Afr. J. Biotechnol. 2004, 2, 334–340. [Google Scholar] [CrossRef]
- Hokanson, S.; Szewc-McFadden, A.; Lamboy, W.; McFerson, J. Microsatellite (SSR) markers reveal genetic identities, genetic diversity and relationships in a Malus × domestica Borkh. core subset collection. Theor. Appl. Genet. 1998, 97, 671–683. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kimura, T.; Sawamura, Y.; Kotobuki, K.; Ban, Y.; Hayashi, T.; Matsuta, N. SSRs isolated from apple can identify polymorphism and genetic diversity in pear. Theor. Appl. Genet. 2001, 102, 865–870. [Google Scholar] [CrossRef]
- Vaiman, D.; Mercier, D.; Moazami-Goudarzi, K.; Eggen, A.; Ciampolini, R.; Lépingle, A.; Velmala, R.; Kaukinen, J.; Varvio, S.; Martin, P.; et al. A set of 99 cattle microsatellites: Characterization, synteny mapping, and polymorphism. Mamm. Genome 1994, 5, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhang, Y.; Zeng, B.; Yin, G.; Zhang, X.; Ji, Y.; Huang, L.; Jiang, X.; Liu, X.; Peng, Y.; Zeng, B. Genetic Diversity and Association of EST-SSR and SCoT Markers with Rust Traits in Orchardgrass (Dactylis glomerata L.). Molecules 2016, 21, 66. [Google Scholar] [CrossRef] [PubMed]
- Nevo, E. Genetic diversity inn wild cereals: Regional and local studies and their bearing on conservation in situ. Genet. Resour. Crop Evol. 1998, 45, 355–370. [Google Scholar] [CrossRef]
- Jalut, G.; Amat, A.E.; Bonnet, L.; Gauquenlin, T.; Fontugne, M. Holocene climatic changes in the Western Mediterranean, from south-east France to south-east Spain. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2000, 160, 255–290. [Google Scholar] [CrossRef]
- Amirouche, N.; Misset M, T. Morphological variation and distribution of cytotypes in the diploid-tetraploid complex of the genus Dactylis L. (Poaceae) from Algeria. Plant Syst. Evol. 2007, 264, 157–174. [Google Scholar] [CrossRef]
- Doyle, J. DNA protocols for plants. In Molecular Techniques in Taxonomy; Godfrey, M., Andrew, W.B., Young, J., Perter, W., Eds.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 283–293. [Google Scholar]
- Huang, L.; Yan, H.; Zhao, X.; Zhang, X.; Wang, J.; Frazier, T.; Yin, G.; Huang, X.; Yan, D.; Zhang, W.; et al. Identifying differentially expressed genes under heat stress and developing molecular markers in orchardgrass (Dactylis glomerata L.) through transcriptome analysis. Mol. Ecol. Res. 2015, 15, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Liu, F.; Zhu, Z.; Tan, L.; Fu, Y.; Sun, C.; Cai, H. Construction of a simple sequence repeat marker-based genetic linkage map in the autotetraploid forage grass Dactylis glomerata L. Grassl. Sci. 2011, 57, 158–167. [Google Scholar] [CrossRef]
- Xie, W.-G.; Zhang, X.-Q.; Ma, X.; Cai, H.-W.; Huang, L.-K.; Peng, Y.; Zeng, B. Diversity comparison and phylogenetic relationships of cocksfoot (Dactylis glomerata L.) germplasm as revealed by SSR markers. Can. J. Plant Sci. 2010, 90, 13–21. [Google Scholar] [CrossRef]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef] [PubMed]
- Archak, S.; Gaikwad, A.B.; Gautam, D.; Rao, E.V.; Swamy, K.R.; Karihaloo, J.L. Comparative assessment of DNA fingerprinting techniques (RAPD, ISSR, AFLP) for genetic analysis of cashew (Anacardium occidentale L.) accessions of India. Genome 2003, 46, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Lewontin, R.C. Testing the theory of natural selection. Nature 1972, 236, 181–182. [Google Scholar]
- Zhang, F. DCFA 1.1, a Program Companied with AMOVA to Compute the Matrix of Distance; Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Science: Beijing, China, 2001. [Google Scholar]
- Dice, L.R. Measures of the amount of ecologic association between species. Ecology 1945, 26, 297–302. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-pc: Numerical taxonomy and multivariate analysis system. Applied Biostatistics, 1992. [Google Scholar]
- Perrier, X.; Jacquemoud-Collet, J.P. DARwin Software. Available online: http://darwin.cirad.fr/ (accessed on 27 October 2016).
