
The Complexity of Targeting PI3K-Akt-mTOR Signalling in Human Acute Myeloid Leukaemia: The Importance of Leukemic Cell Heterogeneity, Neighbouring Mesenchymal Stem Cells and Immunocompetent Cells


Abstract
:1. Introduction
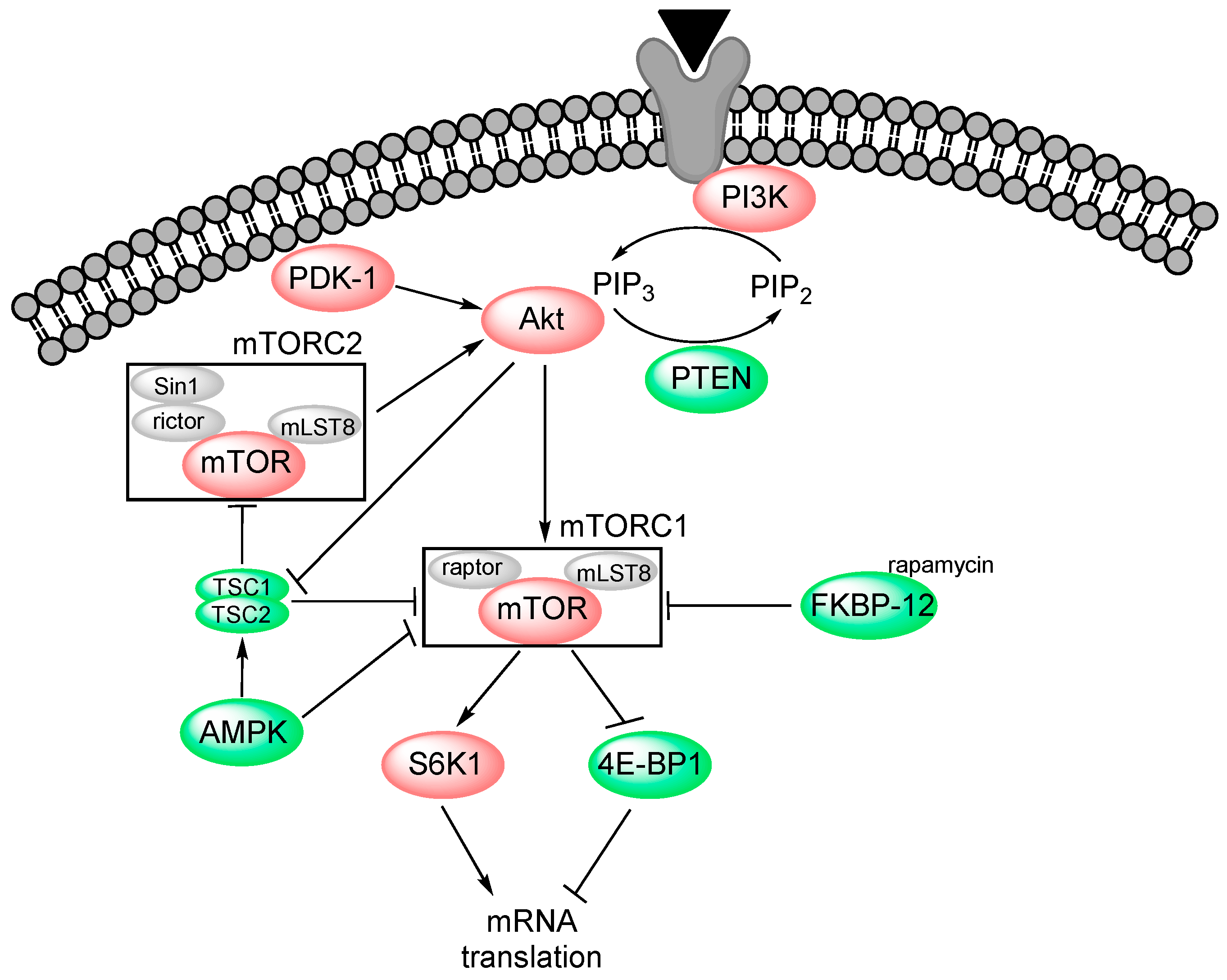
2. PI3K-Akt-mTOR Signalling
2.1. PI3K
2.2. Akt (Protein Kinase B)
2.3. mTOR
2.4. Pharmacological Targeting of the PI3K-Akt-mTOR Pathway
3. Direct Effects of PI3K-Akt-mTOR Inhibition on AML Cells: Patient Heterogeneity and Resistance to Treatment
3.1. Dysregulation of PI3K-Akt-mTOR in Human Malignancies—General Comments
3.2. PI3K-Akt-mTOR Targeting in Human AML
- 50%–80% of AML patients display Akt that is phosphorylated at T308, S473 or both. This upregulation has been detected not only in bulk AML cells but also in the more immature leukemic stem cells [36,37]. Several studies suggest that both overall and disease-free survival is shorter in patients with PI3K-Akt-mTOR pathway upregulation [36]. In contrast, constitutive activation of the upstream PI3K may represent a favourable prognostic parameter [40].
- The causes for activation of PI3K-Akt-mTOR signalling can be mutations in the FMS-like tyrosine kinase 3 (Flt3), proto-oncogene c-Kit (CD117) or K-Ras genes, overexpression of PI3K or PDK-1, low levels of protein phosphatase 2 (PP2A), autocrine or paracrine release of growth factors (e.g., IGF-1, platelet-derived growth factor/PDGF or the chemokine CXCL12), stromal/fibronectin-induced upregulation of integrin-linked kinase 1 (ILK1), or PTEN loss [36,37]. Activating mutations in PI3K or Akt, however, are uncommon also in AML [36].
- Patients are heterogeneous with regard to the effect of PI3K-Akt-mTOR inhibitors on AML cell proliferation; although an antiproliferative effect is observed for most patients, no effect or even growth enhancement is seen for a subset of patients [27]. This adverse effect is possibly associated with differences in cell cycle regulation.
- There seems to exist several escape mechanisms to inhibition of this pathway [1]. Firstly, induction of autophagy during treatment may represent a mechanism of resistance, and combination of PI3K-Akt-mTOR and autophagy inhibitors has therefore been suggested. Secondly, paradoxical Akt phosphorylation during treatment may induce expression and autophosphorylation of the receptors for insulin, IGF-1 and PDGF resulting in increased pathway activation. This feedback effect can be blocked by PDGFR/IGF-1R/Flt3 inhibition. Thirdly, activation of MAPK-interacting kinases can increase eukaryotic translation initiation factor E4 (eIF4E) phosphorylation and thereby trigger synthesis of pro-survival proteins. Finally, increased signalling of alternative pathways (e.g., ERK upregulation) can also be seen. These observations clearly illustrate the intracellular complexity of PI3K-Akt-mTOR inhibition.
- New mTOR inhibitors seem to target both TORC1 and TORC2, whereas the earlier inhibitors targeted mainly TORC1; the more recent inhibitors may thereby have a stronger effect [23].
- 5′ AMP-activated protein kinase (AMPK) is an inhibitor of mTORC1; directly through inhibition of raptor and indirectly through activation of the TSC1/TSC2 complex [41]. At starvation, AMPK initiates increased fatty acid oxidation and also autophagy, and AMPK activation/agonists have a cytotoxic effect in AML cells [25].
- The combination of conventional chemotherapy with PI3K-Akt-mTOR inhibitors seems to have an acceptable toxicity, but further clinical studies are needed to clarify whether there are additive or synergistic antileukemic effects [38].
4. PI3K-Akt-mTOR in MSCs
4.1. Identification, Differentiation and Function of Bone Marrow MSCs
4.2. MSC Contributions to Stem Cell Niches in the Bone Marrow
5. The AML-Supporting Effects of Mesenchymal Stem Cells: Contributions from Cell-Cell Contact and Distant Effects Mediated through the Local Cytokine Network
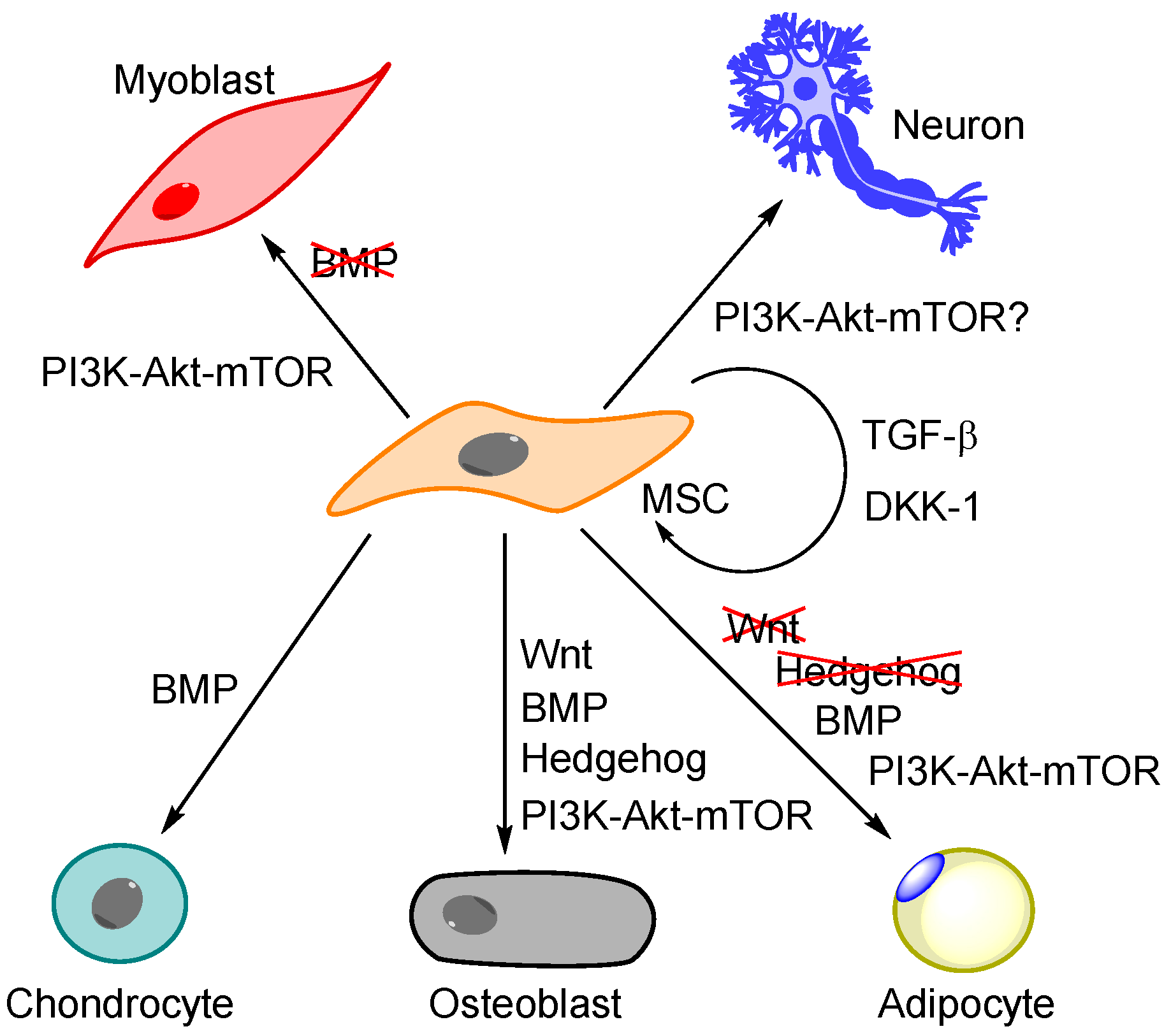
5.1. Regulation of MSC Differentiation Is More Than PI3K-Akt-mTOR—The Importance of BMP, Wnt and Hedgehog Signalling
5.2. The General Effect of PI3K-Akt-mTOR Signalling on MSC Differentiation
5.3. The Unique Membrane Molecule Profile of MSCs: Possible Molecular Mechanisms for Communication with Neighbouring Cells through Direct Cell-Cell Contact and via the Local Cytokine Network
5.4. The Functional Importance of PI3K-Akt-mTOR Signalling in MSCs: The Effects on MSC Differentiation Are Only a Part of a More Extensive and Complex Biological Impact
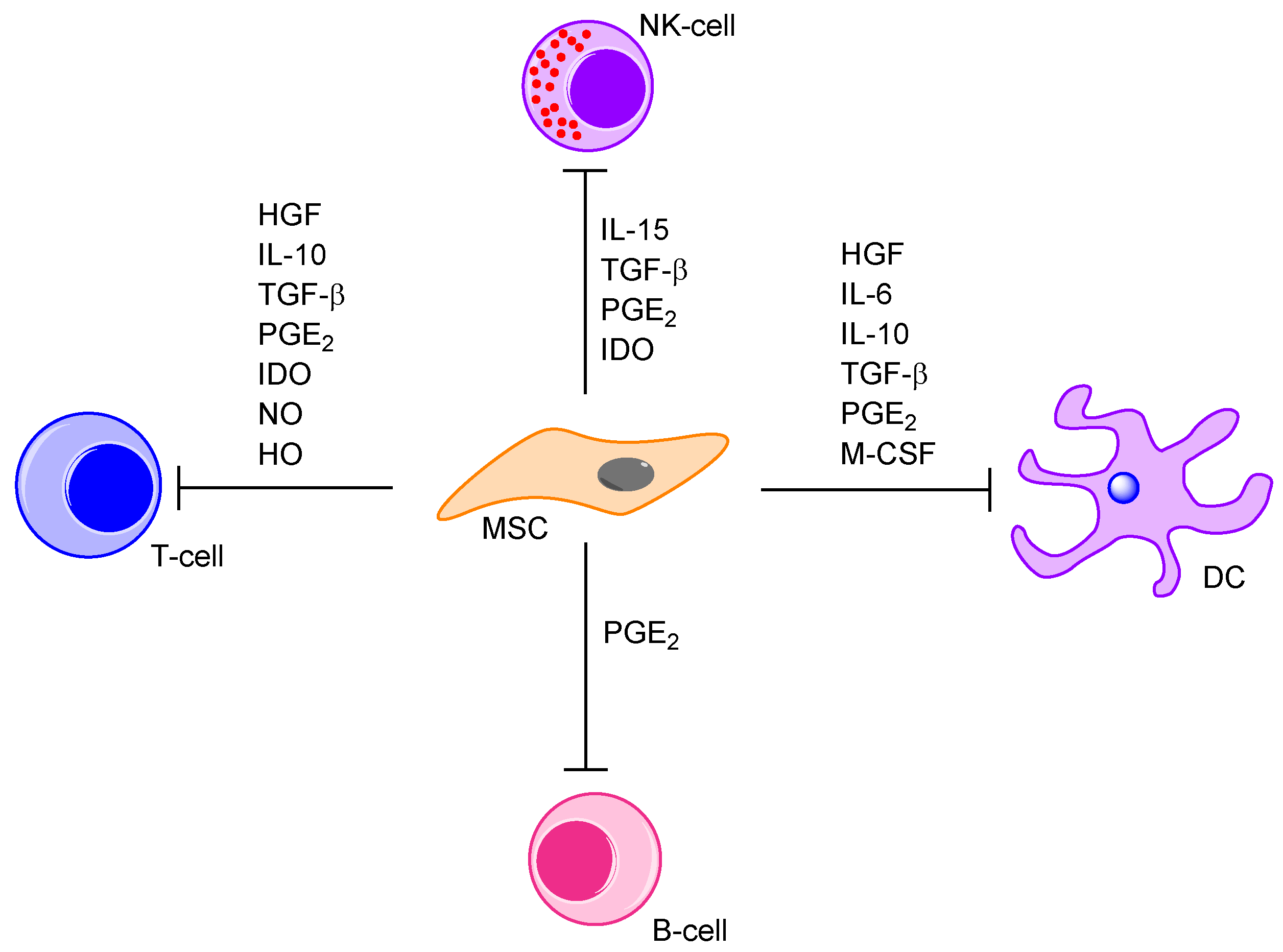
5.5. Cytokine-Mediated Communication between MSCs and the Neighbouring Bone Marrow Cells; a Part of the AML-Supporting Effects by the MSCs
6. Direct Effects of PI3K-Akt-mTOR Inhibition on Immunocompetent Cells and the Dual Function of Monocytes/Macrophages as Immunocompetent Cells and Members of the Stem Cell Niches
6.1. PI3K-Akt-mTOR, MSCs and Immunocompetent Cells; Regulation of Allogeneic and Autologous Antileukemic Immune Reactivity
6.2. The Role of AKT/mTOR as Regulators of Macrophage Metabolism and Cytokine Release
- Increased glycolysis in response to TLR stimulation can be mediated by Akt independent of mTORC1.
- 4E-BP1 and 6SK1 have important roles in controlling the synthesis of both cytokines as well as HIF-1α and IRF-7 that are necessary for the cytokine synthesis.
- Activation of sterol regulatory element-binding protein 1 (SRBP1) is important for synthesis of lipid mediators and cytokines, and also for activation of the pentose-phosphate pathway that is required for adequate respiratory burst.
- mTORC1 is critical for control of glutamine metabolism that again is a regulator of the hexosamine pathway and the processes securing sufficient succinate accumulation.
6.3. The Role of Akt/mTOR in Macrophage Polarization—The Importance for AML Cells and Immunoregulation
6.4. The Effect of Pharmacological Inhibition of Akt/mTOR in Macrophages
7. Summarizing Discussion
7.1. The Complexity of PI3K-Akt-mTOR Signalling—What Is the Optimal Molecular Target for Pathway Inhibition?
7.2. The Complexity and Heterogeneity of Human AML; the Consequences from PI3K-Akt-mTOR Inhibition in the Leukemic Cells with Regard to Communication with Neighbouring Cells May Differ among Patients
7.3. The Complex Effects of PI3K-Akt-mTOR Signalling in MSCs
7.4. The Complex Effects of PI3K-Akt-mTOR Signalling in Macrophages and the Role of Other Immunocompetent Cells
8. Final Conclusions
Acknowledgments
Conflicts of Interest
References
- Carneiro, B.A.; Kaplan, J.B.; Altman, J.K.; Giles, F.J.; Platanias, L.C. Targeting mTOR signaling pathways and related negative feedback loops for the treatment of acute myeloid leukemia. Cancer Biol. Ther. 2015, 16, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Zhao, J.; Xu, G.; Li, Y.; Zhang, W. mTOR and the differentiation of mesenchymal stem cells. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, A.; Trumpp, A. The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J. Exp. Med. 2011, 208, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Morgensztern, D.; McLeod, H.L. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs 2005, 16, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, M.A.; Pearson, R.B.; Hannan, R.D.; Sheppard, K.E. AKT-independent PI3-K signaling in cancer—emerging role for SGK3. Cancer Manag. Res. 2013, 5, 281–292. [Google Scholar] [PubMed]
- Steelman, L.S.; Abrams, S.L.; Whelan, J.; Bertrand, F.E.; Ludwig, D.E.; Basecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 2008, 22, 686–707. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, M.A.; Alessi, D.R. PKB/Akt: A key mediator of cell proliferation, survival and insulin responses? J. Cell Sci. 2001, 114, 2903–2910. [Google Scholar] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Hers, I.; Vincent, E.E.; Tavare, J.M. Akt signalling in health and disease. Cell. Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Caudwell, F.B.; Andjelkovic, M.; Hemmings, B.A.; Cohen, P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 1996, 399, 333–338. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Hall, M.N. mTORC2 caught in a SINful Akt. Dev. Cell 2006, 11, 433–434. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Chen, J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Edinger, A.L.; Thompson, C.B. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol. Biol. Cell 2002, 13, 2276–2288. [Google Scholar] [CrossRef] [PubMed]
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 2000, 10, 47–50. [Google Scholar] [CrossRef]
- Kim, J.A.; Shim, J.S.; Lee, G.Y.; Yim, H.W.; Kim, T.M.; Kim, M.; Leem, S.H.; Lee, J.W.; Min, C.K.; Oh, I.H. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015, 75, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.K.; Sassano, A.; Platanias, L.C. Targeting mTOR for the treatment of AML. New agents and new directions. Oncotarget 2011, 2, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Heidari, N.; Mediani, L.; Capitani, S.; Shahjahani, M.; Ahmadzadeh, A.; Saki, N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell. Mol. Life Sci. 2015, 72, 2337–2347. [Google Scholar] [CrossRef] [PubMed]
