
On the Traceability of Commercial Saffron Samples Using 1H-NMR and FT-IR Metabolomics


 ,
, 
Abstract
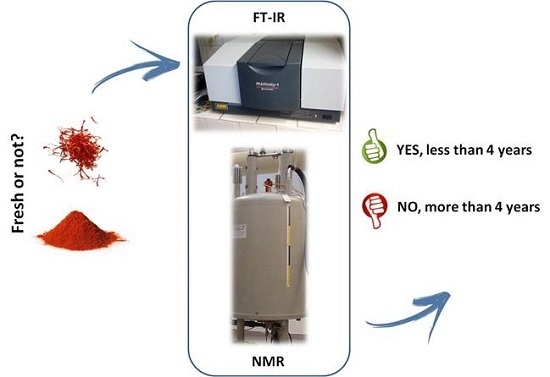
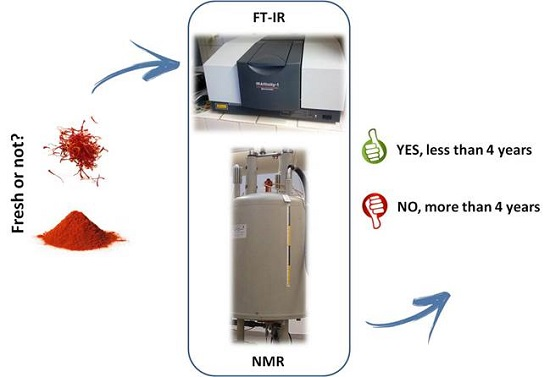
:
1. Introduction
2. Results and Discussion
2.1. UV–VIS and HPLC–DAD Analysis
2.2. FT-IR Analysis
2.3. NMR Analysis
3. Experimental Section
3.1. Saffron Samples
3.2. Standards, Reagents, and Solvents
3.3. Extraction of Crocetin Esters and Picrocrocin
3.4. UV–VIS Spectrophotometric Examination
3.5. Liquid Chromatographic Analysis
3.6. FT-IR Analysis
3.6.1. Sample Preparation
3.6.2. Data Acquisition and Pre-Processing
3.6.3. Multivariate Statistical Analysis
3.7. 1H-NMR Analysis
3.7.1. Sample Preparation
3.7.2. 1H-NMR Data Acquisition
3.7.3. 1H-NMR Data Processing
3.7.4. Multivariate Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DModX | Distance to the model |
| DMSO | Dimethylsulfoxide |
| DRIFTS | Diffuse Reflectance Infrared Fourier Transform Spectroscopy |
| FT-IR | Fourier Transform Infrared Spectroscopy |
| g | gravitational force |
| HPLC-DAD | High performance liquid chromatography-diode array detector |
| MIR | Mid-Infrared |
| NIR | Near-infrared |
| NMR | Nuclear Magnetic Resonance |
| OPLS-DA | Orthogonal Projection to Latent Structures-Discriminant Analysis |
| PCA | Principal Component Analysis |
| PDO | Protected Designation of Origin |
| PLS-DA | Projection to Latent Structures-Discriminant Analysis |
| rpm | Revolutions per minute |
| trans-4-GG | trans-4-digentiobiosyl ester of crocetin |
| UV-Vis | Ultraviolet-visible spectroscopy |
References
- Ordoudi, S.; Tsimidou, M. Saffron quality: Effect of agricultural practices, processing and storage. In Practices and Quality Assessment of Food, 1st ed.; Dris, R., Jain, S.M., Eds.; Kluwer Academic Publisher: Dordrecht, The Netherlands, 2004; Volume 1, pp. 209–260. [Google Scholar]
- Moore, J.C.; Spink, J.; Lipp, M. Development and application of a database of food ingredient fraud and economically motivated adulteration from 1980 to 2010. J. Food Sci. 2012, 77, R118–R126. [Google Scholar] [CrossRef] [PubMed]
- COST Action FA 1101: Omics Technologies for Crop Improvement, Traceability, Determination of Authenticity, Adulteration and Origin in Saffron. Available online: http://www.cost.eu/COST_Actions/fa/Actions/FA1101 (accessed on 24 February 2016).