- Pritchard, J.K.; Stephens, M.; Rosenberg N, A.; Donnelly, P. Association mapping in structured populations. Am. J. Hum. Genet. 2000, 67, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.



{kind=link}
{kind=link}
| Primer Code | TNB | NPB | PPB (%) | PIC | Primer Code | TNB | NPB | PPB (%) | PIC |
|---|---|---|---|---|---|---|---|---|---|
| FOG215 | 11 | 11 | 100 | 0.916 | IT-ISJ01F03R | 8 | 8 | 100 | 0.616 |
| FOG238 | 9 | 9 | 100 | 0.768 | IT-ISJ01F04R | 8 | 8 | 100 | 0.874 |
| FOG258 | 13 | 13 | 100 | 0.909 | IT-ISJ01F05R | 5 | 5 | 100 | 0.957 |
| FOG296 | 6 | 6 | 100 | 0.869 | IT-ISJ01F08R | 6 | 6 | 100 | 0.710 |
| FOG362 | 10 | 10 | 100 | 0.962 | IT-ISJ03F41R | 9 | 9 | 100 | 0.833 |
| FOG402 | 8 | 8 | 100 | 0.861 | IT-ISJ03F47R | 7 | 7 | 100 | 0.741 |
| FOG514 | 11 | 11 | 100 | 0.959 | IT-ISJ03F59R | 10 | 10 | 100 | 0.728 |
| FOG515 | 10 | 10 | 100 | 0.940 | IT-ISJ04F04R | 6 | 6 | 100 | 0.626 |
| FOG537 | 7 | 7 | 100 | 0.938 | IT-ISJ05F20R | 7 | 7 | 100 | 0.835 |
| FOG591 | 10 | 10 | 100 | 0.916 | IT-ISJ05F36R | 3 | 3 | 100 | 0.924 |
| FOG634 | 9 | 9 | 100 | 0.928 | IT-ISJ06F50R | 3 | 3 | 100 | 0.911 |
| FOG655 | 8 | 8 | 100 | 0.910 | IT-ISJ07F05R | 7 | 7 | 100 | 0.814 |
| FOG707 | 8 | 8 | 100 | 0.956 | IT-ISJ07F11R | 7 | 7 | 100 | 0.727 |
| FOG112 | 10 | 10 | 100 | 0.905 | IT-ISJ07F12R | 8 | 8 | 100 | 0.852 |
| FOG393 | 8 | 8 | 100 | 0.948 | IT-ISJ07F15R | 5 | 5 | 100 | 0.545 |
| FOG455 | 10 | 10 | 100 | 0.872 | Total | 99 | |||
| FOG598 | 8 | 8 | 100 | 0.920 | Mean | 6.600 | 0.780 | ||
| FOG624 | 9 | 9 | 100 | 0.871 | |||||
| FOG824 | 9 | 9 | 100 | 0.875 | |||||
| FOG831 | 13 | 13 | 100 | 0.952 | |||||
| DGSSR14primer12 | 9 | 9 | 100 | 0.914 | |||||
| Total | 196 | ||||||||
| Mean | 9.333 | 0.909 |
| Items | SSR | IT-ISJ |
|---|---|---|
| No. of primers | 21 | 15 |
| No. of total loci | 196 | 99 |
| No. of average loci per primer | 9.33 | 6.6 |
| Percentage of polymorphic bands (PPB) | 100 | 100 |
| Average band informativeness (Ibav) | 0.49 | 0.54 |
| Effective multiplex ratio (EMR) | 9.33 | 6.60 |
| Marker index (MI) | 4.58 | 3.54 |
| Subspecies | Na | Ne | He | I | Distribution of Genetic Diversity | |
|---|---|---|---|---|---|---|
| D. glomerata subsp. santai | 1.898 | 1.409 | 0.257 | 0.401 | It | 0.448 |
| D. glomerata subsp. smithii | 1.871 | 1.372 | 0.235 | 0.370 | Is | 0.321 |
| D. glomerata subsp. woronowii | 1.807 | 1.344 | 0.217 | 0.340 | Is/It | 0.717 |