- Hauge, M.; Bruserud, Ø.; Hatfield, K.J. Targeting of cell metabolism in human acute myeloid leukemia—More than targeting of isocitrate dehydrogenase mutations and PI3K/AKT/mTOR signaling? Eur. J. Haematol. 2016, 96, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Loh, K.; Yap, Y.S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol. Med. 2015, 12, 342–354. [Google Scholar] [PubMed]
- Reikvam, H.; Tamburini, J.; Skrede, S.; Holdhus, R.; Poulain, L.; Ersvær, E.; Hatfield, K.J.; Bruserud, Ø. Antileukaemic effect of PI3K-mTOR inhibitors in acute myeloid leukaemia-gene expression profiles reveal CDC25B expression as determinate of pharmacological effect. Br. J. Haematol. 2014, 164, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Roda, D.; Yap, T.A. Strategies for modern biomarker and drug development in oncology. J. Hematol. Oncol. 2014, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Hollestelle, A.; Elstrodt, F.; Nagel, J.H.; Kallemeijn, W.W.; Schutte, M. Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol. Cancer Res. 2007, 5, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Maiello, M.R.; Campiglio, M.; Napolitano, M.; Mancino, M.; Carotenuto, A.; Viglietto, G.; Menard, S. The MEK/MAPK pathway is involved in the resistance of breast cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J. Cell. Physiol. 2006, 207, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; McCubrey, J.A. The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget 2010, 1, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chapuis, N.; Tamburini, J.; Bardet, V.; Cornillet-Lefebvre, P.; Willems, L.; Green, A.; Mayeux, P.; Lacombe, C.; Bouscary, D. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica 2010, 95, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Tasian, S.K.; Teachey, D.T.; Rheingold, S.R. Targeting the PI3K/mTOR pathway in pediatric hematologic malignancies. Front. Oncol. 2014, 4, 108. [Google Scholar] [CrossRef] [PubMed]
- Vachhani, P.; Bose, P.; Rahmani, M.; Grant, S. Rational combination of dual PI3K/mTOR blockade and Bcl-2/-xL inhibition in AML. Physiol. Genom. 2014, 46, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Tamburini, J.; Elie, C.; Bardet, V.; Chapuis, N.; Park, S.; Broet, P.; Cornillet-Lefebvre, P.; Lioure, B.; Ugo, V.; Blanchet, O.; et al. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood 2007, 110, 1025–1028. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Kim, J.; Guan, K.L. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Piatetzky-Shapiro, I.I.; Petrakova, K.V. Osteogenesis in transplants of bone marrow cells. J. Embryol. Exp. Morphol. 1966, 16, 381–390. [Google Scholar] [PubMed]
- Friedenstein, A.J.; Gorskaja, U.F.; Kulagina, N.N. Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp. Hematol. 1976, 4, 267–274. [Google Scholar] [PubMed]
- Jones, E.; McGonagle, D. Human bone marrow mesenchymal stem cells in vivo. Rheumatology (Oxford) 2008, 47, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Xie, N.; Li, W.; Yuan, B.; Shi, Y.; Wang, Y. Immunobiology of mesenchymal stem cells. Cell Death Differ. 2014, 21, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Chamberlain, G.; Ashton, B.A.; Middleton, J. Recent advances into the understanding of mesenchymal stem cell trafficking. Br. J. Haematol. 2007, 137, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W.; Ho, A.D. Mesenchymal stem cell preparations—Comparing apples and oranges. Stem Cell Rev. 2007, 3, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Eggenhofer, E.; Luk, F.; Dahlke, M.H.; Hoogduijn, M.J. The life and fate of mesenchymal stem cells. Front. Immunol. 2014, 5, 148. [Google Scholar] [CrossRef] [PubMed]
- Bergfeld, S.A.; DeClerck, Y.A. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 2010, 29, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.E.; Garcia, C.B. Reprogramming of mesenchymal stem cells by oncogenes. Semin. Cancer Biol. 2015, 32, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.J.; Tuan, R.S.; Cheung, K.M.; Leung, V.Y. The surface markers and identity of human mesenchymal stem cells. Stem Cells 2014, 32, 1408–1419. [Google Scholar] [CrossRef] [PubMed]
- Shi, C. Recent progress toward understanding the physiological function of bone marrow mesenchymal stem cells. Immunology 2012, 136, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Barzilay, R.; Melamed, E.; Offen, D. Introducing transcription factors to multipotent mesenchymal stem cells: Making transdifferentiation possible. Stem Cells 2009, 27, 2509–2515. [Google Scholar] [CrossRef] [PubMed]
- Deans, R.J.; Moseley, A.B. Mesenchymal stem cells: Biology and potential clinical uses. Exp. Hematol. 2000, 28, 875–884. [Google Scholar] [CrossRef]
- Morandi, F.; Raffaghello, L.; Bianchi, G.; Meloni, F.; Salis, A.; Millo, E.; Ferrone, S.; Barnaba, V.; Pistoia, V. Immunogenicity of human mesenchymal stem cells in HLA-class I-restricted T-cell responses against viral or tumor-associated antigens. Stem Cells 2008, 26, 1275–1287. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I.; Bruder, S.P. Mesenchymal stem cells: Building blocks for molecular medicine in the 21st century. Trends Mol. Med. 2001, 7, 259–264. [Google Scholar] [CrossRef]
- Kopen, G.C.; Prockop, D.J.; Phinney, D.G. Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proc. Natl. Acad. Sci. USA 1999, 96, 10711–10716. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Fukuda, K.; Miyoshi, S.; Konishi, F.; Kodama, H.; Pan, J.; Sano, M.; Takahashi, T.; Hori, S.; Abe, H.; et al. Cardiomyocytes can be generated from marrow stromal cells in vitro. J. Clin. Investig. 1999, 103, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, G.; Cusella-De Angelis, G.; Coletta, M.; Paolucci, E.; Stornaiuolo, A.; Cossu, G.; Mavilio, F. Muscle regeneration by bone marrow-derived myogenic progenitors. Science 1998, 279, 1528–1530. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Relan, N.K.; Przywara, D.A.; Schuger, L. Embryonic mesenchymal cells share the potential for smooth muscle differentiation: Myogenesis is controlled by the cell’s shape. Development 1999, 126, 3027–3033. [Google Scholar] [PubMed]
- Baksh, D.; Song, L.; Tuan, R.S. Adult mesenchymal stem cells: Characterization, differentiation, and application in cell and gene therapy. J. Cell. Mol. Med. 2004, 8, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.; Riminucci, M.; Gronthos, S.; Robey, P.G. Bone marrow stromal stem cells: Nature, biology, and potential applications. Stem Cells 2001, 19, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Rameshwar, P. IFNgamma and B7-H1 in the immunology of mesenchymal stem cells. Cell Res. 2008, 18, 805–806. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.; Andreeff, M.; Marini, F. The participation of mesenchymal stem cells in tumor stroma formation and their application as targeted-gene delivery vehicles. Handb. Exp. Pharmacol. 2007, 263–283. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Simmons, P.J.; Torok-Storb, B. Identification of stromal cell precursors in human bone marrow by a novel monoclonal antibody, STRO-1. Blood 1991, 78, 55–62. [Google Scholar] [PubMed]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef] [PubMed]
- Kfoury, Y.; Scadden, D.T. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell 2015, 16, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Colmone, A.; Sipkins, D.A. Beyond angiogenesis: The role of endothelium in the bone marrow vascular niche. Transl. Res. 2008, 151, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Doan, P.L.; Chute, J.P. The vascular niche: Home for normal and malignant hematopoietic stem cells. Leukemia 2012, 26, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Ryningen, A.; Wergeland, L.; Glenjen, N.I.; Gjertsen, B.T. Osteoblasts increase proliferation and release of pro-angiogenic interleukin 8 by native human acute myelogenous leukemia blasts. Haematologica 2004, 89, 391–402. [Google Scholar] [PubMed]
- Glenjen, N.I.; Hatfield, K.; Bruserud, Ø. Coculture of native human acute myelogenous leukemia blasts with fibroblasts and osteoblasts results in an increase of vascular endothelial growth factor levels. Eur. J. Haematol. 2005, 74, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, K.; Ryningen, A.; Corbascio, M.; Bruserud, Ø. Microvascular endothelial cells increase proliferation and inhibit apoptosis of native human acute myelogenous leukemia blasts. Int. J. Cancer 2006, 119, 2313–2321. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, K.J.; Olsnes, A.M.; Gjertsen, B.T.; Bruserud, Ø. Antiangiogenic therapy in acute myelogenous leukemia: Targeting of vascular endothelial growth factor and interleukin 8 as possible antileukemic strategies. Curr. Cancer Drug Targets 2005, 5, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Ryningen, A.; Wergeland, L.; Glenjen, N.; Gjertsen, B.T.; Bruserud, Ø. In vitro crosstalk between fibroblasts and native human acute myelogenous leukemia (AML) blasts via local cytokine networks results in increased proliferation and decreased apoptosis of AML cells as well as increased levels of proangiogenic Interleukin 8. Leuk. Res. 2005, 29, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Sipkins, D.A.; Wei, X.; Wu, J.W.; Runnels, J.M.; Cote, D.; Means, T.K.; Luster, A.D.; Scadden, D.T.; Lin, C.P. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 2005, 435, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Yagi, H.; Soto-Gutierrez, A.; Parekkadan, B.; Kitagawa, Y.; Tompkins, R.G.; Kobayashi, N.; Yarmush, M.L. Mesenchymal stem cells: Mechanisms of immunomodulation and homing. Cell Transplant. 2010, 19, 667–679. [Google Scholar] [CrossRef] [PubMed]
