
Cryptolepine, a Plant Alkaloid, Inhibits the Growth of Non-Melanoma Skin Cancer Cells through Inhibition of Topoisomerase and Induction of DNA Damage

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
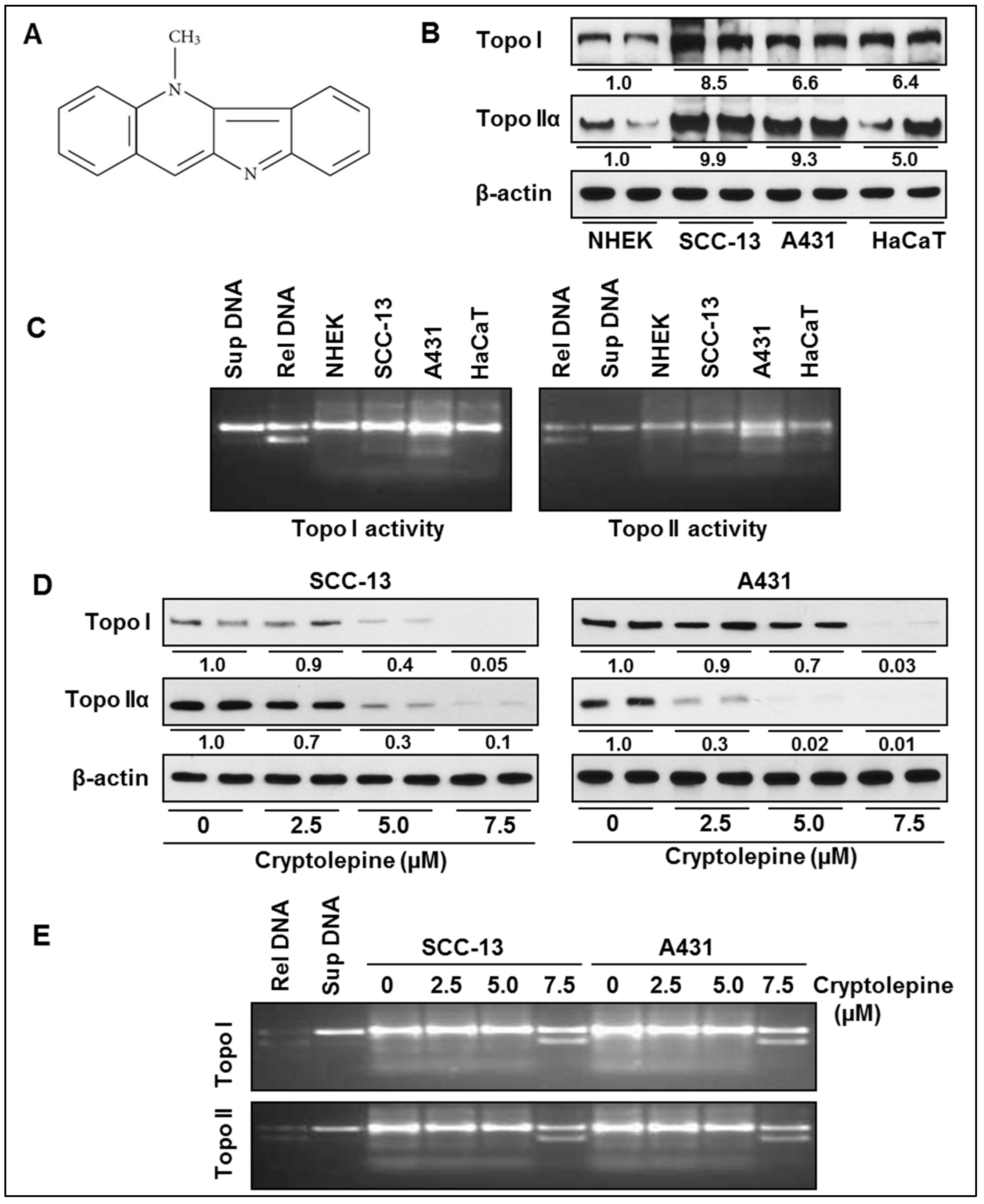
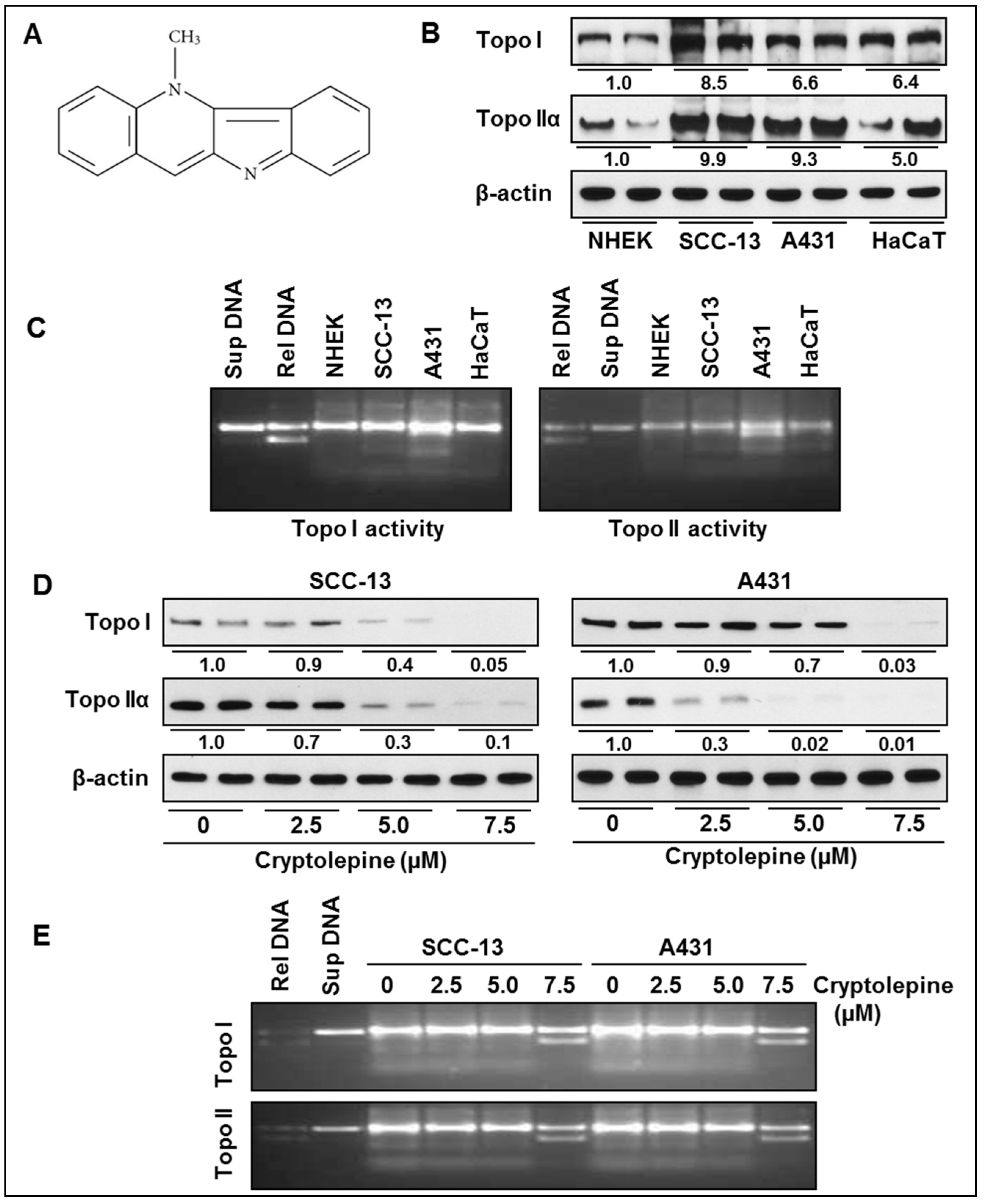
2.1. Basal Expression and Activity of Topoisomerases in NMSC Cells
2.2. Cryptolepine Inhibits Topoisomerase Expression and Activity in NMSC Cells
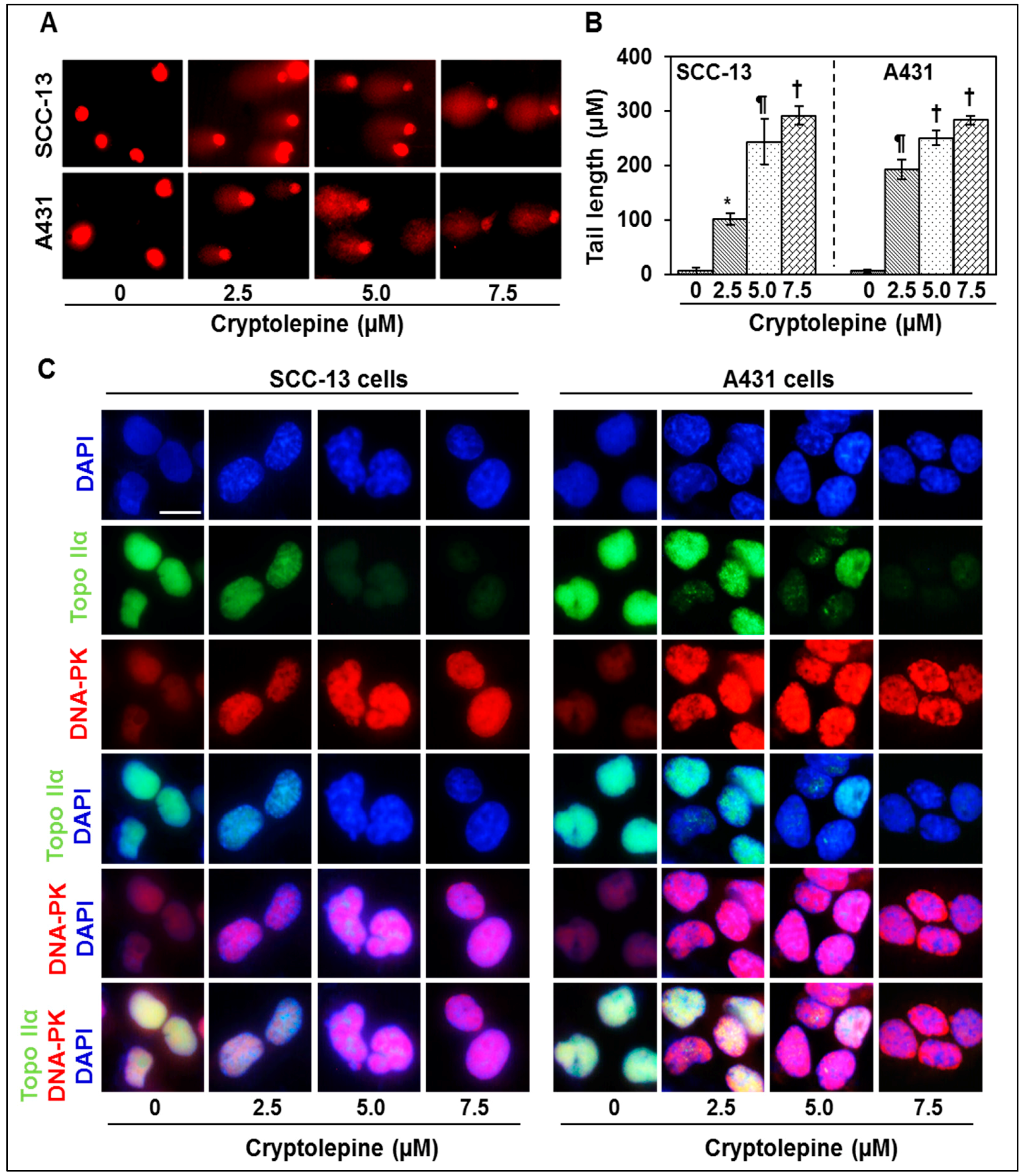
2.3. Cryptolepine Induces DNA Damage in NMSC Cells
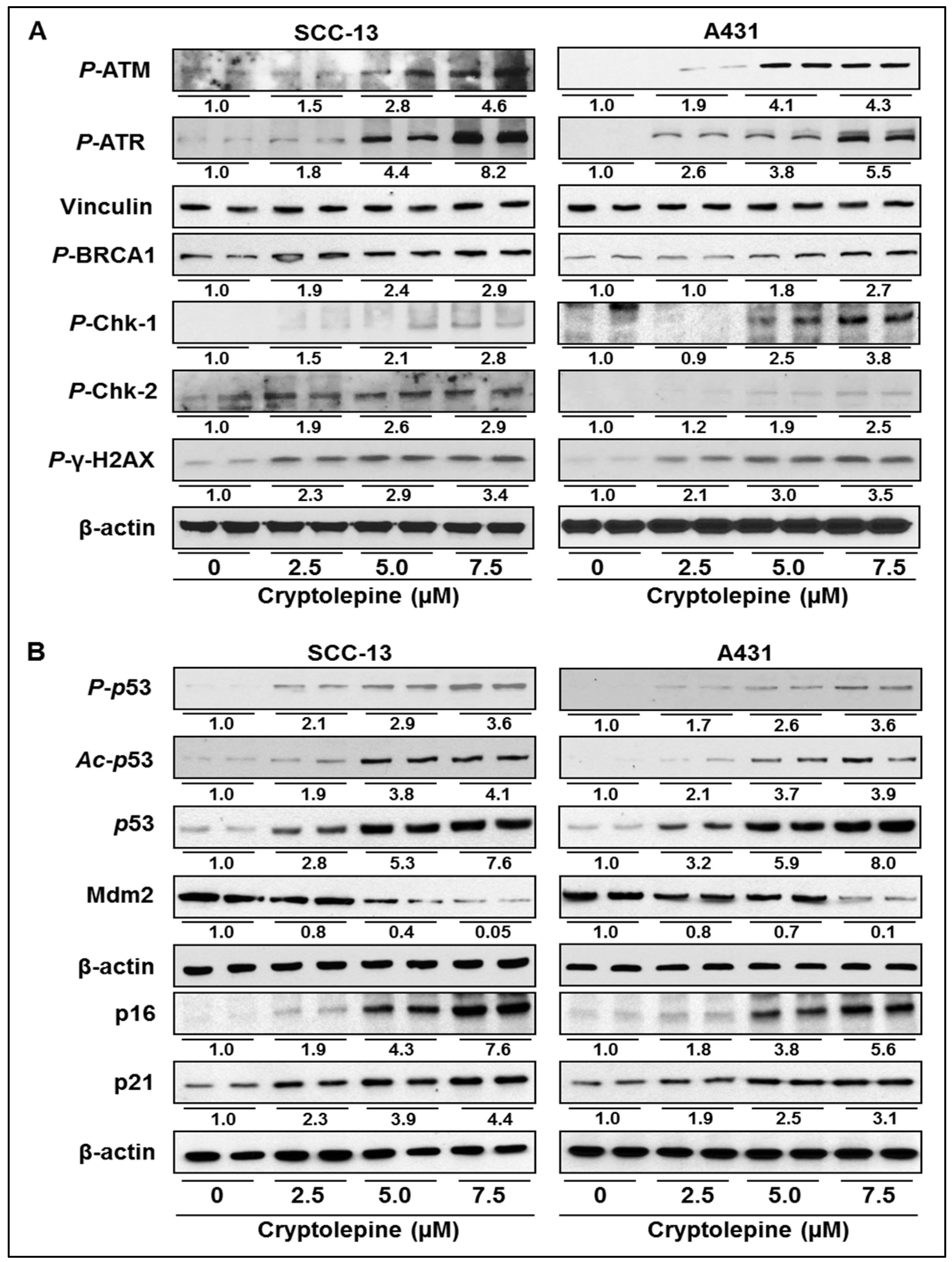
2.4. Cryptolepine Enhances the Expression of DNA Damage Response Mediator and Effector Cascade in NMSC Cell
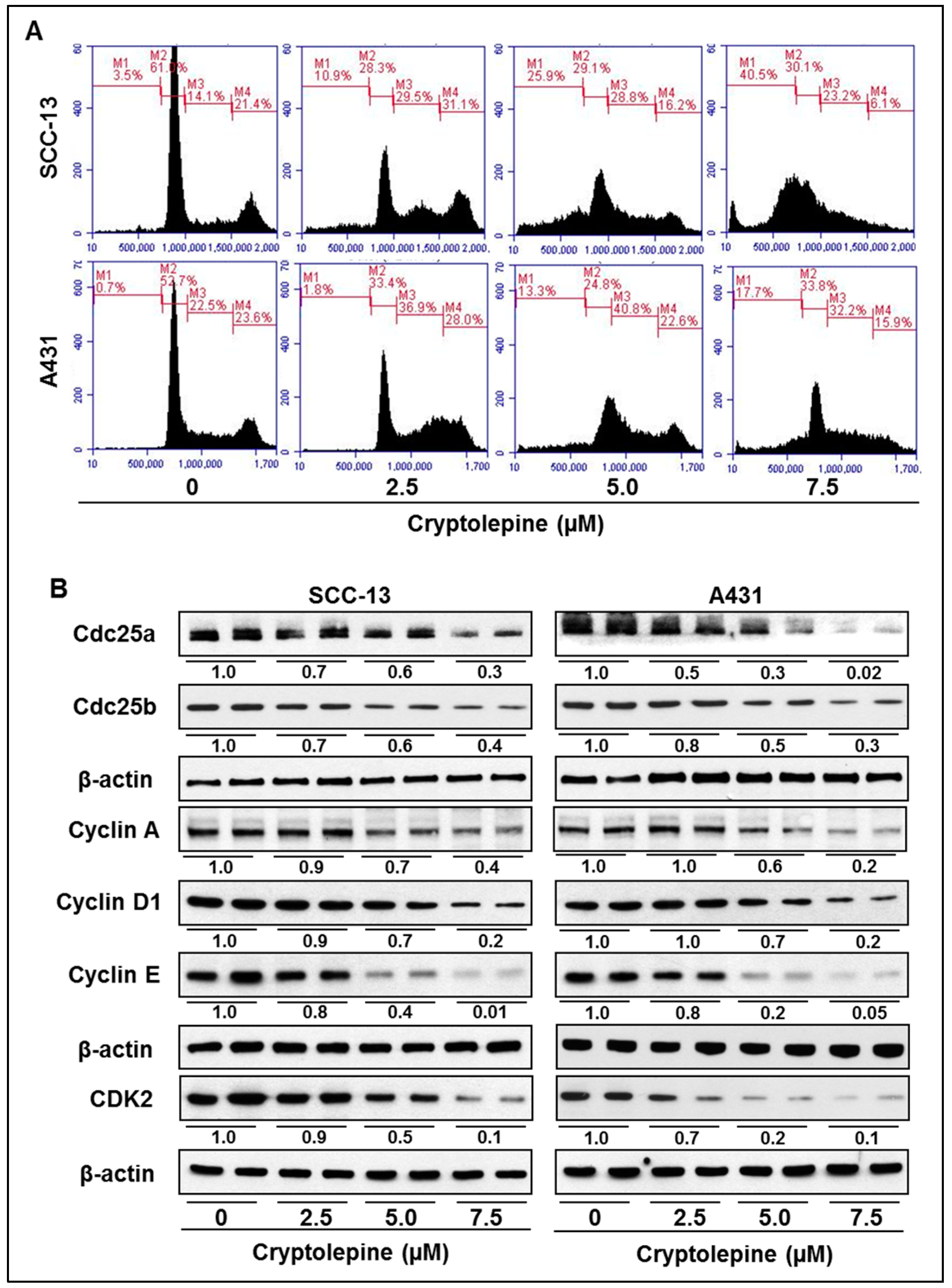
2.5. Cryptolepine Induces S-Phase Cell Cycle Arrest in NMSC Cells
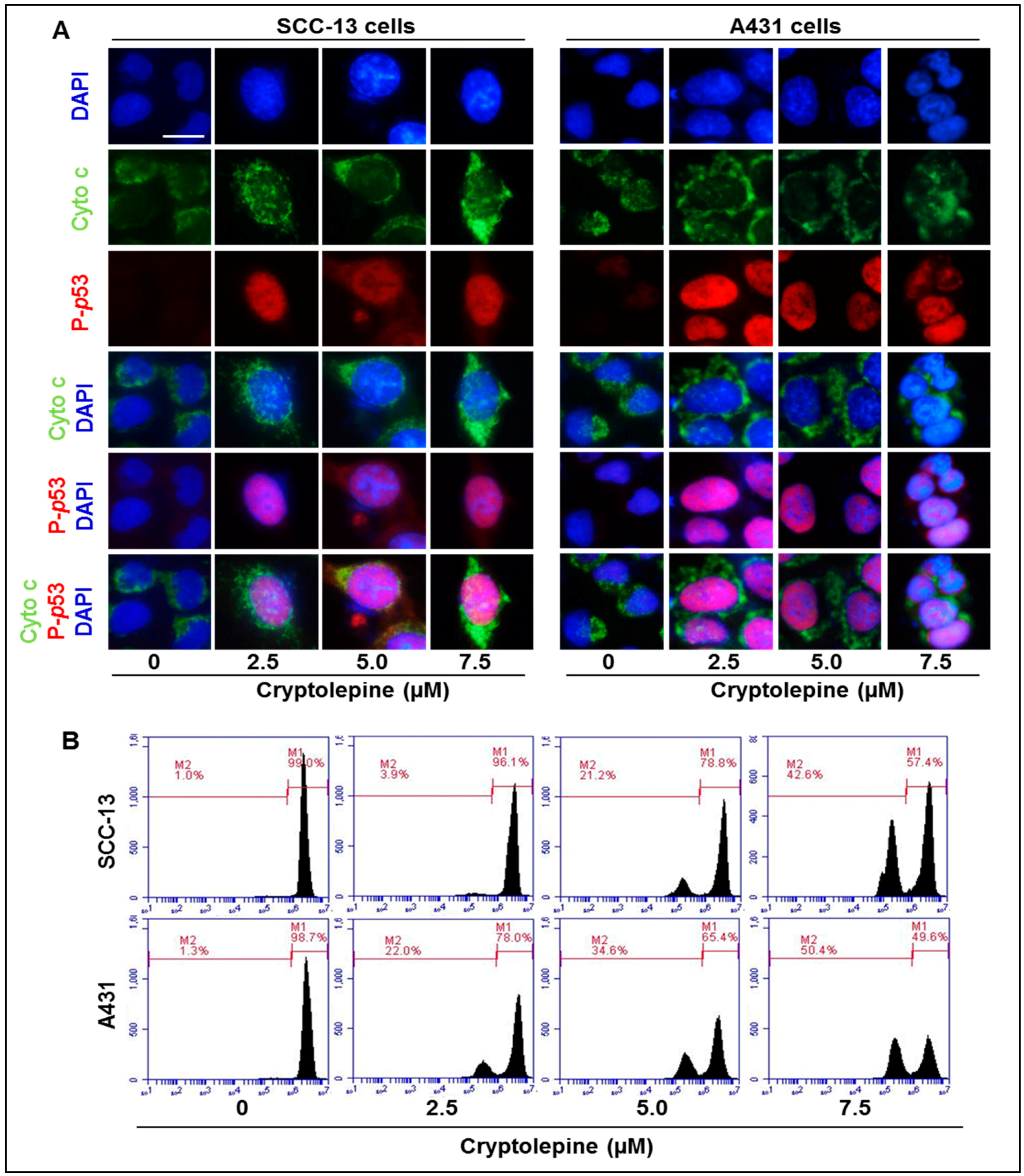
2.6. Cryptolepine Induces Disruption of Mitochondrial Membrane Potential in NMSC Cells
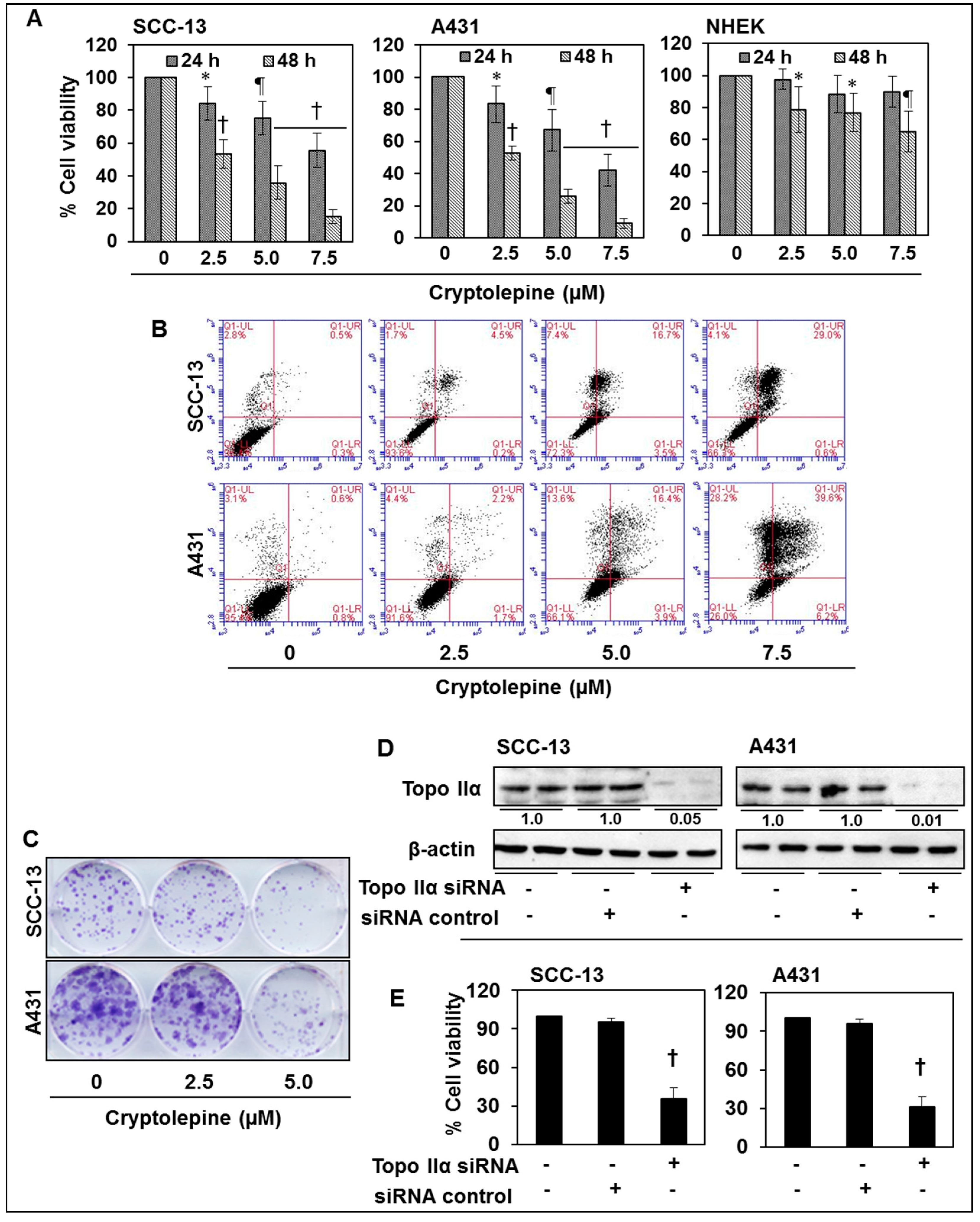
2.7. Cryptolepine Inhibits Cell Viability and Induces Apoptotic Cell Death in NMSC Cells
2.8. siRNA Knock-Down of Topo IIα in NMSC Cells Results in Inhibition of Cell Viability
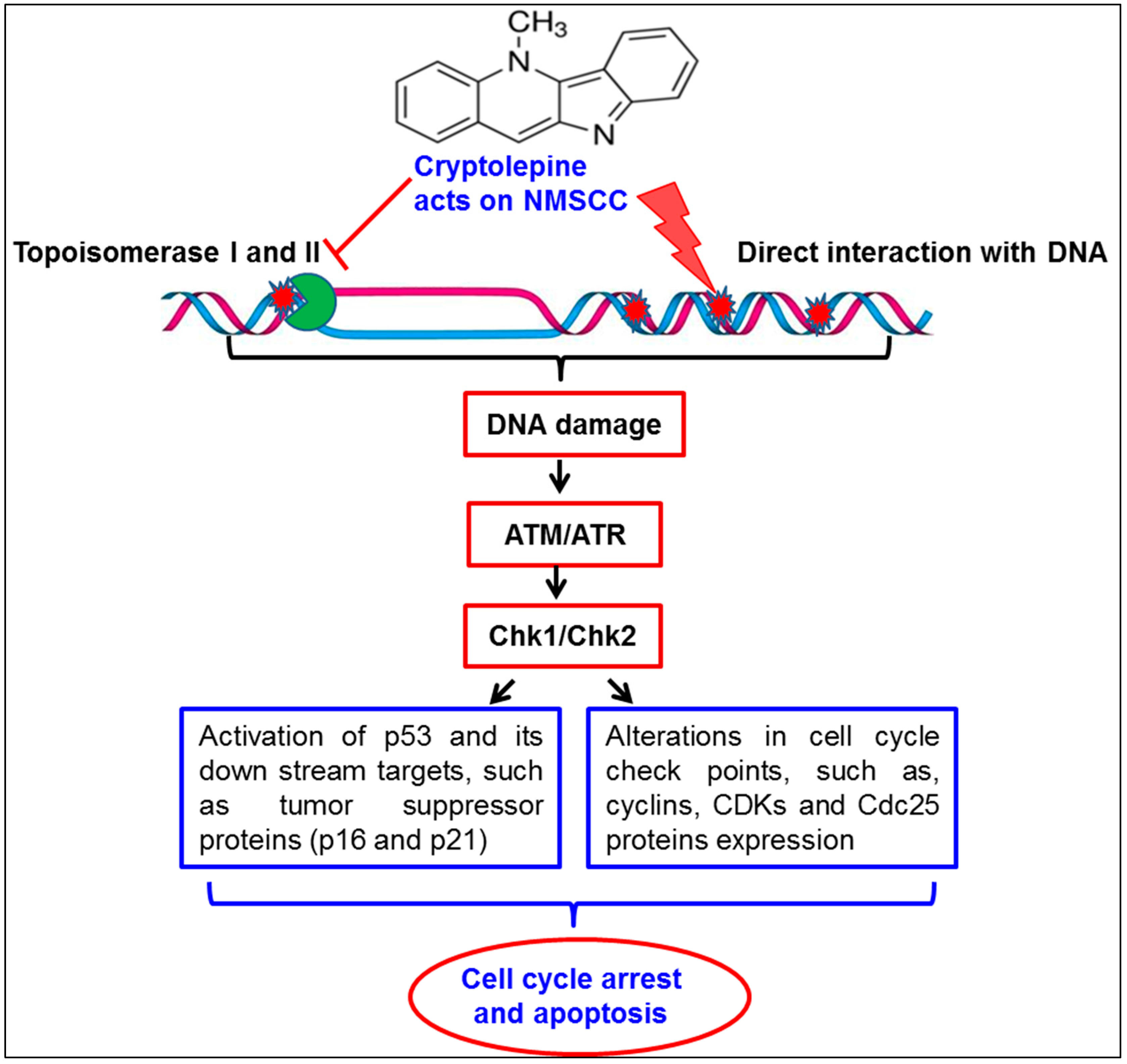
3. Discussion
4. Materials and Methods
4.1. Cryptolepine, Chemicals, Reagents and Antibodies
4.2. Cells and Cell Culture Conditions
4.3. Preparation of Topoisomerase (I & II) Extract from Cells
4.4. Topoisomerase I Enzyme Activity Assay
4.5. Topoisomerase II Enzyme Activity Assay
4.6. Knockdown of Topo IIα Expression in NMSC Cells Using siRNA
4.7. Analysis of DNA Damage by Comet Assay
4.8. Preparation of Cell Lysates and Western Blot Analysis
4.9. Immunofluorescence Staining
4.10. Cell Cycle Analysis