| D. glomerata subsp. lusitanica | 1.471 | 1.213 | 0.135 | 0.211 | S' | 0.283 |
| D. glomerata subsp. himalayensis | 1.556 | 1.255 | 0.159 | 0.248 | ||
| D. glomerata. subsp. glomerata | 1.536 | 1.264 | 0.162 | 0.251 | ||
| D. glomerata subsp. lobata | 1.881 | 1.368 | 0.232 | 0.366 | ||
| D. glomerata subsp. hispanica | 1.993 | 1.420 | 0.269 | 0.425 | ||
| D. glomerata subsp. marina | 1.519 | 1.307 | 0.184 | 0.276 | ||
| Mean | 1.726 | 1.328 | 0.205 | 0.321 | ||
| Total | 2.000 | 1.433 | 0.283 | 0.448 | ||
| Source of Variation | Sum of Squares | Variance Component | Total Variation (%) | Degree of Freedoms | p-Values |
|---|---|---|---|---|---|
| 20 accessions | |||||
| Variance among accessions | 2132.571 | 10.892 | 0.191 | 19 | <0.001 |
| Variance within accessions | 8661.510 | 46.072 | 0.809 | 176 | <0.001 |
| D. glomerata subsp. hispanica | |||||
| Variance among countries | 1384.837 | 11.434 | 0.108 | 6 | <0.001 |
| Variance within countries | 6139.533 | 94.454 | 0.892 | 63 | <0.001 |
| D. glomerata subsp. lobata | |||||
| Variance among countries | 165.050 | 8.970 | 0.165 | 2 | <0.001 |
| Variance within countries | 1272.750 | 45.455 | 0.835 | 27 | <0.001 |
| D. glomerata subsp. smithii | |||||
| Variance among countries | 144.800 | 10.340 | 0.200 | 2 | <0.001 |
| Variance within countries | 745.200 | 41.400 | 0.800 | 17 | <0.001 |
| D. glomerata subsp. woronowii | |||||
| Variance among countries | 141.300 | 10.508 | 0.225 | 2 | <0.001 |
| Variance within countries | 652.000 | 36.222 | 0.775 | 17 | <0.001 |
| Code | Name | Taxon | Origin and Locality | Distribution Range | Climate Type |
|---|---|---|---|---|---|
| 1 | PI237605 | D. glomerata subsp. santai | Mountain Tessala, near Sidi Abbes, Algeria | Unknown | Mediterranean |
| 2 | PI368880 | D. glomerata subsp. santai | Mountain Tessala, north of Sidi Abbes, Algeria | Unknown | Mediterranean |
| 3 | PI441034 | D. glomerata subsp. smithii | United Kingdom (Portugal-madeira Islands; spain-Canary Islands) | Africa | Subtropical |
| 4 | PI237607 | D. glomerata subsp. smithii | Tenerife, Canary Islands, Spain | Africa | Subtropical |
| 5 | PI538922 | D. glomerata subsp. woronowii | Russian Federation, Leningrad | Asia-Temperate | Mediterranean |
| 6 | PI237610 | D. glomerata subsp. woronowii | Tehran, Iran | Asia-Temperate | Mediterranean |
| 7 | PI237602 | D. glomerata subsp. lusitanica | Near Sintra, Algueirao, Portugal | Europe | Mediterranean |
| 8 | PI295271 | D. glomerata subsp. himalayensis | India | Asia-Tropical | Temperate |