- James, A.W. Review of signaling pathways governing MSC osteogenic and adipogenic differentiation. Scientifica (Cairo) 2013, 2013, 684736. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J. Immune regulation by mesenchymal stem cells: Two sides to the coin. Tissue Antigens 2007, 69, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wang, X.F. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009, 19, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.; Shen, X.; Liu, I.; Semenov, M.; He, X.; Wang, X.F. Smad3-dependent nuclear translocation of beta-catenin is required for TGF-beta1-induced proliferation of bone marrow-derived adult human mesenchymal stem cells. Genes Dev. 2006, 20, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Hayashi, M.; Komiya, S.; Imamura, T.; Miyazono, K. Endogenous TGF-beta signaling suppresses maturation of osteoblastic mesenchymal cells. EMBO J. 2004, 23, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.; Boucher, S.; Koh, S.; Sastry, K.S.; Chase, L.; Lakshmipathy, U.; Choong, C.; Yang, Z.; Vemuri, M.C.; Rao, M.S.; et al. PDGF, TGF-beta, and FGF signaling is important for differentiation and growth of mesenchymal stem cells (MSCs): Transcriptional profiling can identify markers and signaling pathways important in differentiation of MSCs into adipogenic, chondrogenic, and osteogenic lineages. Blood 2008, 112, 295–307. [Google Scholar] [PubMed]
- Liu, Z.; Tang, Y.; Qiu, T.; Cao, X.; Clemens, T.L. A dishevelled-1/Smad1 interaction couples WNT and bone morphogenetic protein signaling pathways in uncommitted bone marrow stromal cells. J. Biol. Chem. 2006, 281, 17156–17163. [Google Scholar] [CrossRef] [PubMed]
- Ghosh-Choudhury, N.; Abboud, S.L.; Nishimura, R.; Celeste, A.; Mahimainathan, L.; Choudhury, G.G. Requirement of BMP-2-induced phosphatidylinositol 3-kinase and Akt serine/threonine kinase in osteoblast differentiation and Smad-dependent BMP-2 gene transcription. J. Biol. Chem. 2002, 277, 33361–33368. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.A.; Israel, D.I.; Kelly, S.; Luxenberg, D.P. Bone morphogenetic protein-2 causes commitment and differentiation in C3H10T1/2 and 3T3 cells. Growth Factors 1993, 9, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Keating, A. Mesenchymal stromal cells. Curr. Opin. Hematol. 2006, 13, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chen, Z.; Zhang, J.; Zhang, L.; Ke, H.; Huang, L.; Peng, Y.; Zhang, X.; Li, S.; Lahn, B.T.; et al. Critical role of phosphoinositide 3-kinase cascade in adipogenesis of human mesenchymal stem cells. Mol. Cell. Biochem. 2008, 310, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Carnevalli, L.S.; Masuda, K.; Frigerio, F.; Le Bacquer, O.; Um, S.H.; Gandin, V.; Topisirovic, I.; Sonenberg, N.; Thomas, G.; Kozma, S.C. S6K1 plays a critical role in early adipocyte differentiation. Dev. Cell 2010, 18, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Fitter, S.; Dutta, A.K.; Matthews, M.P.; Walkley, C.R.; Hall, M.N.; Ruegg, M.A.; Gronthos, S.; Zannettino, A.C. Brief report: The differential roles of mTORC1 and mTORC2 in mesenchymal stem cell differentiation. Stem Cells 2015, 33, 1359–1365. [Google Scholar] [CrossRef] [PubMed]
- Cashman, T.J.; Gouon-Evans, V.; Costa, K.D. Mesenchymal stem cells for cardiac therapy: Practical challenges and potential mechanisms. Stem Cell Rev. 2013, 9, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, R.M.; Khan, S.; Barry, F.P.; O’Brien, T.; Kerin, M.J. Advances in mesenchymal stem cell-mediated gene therapy for cancer. Stem Cell Res. Ther. 2010, 1, 25. [Google Scholar] [CrossRef] [PubMed]
- Sordi, V.; Malosio, M.L.; Marchesi, F.; Mercalli, A.; Melzi, R.; Giordano, T.; Belmonte, N.; Ferrari, G.; Leone, B.E.; Bertuzzi, F.; et al. Bone marrow mesenchymal stem cells express a restricted set of functionally active chemokine receptors capable of promoting migration to pancreatic islets. Blood 2005, 106, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Honczarenko, M.; Le, Y.; Swierkowski, M.; Ghiran, I.; Glodek, A.M.; Silberstein, L.E. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells 2006, 24, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.A.; Kinsey, S.E.; English, A.; Jones, R.A.; Straszynski, L.; Meredith, D.M.; Markham, A.F.; Jack, A.; Emery, P.; McGonagle, D. Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum. 2002, 46, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Sohni, A.; Verfaillie, C.M. Mesenchymal stem cells migration homing and tracking. Stem Cells Int. 2013, 2013, 130763. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.A.; English, A.; Kinsey, S.E.; Straszynski, L.; Emery, P.; Ponchel, F.; McGonagle, D. Optimization of a flow cytometry-based protocol for detection and phenotypic characterization of multipotent mesenchymal stromal cells from human bone marrow. Cytom. B Clin. Cytom. 2006, 70, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Rüster, B.; Gottig, S.; Ludwig, R.J.; Bistrian, R.; Muller, S.; Seifried, E.; Gille, J.; Henschler, R. Mesenchymal stem cells display coordinated rolling and adhesion behavior on endothelial cells. Blood 2006, 108, 3938–3944. [Google Scholar] [CrossRef] [PubMed]
- Von Lüttichau, I.; Notohamiprodjo, M.; Wechselberger, A.; Peters, C.; Henger, A.; Seliger, C.; Djafarzadeh, R.; Huss, R.; Nelson, P.J. Human adult CD34-progenitor cells functionally express the chemokine receptors CCR1, CCR4, CCR7, CXCR5, and CCR10 but not CXCR4. Stem Cells Dev. 2005, 14, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Krampera, M.; Glennie, S.; Dyson, J.; Scott, D.; Laylor, R.; Simpson, E.; Dazzi, F. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood 2003, 101, 3722–3729. [Google Scholar] [CrossRef] [PubMed]
- Wynn, R.F.; Hart, C.A.; Corradi-Perini, C.; O’Neill, L.; Evans, C.A.; Wraith, J.E.; Fairbairn, L.J.; Bellantuono, I. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood 2004, 104, 2643–2645. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Ylostalo, J.; Lynch, P.J.; Smith, J.; Perry, A.; Peister, A.; Wang, M.Y.; Prockop, D.J. Differentiation, cell fusion, and nuclear fusion during ex vivo repair of epithelium by human adult stem cells from bone marrow stroma. Proc. Natl. Acad. Sci. USA 2003, 100, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Case, N.; Thomas, J.; Sen, B.; Styner, M.; Xie, Z.; Galior, K.; Rubin, J. Mechanical regulation of glycogen synthase kinase 3beta (GSK3beta) in mesenchymal stem cells is dependent on Akt protein serine 473 phosphorylation via mTORC2 protein. J. Biol. Chem. 2011, 286, 39450–39456. [Google Scholar] [CrossRef] [PubMed]
- Chow, D.C.; Wenning, L.A.; Miller, W.M.; Papoutsakis, E.T. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. I. Krogh’s model. Biophys. J. 2001, 81, 675–684. [Google Scholar] [CrossRef]
- Feng, X.; Huang, D.; Lu, X.; Feng, G.; Xing, J.; Lu, J.; Xu, K.; Xia, W.; Meng, Y.; Tao, T.; et al. Insulin-like growth factor 1 can promote proliferation and osteogenic differentiation of human dental pulp stem cells via mTOR pathway. Dev. Growth Differ. 2014, 56, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Fiegl, M.; Samudio, I.; Clise-Dwyer, K.; Burks, J.K.; Mnjoyan, Z.; Andreeff, M. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood 2009, 113, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Fujio, K.; Komai, T.; Inoue, M.; Morita, K.; Okamura, T.; Yamamoto, K. Revisiting the regulatory roles of the TGF-beta family of cytokines. Autoimmun. Rev. 2016, 15, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Gharibi, B.; Farzadi, S.; Ghuman, M.; Hughes, F.J. Inhibition of Akt/mTOR attenuates age-related changes in mesenchymal stem cells. Stem Cells 2014, 32, 2256–2266. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Tan, W.; Ji, J.; Feng, G.; Meng, Y.; Da, Z.; Guo, G.; Xia, Y.; Zhu, X.; Shi, G.; et al. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging 2016, 8, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.S.; Rameshwar, P.; Chang, V.; Bandari, P. Oxygen saturation in the bone marrow of healthy volunteers. Blood 2002, 99, 394. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, K.J.; Bedringsaas, S.L.; Ryningen, A.; Gjertsen, B.T.; Bruserud, Ø. Hypoxia increases HIF-1alpha expression and constitutive cytokine release by primary human acute myeloid leukaemia cells. Eur. Cytokine Netw. 2010, 21, 154–164. [Google Scholar] [PubMed]
- Kim, J.; Jung, Y.; Sun, H.; Joseph, J.; Mishra, A.; Shiozawa, Y.; Wang, J.; Krebsbach, P.H.; Taichman, R.S. Erythropoietin mediated bone formation is regulated by mTOR signaling. J. Cell. Biochem. 2012, 113, 220–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Ryu, J.M.; Jung, Y.H.; Oh, S.Y.; Lee, S.J.; Han, H.J. Novel pathway for hypoxia-induced proliferation and migration in human mesenchymal stem cells: Involvement of HIF-1alpha, FASN, and mTORC1. Stem Cells 2015, 33, 2182–2195. [Google Scholar] [CrossRef] [PubMed]