4.11. Mitochondrial Membrane Potential Analysis
4.12. MTT Assay For Cell Viability
4.13. Apoptotic Cell Death Analysis
4.14. Cell Colony Formation Assay
4.15. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ansah, C.; Khan, A.; Gooderham, N.J. In vitro genotoxicity of the West African anti-malarial herbal Cryptolepis sanguinolenta and its major alkaloid cryptolepine. Toxicology 2005, 208, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Ansah, C.; Mensah, K.B. A review of the anticancer potential of the antimalarial herbal Cryptolepis sanguinolenta and its major alkaloid cryptolepine. Ghana Med. J. 2013, 47, 137–147. [Google Scholar] [PubMed]
- Kirby, G.C.; Paine, A.; Warhurst, D.C.; Noamese, B.K.; Phillipson, J.D. In vitro and in vivo antimalarial activity of cryptolepine, a plant-derived indoloquinoline. Phytother. Res. 1995, 9, 359–363. [Google Scholar] [CrossRef]
- Gibbons, S.; Fallah, F.; Wright, C.W. Cryptolepine hydrochloride: A potent antimycobacterial alkaloid derived from Cryptolepis sanguinolenta. Phytother. Res. 2003, 17, 434–436. [Google Scholar] [CrossRef] [PubMed]
- Sawer, I.K.; Berry, M.I.; Brown, M.W.; Ford, J.L. The effect of cryptolepine on the morphology and survival of Escherichia coli, Candida albicans and Saccharomyces cerevisiae. J. Appl. Bacteriol. 1995, 79, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Oyekan, A.O.; Botting, J.H.; Noamesi, B.K. Cryptolepine inhibits platelet aggregation in vitro and in vivo and stimulates fibrinolysis ex vivo. Gen. Pharmacol. 1988, 19, 233–237. [Google Scholar] [CrossRef]