| 9 | PI538920 | D. glomerata subsp. glomerata | Russian Federation | Unknown | Temperate |
| 10 | PI316209 | D. glomerata subsp. lobata | Bulgaria | Europe | Temperate |
| 11 | PI372621 | D. glomerata subsp. lobata | Bremen, Germany | Europe | Temperate |
| 12 | PI283242 | D. glomerata subsp. lobata | Germany | Europe | Temperate |
| 13 | PI231517 | D. glomerata subsp. hispanica | Midelt, Morocco | Africa | Mediterranean |
| 14 | PI265568 | D. glomerata subsp. hispanica | Spain | Europe (Canry Islands) Africa (Baleares) | Mediterranean |
| 15 | PI265567 | D. glomerata subsp. hispanica | France | Europe | Mediterranean |
| 16 | PI224599 | D. glomerata subsp. hispanica | Ruhama, Israel | Unknown | Mediterranean |
| 17 | PI277836 | D. glomerata subsp. hispanica | Turkey | Asia-Temperate | Mediterranean |
| 18 | PI231550 | D. glomerata subsp. hispanica | Agrinion, Greece | Europe | Mediterranean |
| 19 | PI231541 | D. glomerata subsp. hispanica | Nazare, Portugal | Europe | Mediterranean |
| 20 | PI577065 | D. glomerata subsp. marina | Sao Vivent, Portugal | Europe | Mediterranean |
| Primer Name | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|
| FOG215 | CAGTGACTACCGTCGTTACTC | TTGCTGCAAGGAAAATTC |
| FOG238 | GTCACCTAAGCCATAGCAAG | ACTTCTGTGTTGGTACCGAC |
| FOG258 | GCAGTATGGTGCTCTCTCTT | CACTCGTTCAGATCGTCC |
| FOG296 | ATGGAAGTTTCCTGGAATG | GAAGCAGAGTAGAGCCCAC |
| FOG362 | ATTGCATGGTTCTGCACT | GTGAGTATGCGTGTTTGCTA |
| FOG402 | TCCTTATGAAATGAATGAATGA | AAGAACTGGACATATACTTGGG |
| FOG514 | CTGATTCGATATGAATGCTTC | ACATGATTGAGAAACGGAAC |
| FOG515 | GATGAAGGAACTGCTGGAT | ACACCAGACCCTAAACAGC |
| FOG537 | TAAATCTTGCACTTATCTGTGC | AACTGTACTCTCTCACACCCTC |
| FOG591 | CTCATGCAAGATATGGCAC | AAGATCAGGTTTGAACCTCTC |
| FOG634 | GTGCGTCTTTTAAATGGTATG | GAGCCTCCCTAACCCTAGTA |
| FOG655 | GAGATGAGCCATGATTCATT | GTGCTTGCTTGATTCACC |
| FOG707 | CGCTAGAACCTCCCTACAC | CAAAAGATCTTCAACGCTG |
| FOG112 | TAAGAATCGATCCTCCCG | ACCTTCTTCCACTCCGTC |
| FOG393 | GGGGAGGTACCCACTTCT | TTACCCAATCTAAGATCTTTGC |
| FOG455 | GTGAGGTTGTGGAAAGTGAC | CAAAACTAACGCCGTTAACT |
| FOG598 | GCCAGGTCAGTCACTCAC | AGAAAATTCCCCAACAGC |
| FOG624 | CATCAGGGTAGTATCCCGTA | AGTTCCTTCTCCTCTGCTTT |
| FOG824 | TAGTGGAATGTCAAGAAAATGA | AAAATGTCCTTGTTCCCAG |
| FOG831 | TAAAGCATATGCAACAATGC | TGCTAAAGCCTTTTACAGCT |
| DGSSR14 primer12 | AGGCCTTCTTGCACTGGTAC | GAGTTCACTGAGGCCGAGAG |
| Primer Name | 5′→3′ Sequence | Primer Name | 5′→3′ Sequence |