- Levy, O.; Ruvinov, E.; Reem, T.; Granot, Y.; Cohen, S. Highly efficient osteogenic differentiation of human mesenchymal stem cells by eradication of STAT3 signaling. Int. J. Biochem. Cell Biol. 2010, 42, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Cheng, P.; Liang, M.K.; Chen, Y.S.; Lu, Q.; Wang, J.Y.; Xia, Z.Y.; Zhou, H.D.; Cao, X.; Xie, H.; et al. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J. Clin. Investig. 2015, 125, 1509–1522. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hao, H.; Huang, H.; Tong, C.; Ti, D.; Dong, L.; Chen, D.; Zhao, Y.; Liu, H.; Han, W.; et al. Hypoxia regulates the therapeutic potential of mesenchymal stem cells through enhanced autophagy. Int. J. Low Extrem. Wounds 2015, 14, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kou, X.; Chen, C.; Yu, W.; Su, Y.; Kim, Y.; Shi, S.; Liu, Y. Chronic high dose alcohol induces osteopenia via activation of mTOR signaling in bone marrow mesenchymal stem cells. Stem Cells 2016, 34, 2157–2168. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.K.; Fitter, S.; Bong, L.F.; Drew, J.J.; Gronthos, S.; Shepherd, P.R.; Zannettino, A.C. NVP-BEZ235, a dual pan class I PI3 kinase and mTOR inhibitor, promotes osteogenic differentiation in human mesenchymal stromal cells. J. Bone Miner. Res. 2010, 25, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Pantovic, A.; Krstic, A.; Janjetovic, K.; Kocic, J.; Harhaji-Trajkovic, L.; Bugarski, D.; Trajkovic, V. Coordinated time-dependent modulation of AMPK/Akt/mTOR signaling and autophagy controls osteogenic differentiation of human mesenchymal stem cells. Bone 2013, 52, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, R.A.; Andrianifahanana, M.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Blenis, J.; Leof, E.B. Distinct roles for mammalian target of rapamycin complexes in the fibroblast response to transforming growth factor-beta. Cancer Res. 2009, 69, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Nepstad, I.; Bruserud, Ø.; Hatfield, K.J. Pharmacological targeting of the PI3K/mTOR pathway alters the release of angioregulatory mediators both from primary human acute myeloid leukemia cells and their neighboring stromal cells. Oncotarget 2013, 4, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Roforth, M.M.; Farr, J.N.; Fujita, K.; McCready, L.K.; Atkinson, E.J.; Therneau, T.M.; Cunningham, J.M.; Drake, M.T.; Monroe, D.G.; Khosla, S. Global transcriptional profiling using RNA sequencing and DNA methylation patterns in highly enriched mesenchymal cells from young versus elderly women. Bone 2015, 76, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.M.; Lee, M.Y.; Yun, S.P.; Han, H.J. High glucose regulates cyclin D1/E of human mesenchymal stem cells through TGF-beta1 expression via Ca2+/PKC/MAPKs and PI3K/Akt/mTOR signal pathways. J. Cell. Physiol. 2010, 224, 59–70. [Google Scholar] [PubMed]
- Rölfing, J.H.; Baatrup, A.; Stiehler, M.; Jensen, J.; Lysdahl, H.; Bünger, C. The osteogenic effect of erythropoietin on human mesenchymal stromal cells is dose-dependent and involves non-hematopoietic receptors and multiple intracellular signaling pathways. Stem Cell Rev. 2014, 10, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Sen, B.; Xie, Z.; Case, N.; Thompson, W.R.; Uzer, G.; Styner, M.; Rubin, J. mTORC2 regulates mechanically induced cytoskeletal reorganization and lineage selection in marrow-derived mesenchymal stem cells. J. Bone Miner. Res. 2014, 29, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Song, B.Q.; Chi, Y.; Li, X.; Du, W.J.; Han, Z.B.; Tian, J.J.; Li, J.J.; Chen, F.; Wu, H.H.; Han, L.X.; et al. Inhibition of Notch signaling promotes the adipogenic differentiation of mesenchymal stem cells through autophagy activation and PTEN-PI3K/AKT/mTOR pathway. Cell. Physiol. Biochem. 2015, 36, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, X.; Zhu, W.; Jiang, Z.; Zhou, Y.; Chen, P.; Wang, J. Increased leptin by hypoxic-preconditioning promotes autophagy of mesenchymal stem cells and protects them from apoptosis. Sci. China Life Sci. 2014, 57, 171–180. [Google Scholar] [CrossRef] [PubMed]
- White, E.S.; Sagana, R.L.; Booth, A.J.; Yan, M.; Cornett, A.M.; Bloomheart, C.A.; Tsui, J.L.; Wilke, C.A.; Moore, B.B.; Ritzenthaler, J.D.; et al. Control of fibroblast fibronectin expression and alternative splicing via the PI3K/Akt/mTOR pathway. Exp. Cell Res. 2010, 316, 2644–2653. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Peng, Y.; Gao, D.; Feng, C.; Yuan, X.; Li, H.; Wang, Y.; Yang, L.; Huang, S.; Fu, X. Mesenchymal stem cells suppress fibroblast proliferation and reduce skin fibrosis through a TGF-beta3-dependent activation. Int. J. Low Extrem. Wounds 2015, 14, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Dong, P.; Fu, X.; Li, Q.; Ma, S.; Wu, D.; Kang, N.; Liu, X.; Yan, L.; Xiao, R. CD49f acts as an inflammation sensor to regulate differentiation, adhesion, and migration of human mesenchymal stem cells. Stem Cells 2015, 33, 2798–2810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, Y.J.; Wang, H.; Dong, Q.T.; Wang, T.J.; Qian, H.Y.; Xu, H. Autophagy activation: A novel mechanism of atorvastatin to protect mesenchymal stem cells from hypoxia and serum deprivation via AMP-activated protein kinase/mammalian target of rapamycin pathway. Stem Cells Dev. 2012, 21, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, M.; Wang, Y.; Wang, L.; Jin, Z.; Ding, L.; Zhang, L.; Zhang, L.; Jiang, W.; Gao, G.; et al. Autophagy regulates the apoptosis of bone marrow-derived mesenchymal stem cells under hypoxic condition via AMP-activated protein kinase/mammalian target of rapamycin pathway. Cell Biol. Int. 2016, 40, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Brenner, A.K.; Hagen, K.M.; Liseth, K.; Skrede, S.; Hatfield, K.J.; Bruserud, Ø. The cytokine-mediated crosstalk between primary human acute myeloid cells and mesenchymal stem cells alters the local cytokine network and the global gene expression profile of the mesenchymal cells. Stem Cell Res. 2015, 15, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef] [PubMed]
- Spaggiari, G.M.; Capobianco, A.; Becchetti, S.; Mingari, M.C.; Moretta, L. Mesenchymal stem cell-natural killer cell interactions: Evidence that activated NK cells are capable of killing MSCs, whereas MSCs can inhibit IL-2-induced NK-cell proliferation. Blood 2006, 107, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J. Mesenchymal stem cells in cancer. Stem Cell Rev. 2008, 4, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Chabannes, D.; Hill, M.; Merieau, E.; Rossignol, J.; Brion, R.; Soulillou, J.P.; Anegon, I.; Cuturi, M.C. A role for heme oxygenase-1 in the immunosuppressive effect of adult rat and human mesenchymal stem cells. Blood 2007, 110, 3691–3694. [Google Scholar] [CrossRef] [PubMed]
- Selmani, Z.; Naji, A.; Zidi, I.; Favier, B.; Gaiffe, E.; Obert, L.; Borg, C.; Saas, P.; Tiberghien, P.; Rouas-Freiss, N.; et al. Human leukocyte antigen-G5 secretion by human mesenchymal stem cells is required to suppress T lymphocyte and natural killer function and to induce CD4+CD25highFOXP3+ regulatory T cells. Stem Cells 2008, 26, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Sotiropoulou, P.A.; Perez, S.A.; Gritzapis, A.D.; Baxevanis, C.N.; Papamichail, M. Interactions between human mesenchymal stem cells and natural killer cells. Stem Cells 2006, 24, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Ozaki, K.; Oh, I.; Meguro, A.; Hatanaka, K.; Nagai, T.; Muroi, K.; Ozawa, K. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood 2007, 109, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.L.; Fu, C.J.; Chen, L.; Qin, J.H.; Zeng, Q.; Yuan, H.F.; Nan, X.; Chen, H.X.; Zhou, J.N.; Lin, Y.L.; et al. Mesenchymal stem cells from primary breast cancer tissue promote cancer proliferation and enhance mammosphere formation partially via EGF/EGFR/Akt pathway. Breast Cancer Res. Treat. 2012, 132, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, A.; Bjerkvig, R. Mesenchymal stem cell signaling in cancer progression. Cancer Treat. Rev. 2013, 39, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Che, N.; Li, X.; Zhang, L.; Liu, R.; Chen, H.; Gao, X.; Shi, S.; Chen, W.; Sun, L. Impaired B cell inhibition by lupus bone marrow mesenchymal stem cells is caused by reduced CCL2 expression. J. Immunol. 2014, 193, 5306–5314. [Google Scholar] [CrossRef] [PubMed]
- Corcione, A.; Benvenuto, F.; Ferretti, E.; Giunti, D.; Cappiello, V.; Cazzanti, F.; Risso, M.; Gualandi, F.; Mancardi, G.L.; Pistoia, V.; et al. Human mesenchymal stem cells modulate B-cell functions. Blood 2006, 107, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Glennie, S.; Soeiro, I.; Dyson, P.J.; Lam, E.W.; Dazzi, F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood 2005, 105, 2821–2827. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Prevosto, C.; Massaro, A.M.; Negrini, S.; Urbani, S.; Pierri, I.; Saccardi, R.; Gobbi, M.; Zocchi, M.R. Interaction between human NK cells and bone marrow stromal cells induces NK cell triggering: Role of NKp30 and NKG2D receptors. J. Immunol. 2005, 175, 6352–6360. [Google Scholar] [CrossRef] [PubMed]