- Bierer, D.E.; Fort, D.M.; Mendez, C.D.; Luo, J.; Imbach, P.A.; Dubenko, L.G.; Jolad, S.D.; Gerber, R.E.; Litvak, J.; Lu, Q.; et al. Ethnobotanical-directed discovery of the antihyperglycemic properties of cryptolepine: Its isolation from Cryptolepis sanguinolenta, synthesis, and in vitro and in vivo activities. J. Med. Chem. 1998, 41, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Olajide, O.A.; Ajayi, A.M.; Wright, C.W. Anti-inflammatory properties of cryptolepine. Phytother. Res. 2009, 23, 1421–1425. [Google Scholar] [CrossRef] [PubMed]
- Olajide, O.A.; Bhatia, H.S.; de Oliveira, A.C.; Wright, C.W.; Fiebich, B.L. Anti-neuroinflammatory properties of synthetic cryptolepine in human neuroblastoma cells: Possible involvement of NF-κB and p38 MAPK inhibition. Eur. J. Med. Chem. 2013, 63, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Olajide, O.A.; Bhatia, H.S.; de Oliveira, A.C.; Wright, C.W.; Fiebich, B.L. Inhibition of Neuroinflammation in LPS-Activated Microglia by Cryptolepine. Evid. Based Complement. Alternat. Med. 2013, 2013, 459723. [Google Scholar] [CrossRef] [PubMed]
- Olajide, O.A.; Heiss, E.H.; Schachner, D.; Wright, C.W.; Vollmar, A.M.; Dirsch, V.M. Synthetic cryptolepine inhibits DNA binding of NF-κB. Bioorg. Med. Chem. 2007, 15, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gooderham, N.J. Mechanisms of induction of cell cycle arrest and cell death by cryptolepine in human lung adenocarcinoma a549 cells. Toxicol. Sci. 2006, 91, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.A.; Sowa, Y.; Murata, H.; Takagi, K.; Nakanishi, R.; Aoki, S.; Yoshikawa, M.; Kobayashi, M.; Sakabe, T.; Kubo, T.; et al. The plant alkaloid cryptolepine induces p21WAF1/CIP1 and cell cycle arrest in a human osteosarcoma cell line. Int. J. Oncol. 