|---|---|---|---|
| Forward primer | Reverse primer | ||
| IT-ISJ01F | GCATGCCAGGTAAGTAAA | IT-ISJ24R | GGAATTCCACCTGCACCT |
| IT-ISJ03F | GCATGCCAGGTAAGTAAG | IT-ISJ26R | GGAATTCCACCTGCACGC |
| IT-ISJ04F | GCATGCCAGGTAAGTAAT | IT-ISJ28R | GGAATTCCACCTGCACGT |
| IT-ISJ05F | GCATGCCAGGTAAGTACA | IT-ISJ29R | GGAATTCCACCTGCACTA |
| IT-ISJ06F | GCATGCCAGGTAAGTACC | IT-ISJ32R | GGAATTCCACCTGCACTT |
| IT-ISJ07F | GCATGCCAGGTAAGTACG | IT-ISJ36R | GGAATTCCACCTGCAGAT |
| Reverse primer | IT-ISJ38R | GGAATTCCACCTGCAGCC | |
| IT-ISJ01R | GGAATTCCACCTGCAAAA | IT-ISJ40R | GGAATTCCACCTGCAGCT |
| IT-ISJ03R | GGAATTCCACCTGCAAAG | IT-ISJ41R | GGAATTCCACCTGCAGGA |
| IT-ISJ04R | GGAATTCCACCTGCAAAT | IT-ISJ42R | GGAATTCCACCTGCAGGC |
| IT-ISJ05R | GGAATTCCACCTGCAACA | IT-ISJ44R | GGAATTCCACCTGCAGGT |
| IT-ISJ06R | GGAATTCCACCTGCAACC | IT-ISJ47R | GGAATTCCACCTGCAGTG |
| IT-ISJ07R | GGAATTCCACCTGCAACG | IT-ISJ50R | GGAATTCCACCTGCATAC |
| IT-ISJ08R | GGAATTCCACCTGCAACT | IT-ISJ51R | GGAATTCCACCTGCATAG |
| IT-ISJ11R | GGAATTCCACCTGCAAGG | IT-ISJ52R | GGAATTCCACCTGCATAT |
| IT-ISJ12R | GGAATTCCACCTGCAAGT | IT-ISJ54R | GGAATTCCACCTGCATCC |
| IT-ISJ13R | GGAATTCCACCTGCAATA | IT-ISJ58R | GGAATTCCACCTGCATGC |
| IT-ISJ15R | GGAATTCCACCTGCAATG | IT-ISJ59R | GGAATTCCACCTGCATGG |
| IT-ISJ20R | GGAATTCCACCTGCACAT | IT-ISJ63R | GGAATTCCACCTGCATTG |
| IT-ISJ22R | GGAATTCCACCTGCACCC | IT-ISJ64R | GGAATTCCACCTGCATTT |
| IT-ISJ23R | GGAATTCCACCTGCACCG | ||
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, D.; Zhao, X.; Cheng, Y.; Ma, X.; Huang, L.; Zhang, X. Phylogenetic and Diversity Analysis of Dactylis glomerata Subspecies Using SSR and IT-ISJ Markers. Molecules 2016, 21, 1459. https://doi.org/10.3390/molecules21111459
Yan D, Zhao X, Cheng Y, Ma X, Huang L, Zhang X. Phylogenetic and Diversity Analysis of Dactylis glomerata Subspecies Using SSR and IT-ISJ Markers. Molecules. 2016; 21(11):1459. https://doi.org/10.3390/molecules21111459
Chicago/Turabian StyleYan, Defei, Xinxin Zhao, Yajuan Cheng, Xiao Ma, Linkai Huang, and Xinquan Zhang. 2016. "Phylogenetic and Diversity Analysis of Dactylis glomerata Subspecies Using SSR and IT-ISJ Markers" Molecules 21, no. 11: 1459. https://doi.org/10.3390/molecules21111459
APA StyleYan, D., Zhao, X., Cheng, Y., Ma, X., Huang, L., & Zhang, X. (2016). Phylogenetic and Diversity Analysis of Dactylis glomerata Subspecies Using SSR and IT-ISJ Markers. Molecules, 21(11), 1459. https://doi.org/10.3390/molecules21111459