- Prevosto, C.; Zancolli, M.; Canevali, P.; Zocchi, M.R.; Poggi, A. Generation of CD4+ or CD8+ regulatory T cells upon mesenchymal stem cell-lymphocyte interaction. Haematologica 2007, 92, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Rosado, M.M.; Bernardo, M.E.; Scarsella, M.; Conforti, A.; Giorda, E.; Biagini, S.; Cascioli, S.; Rossi, F.; Guzzo, I.; Vivarelli, M.; et al. Inhibition of B-cell proliferation and antibody production by mesenchymal stromal cells is mediated by T cells. Stem Cells Dev. 2015, 24, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, S. Role of mesenchymal stem cells in cell life and their signaling. World J. Stem Cells 2014, 6, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; de Rosbo, N.K. The immunomodulatory function of mesenchymal stem cells: Mode of action and pathways. Ann. N. Y. Acad. Sci. 2015, 1351, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Barda-Saad, M.; Rozenszajn, L.A.; Globerson, A.; Zhang, A.S.; Zipori, D. Selective adhesion of immature thymocytes to bone marrow stromal cells: Relevance to T cell lymphopoiesis. Exp. Hematol. 1996, 24, 386–391. [Google Scholar] [PubMed]
- Plumas, J.; Chaperot, L.; Richard, M.J.; Molens, J.P.; Bensa, J.C.; Favrot, M.C. Mesenchymal stem cells induce apoptosis of activated T cells. Leukemia 2005, 19, 1597–1604. [Google Scholar] [CrossRef] [PubMed]
- Touzot, M.; Soulillou, J.P.; Dantal, J. Mechanistic target of rapamycin inhibitors in solid organ transplantation: From benchside to clinical use. Curr. Opin. Organ Transplant. 2012, 17, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Hoogduijn, M.J. Are mesenchymal stromal cells immune cells? Arthritis Res. Ther. 2015, 17, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, H.S.; Lin, J.H.; Hsu, T.W.; Su, K.; Wang, C.W.; Yang, K.Y.; Chiou, S.H.; Hung, S.C. Mesenchymal stem cells enhance lung cancer initiation through activation of IL-6/JAK2/STAT3 pathway. Lung Cancer 2012, 75, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.S.; Yang, S.H.; Lei, Y.P.; Tsai, C.C.; Chen, H.W.; Hsu, C.Y.; Chen, L.L.; Wang, H.W.; Miller, S.A.; Chiou, S.H.; et al. Mesenchymal stem cells promote formation of colorectal tumors in mice. Gastroenterology 2011, 141, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Kucerova, L.; Matuskova, M.; Hlubinova, K.; Altanerova, V.; Altaner, C. Tumor cell behaviour modulation by mesenchymal stromal cells. Mol. Cancer 2010, 9, 129. [Google Scholar] [CrossRef] [PubMed]
- Behl, D.; Porrata, L.F.; Markovic, S.N.; Letendre, L.; Pruthi, R.K.; Hook, C.C.; Tefferi, A.; Elliot, M.A.; Kaufmann, S.H.; Mesa, R.A.; et al. Absolute lymphocyte count recovery after induction chemotherapy predicts superior survival in acute myelogenous leukemia. Leukemia 2006, 20, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Porrata, L.F. Autologous Graft-versus-Tumor Effect: Reality or Fiction? Adv. Hematol. 2016, 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Porrata, L.F.; Litzow, M.R.; Tefferi, A.; Letendre, L.; Kumar, S.; Geyer, S.M.; Markovic, S.N. Early lymphocyte recovery is a predictive factor for prolonged survival after autologous hematopoietic stem cell transplantation for acute myelogenous leukemia. Leukemia 2002, 16, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- De Peppo, G.M.; Marcos-Campos, I.; Kahler, D.J.; Alsalman, D.; Shang, L.; Vunjak-Novakovic, G.; Marolt, D. Engineering bone tissue substitutes from human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 8680–8685. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Garlanda, C.; Jaillon, S.; Marone, G.; Mantovani, A. Tumor associated macrophages and neutrophils in tumor progression. J. Cell. Physiol. 2013, 228, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Mantovani, A.; Sica, A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv. Immunol. 2013, 120, 163–184. [Google Scholar] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A. A broken krebs cycle in macrophages. Immunity 2015, 42, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin. Immunol. 2015, 27, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Fullerton, M.D.; Schertzer, J.D.; Sikkema, S.; Marcinko, K.; Walkley, C.R.; Izon, D.; Honeyman, J.; Chen, Z.P.; van Denderen, B.J.; et al. Hematopoietic AMPK beta1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Investig. 2011, 121, 4903–4915. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.P.; Brown, J.R.; Sag, D.; Zhang, L.; Suttles, J. Adenosine 5′-monophosphate-activated protein kinase regulates IL-10-mediated anti-inflammatory signaling pathways in macrophages. J. Immunol. 2015, 194, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Haubenwallner, S.; Kuttke, M.; Kollmann, I.; Halfmann, A.; Dohnal, A.M.; Chen, L.; Cheng, P.; Hoesel, B.; Einwallner, E.; et al. B Macrophage PTEN regulates expression and secretion of arginase I modulating innate and adaptive immune responses. J. Immunol. 2014, 193, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Rao, J.; Zhu, J.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Wang, X.; Zhai, Y. Myeloid PTEN deficiency protects livers from ischemia reperfusion injury by facilitating M2 macrophage differentiation. J. Immunol. 2014, 192, 5343–5353. [Google Scholar] [CrossRef] [PubMed]
- Arranz, A.; Doxaki, C.; Vergadi, E.; Martinez de la Torre, Y.; Vaporidi, K.; Lagoudaki, E.D.; Ieronymaki, E.; Androulidaki, A.; Venihaki, M.; Margioris, A.N.; et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. USA 2012, 109, 9517–9522. [Google Scholar] [CrossRef] [PubMed]
- Makowski, L.; Boord, J.B.; Maeda, K.; Babaev, V.R.; Uysal, K.T.; Morgan, M.A.; Parker, R.A.; Suttles, J.; Fazio, S.; Hotamisligil, G.S.; et al. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat. Med. 2001, 7, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 2834. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Yu, J.; Luo, Y.; Chen, S.; Wang, W.; Zhao, C.; Sun, Z.; Wu, W.; Guo, W.; Han, Z.; et al. H Tsc1 is a Critical Regulator of Macrophage Survival and Function. Cell. Physiol. Biochem. 2015, 36, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yang, T.; Li, L.; Sun, L.; Hou, Y.; Hu, X.; Zhang, L.; Tian, H.; Zhao, Q.; Peng, J.; et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat. Commun. 2014, 5, 4696. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, W.T.; Pouliot, P.; Bakan, I.; Sabatini, D.M.; Laplante, M. Myeloid-specific Rictor deletion induces M1 macrophage polarization and potentiates in vivo pro-inflammatory response to lipopolysaccharide. PLoS ONE 2014, 9, e95432. [Google Scholar] [CrossRef] [PubMed]
- Baetta, R.; Granata, A.; Canavesi, M.; Ferri, N.; Arnaboldi, L.; Bellosta, S.; Pfister, P.; Corsini, A. Everolimus inhibits monocyte/macrophage migration in vitro and their accumulation in carotid lesions of cholesterol-fed rabbits. J. Pharmacol. Exp. Ther. 2009, 328, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Gao, J.; Zhai, H.; Xiao, J.; Ding, Y.; Hao, J. RAD001 (everolimus) attenuates experimental autoimmune neuritis by inhibiting the mTOR pathway, elevating Akt activity and polarizing M2 macrophages. Exp. Neurol. 2016, 280, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.; Koren, E.; Chan, Y.; Koscec, M.; Sheehy, A.; Kolodgie, F.; Virmani, R.; Feder, D. Effects of everolimus on macrophage-derived foam cell behavior. Cardiovasc. Revasc. Med. 2014, 15, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chang, K.T.; Hung, C.C.; Kuo, C.H.; Hwang, S.J.; Chen, H.C.; Hung, C.H.; Lin, S.F. Effects of the mTOR inhibitor rapamycin on monocyte-secreted chemokines. BMC Immunol. 2014, 15, 37. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Verheye, S.; De Meyer, I.; Timmermans, J.P.; Schrijvers, D.M.; Van Brussel, I.; Bult, H.; De Meyer, G.R. Everolimus triggers cytokine release by macrophages: Rationale for stents eluting everolimus and a glucocorticoid. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Mercalli, A.; Calavita, I.; Dugnani, E.; Citro, A.; Cantarelli, E.; Nano, R.; Melzi, R.; Maffi, P.; Secchi, A.; Sordi, V.; et al. Rapamycin unbalances the polarization of human macrophages to M1. Immunology 2013, 140, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, V.; Arbabi, S.; Garcia, I.A.; Knoll, M.L.; Cuschieri, J.; Bulger, E.M.; Maier, R.V. Role of the mTOR pathway in LPS-activated monocytes: Influence of hypertonic saline. J. Surg. Res. 2011, 171, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Shokri, M.; Bagheri, B.; Garjani, A.; Sohrabi, B.; Habibzadeh, A.; Kazemi, B.; Movassaghpour, A.A. Everolimus-eluting stents reduce monocyte expression of Toll-like receptor 4. Adv. Pharm. Bull. 2015, 5 (Suppl. 1), 643–647. [Google Scholar] [CrossRef] [PubMed]
- Brouard, S.; Puig-Pey, I.; Lozano, J.J.; Pallier, A.; Braud, C.; Giral, M.; Guillet, M.; Londono, M.C.; Oppenheimer, F.; Campistol, J.M.; et al. Comparative transcriptional and phenotypic peripheral blood analysis of kidney recipients under cyclosporin A or sirolimus monotherapy. Am. J. Transplant. 2010, 10, 2604–2614. [Google Scholar] [CrossRef] [PubMed]