2007, 31, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Laryea, D.; Isaksson, A.; Wright, C.W.; Larsson, R.; Nygren, P. Characterization of the cytotoxic activity of the indoloquinoline alkaloid cryptolepine in human tumour cell lines and primary cultures of tumour cells from patients. Investig. New Drugs 2009, 27, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Bonjean, K.; de Pauw-Gillet, M.C.; Defresne, M.P.; Colson, P.; Houssier, C.; Dassonneville, L.; Bailly, C.; Greimers, R.; Wright, C.; Quetin-Leclercq, J.; et al. The DNA intercalating alkaloid cryptolepine interferes with topoisomerase II and inhibits primarily DNA synthesis in B16 melanoma cells. Biochemistry 1998, 37, 5136–5146. [Google Scholar] [CrossRef] [PubMed]
- Dassonneville, L.; Bonjean, K.; de Pauw-Gillet, M.C.; Colson, P.; Houssier, C.; Quetin-Leclercq, J.; Angenot, L.; Bailly, C. Stimulation of topoisomerase II-mediated DNA cleavage by three DNA-intercalating plant alkaloids: Cryptolepine, matadine, and serpentine. Biochemistry 1999, 38, 7719–7726. [Google Scholar] [CrossRef] [PubMed]
- Lisgarten, J.N.; Coll, M.; Portugal, J.; Wright, C.W.; Aymami, J. The antimalarial and cytotoxic drug cryptolepine intercalates into DNA at cytosine-cytosine sites. Nat. Struct. Biol. 2002, 9, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. A journey in the world of DNA rings and beyond. Annu. Rev. Biochem. 2009, 78, 31–54. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Dermatol. 2015, 151, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.C.; Divecha, N.; Lakin, N.D.; Jackson, S.P. DNA-dependent protein kinase and related proteins. Biochem. Soc. Symp. 1999, 64, 91–104. [Google Scholar] [PubMed]