- Gallon, L.; Traitanon, O.; Sustento-Reodica, N.; Leventhal, J.; Ansari, M.J.; Gehrau, R.C.; Ariyamuthu, V.; De Serres, S.A.; Alvarado, A.; Chhabra, D.; et al. Cellular and molecular immune profiles in renal transplant recipients after conversion from tacrolimus to sirolimus. Kidney Int. 2015, 87, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Kawano, Y.; Nakae, J.; Watanabe, N.; Fujisaka, S.; Iskandar, K.; Sekioka, R.; Hayashi, Y.; Tobe, K.; Kasuga, M.; Noda, T.; et al. Loss of Pdk1-Foxo1 signaling in myeloid cells predisposes to adipose tissue inflammation and insulin resistance. Diabetes 2012, 61, 1935–1948. [Google Scholar] [CrossRef] [PubMed]
- Weisser, S.B.; McLarren, K.W.; Voglmaier, N.; van Netten-Thomas, C.J.; Antov, A.; Flavell, R.A.; Sly, L.M. Alternative activation of macrophages by IL-4 requires SHIP degradation. Eur. J. Immunol. 2011, 41, 1742–1753. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Westerterp, M.; Wang, C.; Zhu, Y.; Ai, D. Macrophage mTORC1 disruption reduces inflammation and insulin resistance in obese mice. Diabetologia 2014, 57, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Brenner, A.K.; Reikvam, H.; Bruserud, Ø. A subset of patients with acute myeloid leukemia has leukemia cells characterized by chemokine responsiveness and altered expression of transcriptional as well as angiogenic regulators. Front. Immunol. 2016, 7, 205. [Google Scholar] [CrossRef] [PubMed]
- Reikvam, H.; Øyan, A.M.; Kalland, K.H.; Hovland, R.; Hatfield, K.J.; Bruserud, Ø. Differences in proliferative capacity of primary human acute myelogenous leukaemia cells are associated with altered gene expression profiles and can be used for subclassification of patients. Cell Prolif. 2013, 46, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Ryningen, A.; Olsnes, A.M.; Stordrange, L.; Øyan, A.M.; Kalland, K.H.; Gjertsen, B.T. Subclassification of patients with acute myelogenous leukemia based on chemokine responsiveness and constitutive chemokine release by their leukemic cells. Haematologica 2007, 92, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Maffei, R.; Fiorcari, S.; Martinelli, S.; Potenza, L.; Luppi, M.; Marasca, R. Targeting neoplastic B cells and harnessing microenvironment: The “double face” of ibrutinib and idelalisib. J. Hematol. Oncol. 2015, 8, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, K.R. Therapeutic potential of new B cell-targeted agents in the treatment of elderly and unfit patients with chronic lymphocytic leukemia. J. Hematol. Oncol. 2015, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Tvedt, T.H.A.; Reikvam, H.; Aasebø, E.; Hernandez-Valladares, M.; Berven, F.S.; Selheim, F.; Bruserud, Ø. How should quality of life assessment be integrated in the evaluation of patients with acute myeloid leukemia? Expert Rev. Qual. Life Cancer Care 2016, 1, 373–387. [Google Scholar] [CrossRef]
- Kidd, S.; Spaeth, E.; Klopp, A.; Andreeff, M.; Hall, B.; Marini, F.C. The (in) auspicious role of mesenchymal stromal cells in cancer: Be it friend or foe. Cytotherapy 2008, 10, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef] [PubMed]
- Ghiaur, G.; Wroblewski, M.; Loges, S. Acute myelogenous leukemia and its microenvironment: A molecular conversation. Semin. Hematol. 2015, 52, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Blau, O.; Baldus, C.D.; Hofmann, W.K.; Thiel, G.; Nolte, F.; Burmeister, T.; Turkmen, S.; Benlasfer, O.; Schumann, E.; Sindram, A.; et al. Mesenchymal stromal cells of myelodysplastic syndrome and acute myeloid leukemia patients have distinct genetic abnormalities compared with leukemic blasts. Blood 2011, 118, 5583–5592. [Google Scholar] [PubMed]
- Sun, Z.; Wang, S.; Zhao, R.C. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J. Hematol. Oncol. 2014, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø. Cellular immune responses in acute leukaemia patients with severe chemotherapy-induced leucopenia; characterization of the cytokine repertoire of clonogenic T cells. Cancer Immunol. Immunother. 1998, 46, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Ehninger, G.; Hamann, W.; Pawelec, G. Secretion of IL-2, IL-3, IL-4, IL-6 and GM-CSF by CD4+ and CD8+ TCR alpha beta+ T-cell clones derived early after allogeneic bone marrow transplantation. Scand. J. Immunol. 1993, 38, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Pawelec, G. Human T lymphocyte activation in the presence of acute myelogenous leukaemia blasts; studies of normal polyclonal T cells and T lymphocyte clones derived early after allogenic bone marrow transplantation. Cancer Immunol. Immunother. 1996, 42, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Pawelec, G. Interleukin-13 secretion by normal and posttransplant T lymphocytes; in vitro studies of cellular immune responses in the presence of acute leukaemia blast cells. Cancer Immunol. Immunother. 1997, 45, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, Ø.; Ulvestad, E. Acute myelogenous leukemia blasts as accessory cells during in vitro T lymphocyte activation. Cell. Immunol. 2000, 206, 36–50. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Direct Inhibition of the PI3K-Akt-mTOR Pathway Members |
|---|
| PI3K inhibitors |
| Pan-PI3K inhibitors: buparlisib, pilaralisib, pictilisib |
| Isoform-specific inhibitors: alpelisib, tazelisib, CAL-101, GDC-0941 |
| Others: MVP-BAG956 (PI3K-PDK1), resveratrol (PI3K/Akt) |
| Dual PI3K-mTOR inhibitors |
| NVP-BEZ235, LY3023414, GSK2126458 |
| Akt inhibitors |
| MK-2206, uprosertib, ipatasertib, AZD5363 |
| mTORC1 inhibitors |
| Sirolimus, everolimus, temsirolimus, ridaforolimus |
| Dual mTORC1/2 inhibitors |
| LNK128, AZD8055, MLN0138, CC-223 |
| Indirect Inhibition—Activation of Pathway Inhibitors |
| AMPK agonists: metformin, A-769662GSK621 |
| PTEN activation: l-sercurinine |
| Adhesion Molecules and Other Cell Surface Molecules Involved in Local Cell-Cell or Cell-Matrix Contact | ||
|---|---|---|
| CD90: Thy-1 | A cell surface glycoprotein and member of the immunoglobulin superfamily involved in cell adhesion and cell communication in numerous cell types, including stem cells. | |
| CD29: Integrin β1 [48] | Integrins are heterodimeric proteins that mediate bidirectional communication across the cell membrane. They are made up of α and β subunits. At least 18 α and eight β subunits have been described. This protein is a β subunit. | |
| CD31: PECAM-1 [55] | This is a cell surface protein; it can be a part of intercellular junctions and is probably involved in leukocyte migration and integrin activation. | |
| CD44: HCAM [50] | This cell-surface glycoprotein is a receptor for hyaluronic acid and is involved in cell-cell interactions, cell adhesion and migration. It can also interact with other ligands, such as osteopontin, collagens, and matrix metalloproteinases (MMPs). | |
| CD48a/e/f: Integrins α1/α5/α6 [48,52] | These integrins are members of the immunoglobulin-like receptor family; it does not have a transmembrane domain, however, but is held at the cell surface by a GPI anchor via a C-terminal domain which may be cleaved to yield a soluble form of the receptor. | |
| CD54: ICAM-1 [97] | This cell surface glycoprotein binds to integrins of type CD11a /CD18, or CD11b/CD18. | |
| CD56 NCAM [48] | This cell adhesion protein is a member of the immunoglobulin superfamily, and it is involved in cell-cell as well as cell-matrix interactions. | |
| CD62L/P: L/P-selectin [55] | CD62P: This 140 kDa membrane protein is a calcium-dependent receptor that binds to sialylated forms of carbohydrate antigens. CD62L: This cell surface adhesion molecule can mediate binding of leucocytes. | |
| CD106: VCAM-1 [98] | This member of the Ig superfamily is a cell surface sialoglycoprotein mediating cell-cell adhesion and signal transduction. | |
| CD146: MCAM [98] | Probably acting as a cell adhesion molecule. | |
| Cadherin-11/Cadherin-2 [48] | Cadherin-11 is a type II classical cadherin from the cadherin superfamily, integral membrane proteins that mediate calcium-dependent cell-cell adhesion. Type II (atypical) cadherins are defined based on their lack of a HAV cell adhesion recognition sequence specific to type I cadherins. This cadherin seems to have a specific function in bone development. Cadherin-2 is a classical cadherin, i.e., a calcium-dependent cell adhesion molecule and glycoprotein. | |
| Cell Surface Cytokine Receptors | ||
| CD95: Endoglin RNG | A homodimeric transmembrane protein, it is a component of the transforming growth factor β (TGF-β) receptor complex | |
| CD71: Transferrin receptor protein 1 [47] | This cell surface receptor is necessary for cellular iron uptake by the process of receptor-mediated endocytosis. | |
| CD117: c-Kit [99] | This protein is the receptor for the stem cell factor (SCF). | |
| CD135: Flt3 [55] | This receptor is activated by binding of the Flt3 ligand. | |
| CD166: ALCAM [46] | This protein is a member of a subfamily of immunoglobulin receptors and is a CD6 receptor; it is implicated in cell migration. | |
| CD271: LNGFR [96] | This is the nerve growth factor receptor. | |