- Kemp, M.G.; Lindsey-Boltz, L.A.; Sancar, A. The DNA damage response kinases DNA-dependent protein kinase (DNA-PK) and ataxia telangiectasia mutated (ATM) are stimulated by bulky adduct-containing DNA. J. Biol. Chem. 2011, 286, 19237–19246. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Li, M.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 2259–2264. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.; Tovar, C.; Yang, H.; Carvajal, D.; Vu, B.T.; Xu, Q.; Wahl, G.M.; Heimbrook, D.C.; Vassilev, L.T. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J. Biol. Chem. 2004, 279, 53015–53022. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Chen, Z.; Gunasekera, A.H.; Sowin, T.J.; Rosenberg, S.H.; Fesik, S.; Zhang, H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J. Biol. Chem. 2003, 278, 21767–21773. [Google Scholar] [CrossRef] [PubMed]
- Busino, L.; Chiesa, M.; Draetta, G.F.; Donzelli, M. Cdc25A phosphatase: Combinatorial phosphorylation, ubiquitylation and proteolysis. Oncogene 2004, 23, 2050–2056. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Bossy-Wetzel, E.; Goldstein, J.C.; Fitzgerald, P.; Green, D.R. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J. Biol. Chem. 2000, 275, 7337–7342. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Multiple pathways of cytochrome c release from mitochondria in apoptosis. Biochim. Biophys. Acta 2006, 1757, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Pal, H.C.; Baxter, R.D.; Hunt, K.M.; Agarwal, J.; Elmets, C.A.; Athar, M.; Afaq, F. Fisetin, a phytochemical, potentiates sorafenib-induced apoptosis and abrogates tumor growth in athymic nude mice implanted with BRAF-mutated melanoma cells. Oncotarget 2015, 6, 28296–28311. [Google Scholar] [PubMed]
- Norbury, C.J.; Zhivotovsky, B. DNA damage-induced apoptosis. Oncogene 2004, 23, 2797–2808. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.; Chen, J. The DNA damage response pathways: At the crossroad of protein modifications. Cell Res. 2008, 18, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Benchimol, S. p53-dependent pathways of apoptosis. Cell Death Differ. 2001, 8, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; González, D.; Marcos-Villar, L.; Campagna, M.; Gallego, P.; González-Santamaría, J.; Herranz, D.; Gu, W.; Serrano, M.; Aaronson, S.A.; et al. Acetylation is indispensable for p53 antiviral activity. Cell Cycle 2011, 10, 3701–3715. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Siddik, Z.H.; Huang, Z.; Wang, R.; Koomen, J.; Kobayashi, R.; Khokhar, A.R.; Kuang, J. Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene 2005, 24, 2929–2943. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Borgne, A.; Mulner, O.; Chong, J.P.; Blow, J.J.; Inagaki, N.; Inagaki, M.; Delcros, J.G.; Moulinoux, J.P. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur. J. Biochem. 1997, 243, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Liu, J.; Legerski, R.J. Cyclin E is stabilized in response to replication fork barriers leading to prolonged S phase arrest. J. Biol. Chem. 2009, 284, 35325–35337. [Google Scholar] [CrossRef] [PubMed]