| CD349: Frizzled-9 [52] | Members of the ‘frizzled’ gene family encode transmembrane proteins that are receptors for Wnt signalling proteins. | |
| CCR1/4/6/7/9/10 CXCR4/5/6 CX3CR1 [94,95,100] | These are all chemokine receptors that can bind a wide range of CCL and CXCL chemokines; mediators which are important regulators of cell trafficking, cell cycle progression and cell survival. | |
| Enzymes | ||
| CD73: ecto 5′ nucleotidase | A plasma membrane protein that catalyses the conversion of extracellular nucleotides to membrane-permeable nucleosides. | |
| CD10: Neprilysin [96] | A glycoprotein that is a neutral endopeptidase that cleaves peptides at the amino side of hydrophobic residues and inactivates several peptide hormones | |
| CD13: Alanine aminopeptidase [96] | A plasma membrane protein; the large extracellular carboxy-terminal domain contains a pentapeptide consensus sequence characteristic of members of the zinc-binding metalloproteinase superfamily. The enzyme was thought to be involved in the metabolism of regulatory peptides. | |
| Thrombospondin [48] | This protein has several distinct regions, including a metalloproteinase domain, a disintegrin-like domain, and a thrombospondin type 1 motif. | |
| Additional MSC-Expressed Molecules | ||
| CD157: Stromal cell antigen [95] | This glycosylphosphatidylinositol-anchored molecule can facilitate cell growth. | |
| Nestin [52] | This protein is a member of the intermediate filament protein family. | |
| Sox-2 [50] | This protein is a member of the SRY-related HMG-box (SOX) family of transcription factors required for stem-cell maintenance. | |
| OCT-4 [50] | This transcription factor is important for stem cell pluripotency. | |
| Aging of MSCs: PI3K-Akt-mTOR Inhibition Maintains an Immature State |
| BM MSCs show decreased self-renewal, differentiation and function with aging; inhibition of the PI3K-Akt-mTOR pathway preserves the immature state and prevents the development of the age-related phenotype [109]. Increased expression of the transcription factors NANOG and OCT-1 may be responsible for this. Furthermore, a comparison of BM MSCs for younger (<30 years of age) and elderly (>70 years of age) individuals showed increased expression of genes associated with mTOR signalling [123]. Studies of both murine and human BM MSCs have shown that miR-188 regulates the age-related switch between osteoblast and adipocyte differentiation, and miR-188 then targets rictor [116]. Animal studies have shown decreased BM levels of IGF-1 in aged rats; IGF-1 seems to stimulate osteoblastic MSC differentiation through activation of mTOR and aging of MSCs may thus be caused by decreased mTOR signalling [131]. |
| Metabolic Regulation |
| MSC proliferation is regulated by extracellular glucose levels; this effect is mediated through both the PKC-MAPK and PI3K-Akt-mTOR pathways [124]. Glycogen synthase kinase 3 β is a metabolic regulator; inactivation of this regulator can be caused by signalling through mTORC2 and Akt phosphorylation at S437 [104]. |
| Differentiation of MSCs—General Effects |
| Murine studies suggest that absence of mTORC1 causes reduced capacity of adipocyte differentiation, whereas absence of mTOCR2 causes reduced osteogenic differentiation capacity and accelerated adipogenesis [91,126]. mTORC2 regulates mechanically induced cytoskeletal reorganization (actin stress fibre development) and favours osteogenesis over adipogenesis [126]. The stemness marker CD49f identifies a subset with high proliferative ability and differentiation potential; downregulation of this marker (i.e., knockdown, tumour necrosis factor α, TNF-α, treatment) is associated with decreased differentiation and downregulation through TNF-α is mediated by mTOR [132]. |
| Osteogenic Differentiation of MSCs |
| IGF-1-induced growth enhancement and osteoblastic differentiation of MSCs is inhibited by mTORC1 inhibitor rapamycin; this IGF-1effect is seen for MSCs derived from different tissues including BM [106,131]. The osteogenic effect of erythropoietin is mediated through various intracellular pathways, including signalling through PI3K and mTOR [113,125]. There seems to be a time-dependent modulation of AMPK-Akt-mTOR signalling during osteogenic differentiation with early activation of AMPK/raptor and thereby mTOR/S6K1 inhibition, and later activation of Akt/mTOR [120]. Stat3 activation seems to be a negative regulator of osteogenic differentiation [115]. BMP-2 and -4 stimulate osteogenic differentiation; JAK2 signalling then mediates Stat3 tyrosine phosphorylation whereas serine phosphorylation is mediated through ERK1/2 and mTOR signalling. Stat3 knockdown accelerates and augments osteogenic differentiation. mTOR inhibitors can increase osteogenic differentiation [119], and studies in human BM MSCs suggest that osteopenia can be induced through PI3K-Akt-mTOR and activation of S6K1 [118]. However, the effects of the PI3K-Akt-mTOR pathway are complex and effects of pathway inhibitors are difficult to predict. Induction of osteogenic differentiation has not been detected in all experimental models, and a possible explanation is that the final effect of mTOR inhibitors depends on the experimental model and the biological context [2]. However, the dual PI3K/mTOR inhibitor BEZ235 strongly inhibited osteogenic differentiation in human MSCs [119]. |
| Adipocytic Differentiation of MSCs |
| Adipocytic differentiation is associated with downregulation of Notch gene expression; modulation of PTEN-PI3K-Akt-mTOR signalling seems important for this Notch effect [127]. Insulin, Akt and mTOR signalling is important in adipocyte differentiation and rapamycin can reduce the expression of most adipocyte markers [2]. mTOR is essential for adipocytes to sense nutrient availability and modulation of PPAR-γ activity that is an important regulator of the adipogenic gene expression program [2]. Differentiation of brown adipocytes requires signalling pathways distinct from white adipocytes; mTOR activity is involved in the initial steps but later inhibition through AMPK activation is also necessary [2]. |
| Myogenic Differentiation |
| A recent review concluded that several studies suggest that mTOR is indispensable for myogenesis, but the mechanisms behind this function are largely unknown [2]. |
| Regulation of Autophagy and Senescence in MSCs |
| Autophagy is the natural regulated mechanism that disassembles unnecessary or dysfunctional cellular components. Cellular senescence is the phenomenon by which normal diploid cells cease to divide, but they remain metabolically active and commonly adopt an immunogenic phenotype consisting of a pro-inflammatory secretome. These two processes seem to be regulated by overlapping mechanisms. AMPK is a positive regulator of autophagy in MSCs; autophagy can then be activated through the AMPK-mTOR pathway and protect BM MSCs from stress-induced apoptosis [133]. Animal studies suggest that senescent BM MSCs show upregulation of p53 and downregulation of mTOR. Knockdown of p53 then alleviates senescence, reduces autophagy and upregulates mTOR [134]. Human BM MSCs show downregulation of Notch gene expression during adipocyte differentiation; Notch inhibition will also enhance adipocyte differentiation and at the same time induce autophagy by acting on the PTEN-PI3K-Akt-mTOR pathway [127]. mTOR inhibition can also reverse the senescent phenotype of human MSCs [110]. Thus, the PI3K-Akt-mTOR pathway is involved in the regulation of senescence/autophagy/apoptosis in BM MSCs. |
| Adaptation to the Hypoxic BM Microenvironment |
| The BM microenvironment is hypoxic [105,107,111]; the hypoxia seems to causes upregulation of hypoxia-inducible factor 1α (HIF-1α) in primary human AML cells and increased constitutive release of several cytokines by the leukemic cells [112]. Hypoxia induces autophagy and eventually apoptosis in BM MSCs; at the same time hypoxia seems to activate AMPK-mTOR signalling and inhibition of mTOR will then further increase the hypoxia-induced apoptosis [134]. However, hypoxia stimulated by Toll-like receptor (TLR) ligation show decreased apoptosis in response to hypoxia and at the same time increased autophagy and activated AMPK-mTOR signalling [117]. Furthermore, downregulation of leptin will attenuate hypoxia-induced autophagy [128]. Finally, hypoxia also increases the levels of fatty acid synthetase in umbilical cord MSCs; increased signalling through HIF-1α-fatty acid synthase-mTORC1 then represents an important link between hypoxia-induced lipid metabolism and increased proliferation as well as migration of MSCs [114]. Thus, autophagy seems to protect MSCs against hypoxia-induced apoptosis; AMPK-mTOR signalling seems important for regulation of autophagy and thereby also for the adaptation of MSCs to a hypoxic BM microenvironment together with leptin and possibly p53 (see above). |
| Communication between MSCs and Neighbouring Cells |