- Jirawatnotai, S.; Hu, Y.; Livingston, D.M.; Sicinski, P. Proteomic identification of a direct role for cyclin d1 in DNA damage repair. Cancer Res. 2012, 72, 4289–4293. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. p21 is a universal inhibitor of cyclin kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Bronson, R.T.; Jacks, T. p21 is a critical CDK2 regulator essential for proliferation control in Rb-deficient cells. J. Cell Biol. 1998, 141, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.J.; Keramaris, E.; Rideout, H.J.; Slack, R.S.; Dyson, N.J.; Stefanis, L.; Park, D.S. Cyclin-dependent kinases and P53 pathways are activated independently and mediate Bax activation in neurons after DNA damage. J. Neurosci. 2001, 21, 5017–5026. [Google Scholar] [PubMed]
- Katiyar, S.K.; Mantena, S.K.; Meeran, S.M. Silymarin protects epidermal keratinocytes from ultraviolet radiation-induced apoptosis and DNA damage by nucleotide excision repair mechanism. PLoS ONE 2011, 6, e21410. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Katiyar, S.K. Polyphenols from green tea inhibit the growth of melanoma cells through inhibition of class I histone deacetylases and induction of DNA damage. Genes Cancer 2015, 6, 49–61. [Google Scholar] [PubMed]
- Vaid, M.; Singh, T.; Prasad, R.; Kappes, J.C.; Katiyar, S.K. Therapeutic intervention of proanthocyanidins on the migration capacity of melanoma cells is mediated through PGE2 receptors and β-catenin signaling molecules. Am. J. Cancer Res. 2015, 5, 3325–3338. [Google Scholar] [PubMed]
- Singh, T.; Gupta, N.A.; Xu, S.; Prasad, R.; Velu, S.E.; Katiyar, S.K. Honokiol inhibits the growth of head and neck squamous cell carcinoma by targeting epidermal growth factor receptor. Oncotarget 2015, 6, 21268–21282. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Katiyar, S.K. Down-regulation of miRNA-106b inhibits growth of melanoma cells by promoting G1-phase cell cycle arrest and reactivation of p21/WAF1/Cip1 protein. Oncotarget 2014, 5, 10636–10649. [Google Scholar] [CrossRef] [PubMed]