| Fibroblasts: Fibroblasts can support AML cell proliferation; they also show constitutive release of leukaemia-supporting/angioregulatory cytokines and this release can be altered by PI3K/mTOR inhibition [76]. Conditioned medium from cultures of BM MSCs suppresses fibroblast proliferation; this effect is mediated mainly by TGF-β3 [130]. However, MSCs release a wide range of soluble mediators and other forms of TGF-β that signal through the same receptors [108]; probably, other cytokines/chemokines may also contribute to this effect. PI3K-Akt-mTOR signalling is a downstream effect to TGF-receptors, and this pathway is also important for fibroblast proliferation (both mTORC1 and mTORC2), adherence and release of extracellular matrix molecules [108,121,122,129]. Thus, PI3K-Akt-mTOR targeting may alter this crosstalk between MSCs and fibroblasts both through effects on MSCs and the fibroblasts. Osteoblasts: mTOR/S6K1 signalling is important for osteoblast responses to exogenous cytokines and for the regulation of osteoblast cytokine release [2], including cytokines that can support leukemogenesis and modulate other stromal cells including MSCs [2]. |
| Soluble Mediators Released by MSCs |
|---|
| Interleukins |
| IL-1α/β [47], IL-6 [55,136], IL-10 [139,140], IL-15 [141] |
| Chemokines |
| CCL2 [94,95], CCL3 [94], CCL4 [95], CCL5 [94,95], CCL7 [53], CCL20 [95], CCL26 [53], CXCL1 [53], CXCL2 [53], CXCL5 [53], CXCL8 [94,136], CXCL10 [53], CXCL11 [53], CXCL12 [94,95], CX3CL1 [95] |
| Growth factors |
| Ang-1 [94], VEGF [94,136], TGF-β [139,140,142], PDGF [50], bFGF [50], FGF7 [50], HGF [139,140,142], IGF-1 [45], EGF [45], G-CSF [55], M-CSF [55], GM-CSF [55], SCF [55], LIF [55], IFN-β [136] |
| Other mediators |
| PGE2 [139,140,142] |
| Soluble Mediators Commonly Released by Various Malignant Cells |
| IL-6 [93,143], Ang-1 [144], VEGF [144], TGF-β, BMP-4 [143], Wnt5α [144], Gremlin-1 [144], bFGF [143,144], HGF [143], IGF-I/II [143], EGF [144], CTGF [143], G-CSF [144], CCL5 [93,143], CXCL12 [93,143] |
| T Cells |
|
| NK Cells |
|
| B Cells |
|
| Dendritic Cells |
| Media-TOR | M1/M2 Ratio | Comment | Reference |
|---|---|---|---|
| Akt1 | M1↑M2↓ | Akt is expressed as the three different isoform Akt1, Akt2 and Akt 3. These isoforms contributes differentially to differentiation of monocytes into the two main forms: M1: Reduced responsiveness to TLR ligands, reduced secretion of IL-1βα, IL-6 and TNF-α. M2: C/EBPβ seems important for M2 differentiation, Stat6 also seems to be involved together with miR-155. The Akt1 isoform facilitates differentiation into the M2 phenotype, and Akt2 downregulation upregulates C/EBPβ. Akt1 is important for Akt-mediated effects on ECs whereas Akt2 is important in insulin signalling. | [174] |
| Akt2 | M1↓M2↑ | Akt2 induces differentiation in direction of the pro-inflammatory M1 phenotype. | [174,175] |
| AMPKα1 | M1↓M2↑ | AMPK activation in macrophages results in results in polarization to the anti-inflammatory M2 phenotype. Exposure of macrophages to IL-10 causes AMPK activation, and AMPKα1 is then required for IL-10 activation of PI3K-Akt-mTORC1 and Stat3-mediated anti-inflammatory pathways regulating macrophage polarization. | [171] |
| AMPKβ1 | M1↓M2↑ | Animal studies demonstrated that AMPKβ1 deficient macrophages are M1-activated, i.e., AMPK seems to differentiate macrophages towards an immunosuppressive M2 phenotype. | [170] |
| PTEN | M1↓M2↑ | PTEN is important for the increased release of pro-inflammatory cytokines such as IL-6 by macrophages in response to TLR ligation, and deletion of PTEN then results in diminished inflammatory responses. Furthermore, macrophages isolated from such knockout mice express higher levels of M2 markers, produce lower TNF-α and higher IL-10 levels in response to TLR ligation. Such M2 macrophages also show enhanced Stat3- and Stat6-signalling together with diminished Stat1-signalling pathway activation in response to TLR4 stimulation. | [172,173] |
| PDK1 | M1↑M2↓ | A major characteristic of mice with myeloid PDK1 knockout is increased tissue infiltration of macrophages with the M1 phenotype; the authors concluded that PDK1 regulates macrophage migration through inhibition of FOXO-1 induced CCR2 expression. | [190] |
| Inpp5d | M2↑ | Inositol polyphosphate-5-phosphatase D. The expression of this protein is restricted to haematopoietic cells and it functions as a negative regulator of myeloid cell proliferation and survival. Deficient murine monocytes are more sensitive to IL4-induced induction of the M2 phenotype. | [191] |
| TSC1 | M1↑M2↓ | Knockout of TSC1 in the myeloid lineage causes constitutive mTORC1 activation with downregulation of Akt signalling that is essential for resistance to M2 polarization and increased responsiveness to pro-inflammatory stimuli. Thus, the effect can at least partly be explained by increased mTORC1 activity with a negative feedback on Akt function. The TSC1 deficient cells show impaired migration and reduced expression of chemokine receptors, including CCR2 and CCR5, phagocytosis and reactive oxygen species production is increased and the effect of the knockdown is partially reversed by mTORC1 inhibitors. | [176,177,178] |
| Raptor | M1↓M2↑ | Raptor deficiency reduced inflammatory gene expression in macrophages derived from several organs, including BM macrophages. This seems to be caused by attenuation of Akt inactivation and increased NFκB signalling. | [192] |
| Rictor | M1↑M2↓ | Primary macrophages isolated from myeloid-specific rictor null mice exhibited an exaggerated response to TLRs ligands, and expressed high levels of M1 genes and lower levels of M2 markers. | [179] |
| Hallmark | Drug | Cells | Effect | Reference |
|---|---|---|---|---|
| Cytokine Production | Sirolimus | THP-1 human AML monocytic cell line Normal human monocytes | Sirolimus reduced release of CCL2, CCL3, CCL5 and CXCL8 in both human and murine monocytes; CCL4 was in addition reduced in human cells. There was no effect on TNF-α release. | [183] |
| Everolimus | C57BL/6 murine cells Normal human monocytes | While mTOR inhibition did not lead to any changes during starvation, everolimus significantly increased production of IL-6, CCL2, CCL5 and TNF-α and except for CCL2 this increase was inhibited by MAPK inhibition. | [184] | |
| Rapamycin | Normal human monocytes tested in vivo and in vitro | Rapamycin induced apoptosis of M2- but not M1 polarized cells. M1 polarized: rapamycin reduced the release of CXCR4 and expression of CD206 and CD209; also reduced stem cell growth factor β, CCL4 and CCL13. M2 polarized: rapamycin increased expression of CD86, CCR7, IL-6 and TNF-α; reduced CD206, IL-10, VEGF and CCL18. Stimulation by LPS, Listeria Pam3Cys or flagellin; increased release of IL12p40 and IL-23, reduced TNF-α and IL-6. | [185] | |
| Everolimus | Rat monocytes, in vivo studies | Histological examination of induced experimental neuritis showed that everolimus significantly (i) increased accumulation of M2 cells, spleen M2 cells were also increased; (ii) mRNA levels of INF-γ and IL-17 were reduced whereas they increased for TGF-β and IL-4; (iii) cytokine protein levels showed reduced IL-1α, IFN-γ and CCL5 but increased IL-10 levels. | [181] | |
| Rapamycin | Human, in vitro | Rapamycin decreased IL-6 and IL-10 but did not affect TNF-α release after LPS exposure. | [186] | |
| In vivo migration | Everolimus | In vivo and in vitro studies Monocytes | Everolimus reduced migration of macrophages to atherosclerotic plaque in the carotis wall; in vitro studies showed reduced migration towards CCL2, CXCL3, CXCL8, C5a and N-formylmethionyl-leucyl-phenylalanine. | [180] |
| Foam cells formation | Everolimus | THP-1 foam cells | Decreased viability of foam cells, no effect on release of IL-1β, CXCL8, TNF-α but reduced release of CCL2; increase cellular clustering. | [182] |
| Expression of TLRs | Everolimus | Normal human monocytes, in vitro studies | A significant increase in TLR expression by monocytes was seen in patients with drug eluting stents compared with bare metal stents. | [187] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brenner, A.K.; Andersson Tvedt, T.H.; Bruserud, Ø. The Complexity of Targeting PI3K-Akt-mTOR Signalling in Human Acute Myeloid Leukaemia: The Importance of Leukemic Cell Heterogeneity, Neighbouring Mesenchymal Stem Cells and Immunocompetent Cells. Molecules 2016, 21, 1512. https://doi.org/10.3390/molecules21111512
Brenner AK, Andersson Tvedt TH, Bruserud Ø. The Complexity of Targeting PI3K-Akt-mTOR Signalling in Human Acute Myeloid Leukaemia: The Importance of Leukemic Cell Heterogeneity, Neighbouring Mesenchymal Stem Cells and Immunocompetent Cells. Molecules. 2016; 21(11):1512. https://doi.org/10.3390/molecules21111512
Chicago/Turabian StyleBrenner, Annette K., Tor Henrik Andersson Tvedt, and Øystein Bruserud. 2016. "The Complexity of Targeting PI3K-Akt-mTOR Signalling in Human Acute Myeloid Leukaemia: The Importance of Leukemic Cell Heterogeneity, Neighbouring Mesenchymal Stem Cells and Immunocompetent Cells" Molecules 21, no. 11: 1512. https://doi.org/10.3390/molecules21111512
APA StyleBrenner, A. K., Andersson Tvedt, T. H., & Bruserud, Ø. (2016). The Complexity of Targeting PI3K-Akt-mTOR Signalling in Human Acute Myeloid Leukaemia: The Importance of Leukemic Cell Heterogeneity, Neighbouring Mesenchymal Stem Cells and Immunocompetent Cells. Molecules, 21(11), 1512. https://doi.org/10.3390/molecules21111512