- Pal, H.C.; Sharma, S.; Elmets, C.A.; Athar, M.; Afaq, F. Fisetin inhibits growth, induces G₂/M arrest and apoptosis of human epidermoid carcinoma A431 cells: Role of mitochondrial membrane potential disruption and consequent caspases activation. Exp. Dermatol. 2013, 22, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Vaid, M.; Singh, T.; Prasad, R.; Katiyar, S.K. Bioactive proanthocyanidins inhibit growth and induce apoptosis in human melanoma cells by decreasing the accumulation of β-catenin. Int. J. Oncol. 2016, 48, 624–634. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compound cryptolepine are available from the Sigma-Aldrich company.






© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pal, H.C.; Katiyar, S.K. Cryptolepine, a Plant Alkaloid, Inhibits the Growth of Non-Melanoma Skin Cancer Cells through Inhibition of Topoisomerase and Induction of DNA Damage. Molecules 2016, 21, 1758. https://doi.org/10.3390/molecules21121758
Pal HC, Katiyar SK. Cryptolepine, a Plant Alkaloid, Inhibits the Growth of Non-Melanoma Skin Cancer Cells through Inhibition of Topoisomerase and Induction of DNA Damage. Molecules. 2016; 21(12):1758. https://doi.org/10.3390/molecules21121758
Chicago/Turabian StylePal, Harish C., and Santosh K. Katiyar. 2016. "Cryptolepine, a Plant Alkaloid, Inhibits the Growth of Non-Melanoma Skin Cancer Cells through Inhibition of Topoisomerase and Induction of DNA Damage" Molecules 21, no. 12: 1758. https://doi.org/10.3390/molecules21121758