- Cevallos-Cevallos, J.M.; Reyes-De-Corcuera, J.I. Metabolomics in Food Science. Adv. Food Nutr. Res. 2012, 67, 1–24. [Google Scholar] [PubMed]
- Mannina, L.; Sobolev, A.P.; Viel, S. Liquid state 1H high field NMR in food analysis. Prog. Nucl. Mag. Reson. Spectrosc. 2012, 66, 1–39. [Google Scholar]
- Wishart, D.S. Metabolomics: Applications to food science and nutrition research. Trends Food Sci. Technol. 2008, 19, 482–493. [Google Scholar]
- Consonni, R.; Cagliani, L.R. Nuclear Magnetic Resonance and Chemometrics to Assess Geographical Origin and Quality of Traditional Food Products. Adv. Food Nutr. Res. 2010, 59, 87–165. [Google Scholar] [PubMed]
- Sun, D.-W. Infrared Spectroscopy for food quality analysis and control, 1st ed.; Elsevier: New York, NY, USA, 2009; pp. 83–104. [Google Scholar]
- Karoui, R.; Downey, G.; Blecker, C. Mid-infrared spectroscopy coupled with chemometrics: A tool for the analysis of intact food systems and the exploration of their molecular structure-quality relationships—A review. Chem. Rev. 2010, 110, 6144–6168. [Google Scholar] [CrossRef] [PubMed]
- Engelsen, S.B.; Nørgaard, L. Comparative vibrational spectroscopy for determination of quality parameters in amidated pectins as evaluated by chemometrics. Carbohydr. Polym. 1996, 30, 9–24. [Google Scholar] [CrossRef]
- Reinholds, I.; Bartkevics, V.; Silvis, I.C.J.; van Ruth, S.M.; Esslinger, S. Analytical techniques combined with chemometrics for authentication and determination of contaminants in condiments: A review. J. Food Compos. Anal. 2015, 44, 56–72. [Google Scholar] [CrossRef]
- Assimiadis, M.K.; Tarantilis, P.A.; Polissiou, M.G. UV-Vis, FT-Raman and 1H-NMR spectroscopies of cis-trans carotenoids from saffron (Crocus sativus L.). Appl. Spectrosc. 1998, 52, 519–522. [Google Scholar]
- Van Calsteren, M.R.; Bissonnette, M.C.; Cormier, F.; Dufrense, C.; Ichi, T.; le Blanc, D.; Perreault, J.C.Y.; Roewer, I. Spectroscopic characterization of crocetin derivatives from Crocus sativus and Gardenia jasminoides. J. Agric. Food Chem. 1997, 45, 1055–1061. [Google Scholar]
- Pfister, S.; Meyer, P.; Steck, A.; Pfander, H. Isolation and structure elucidation of carotenoid-glycosyl esters in gardenia fruits (Gardenia jasminoides Ellis) and saffron (Crocus sativus Linnaeus). J. Agric. Food Chem. 1996, 44, 2612–2615. [Google Scholar] [CrossRef]
- Straubinger, M.; Jezussek, M.; Waibel, R.; Winterhalter, P. Novel glycosidic constituents from saffron. J. Agric. Food Chem. 1997, 45, 1678–1681. [Google Scholar] [CrossRef]
- Yilmaz, A.; Nyberg, N.T.; Molgaard, P.; Asili, J.; Jaroszewsk, J.W. 1H NMR metabolic fingerprinting of saffron extracts. Metabolomics 2010, 6, 511–517. [Google Scholar] [CrossRef]
- Cagliani, L.R.; Culeddu, N.; Chessa, M.; Consonni, R. NMR investigations for quality assessment of Italian PDO saffron (Crocus sativus L.). Food Control 2015, 50, 342–348. [Google Scholar]
- Anastasaki, E.; Kanakis, C.; Pappas, C.; Maggi, L.; del Campo, C.P.; Carmona, M.; Alonso, G.L.; Polissiou, M.G. Geographical differentiation of saffron by GC–MS/FID and chemometrics. Eur. Food Res. Technol. 2009, 229, 899–905. [Google Scholar] [CrossRef]
- Zalacain, A.; Ordoudi, S.A.; Díaz-Plaza, E.M.; Carmona, M.; Blázquez, I.; Tsimidou, M.Z.; Alonso, G.L. Near-infrared spectroscopy in saffron quality control: Determination of chemical composition and geographical origin. J. Agric. Food Chem. 2005, 53, 9337–9341. [Google Scholar] [CrossRef] [PubMed]
- Ordoudi, S.A.; de los Mozos Pascual, M.; Tsimidou, M.Z. On the quality control of traded saffron by means of transmission Fourier-transform mid-infrared (FT-MIR) spectroscopy and chemometrics. Food Chem. 2014, 150, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Ordoudi, S.A.; Cagliani, L.R.; Lalou, S.; Naziri, E.; Tsimidou, M.Z.; Consonni, R. 1H-NMR-based metabolomics of saffron reveals markers for its quality deterioration. Food Res. Int. 2015, 70, 1–6. [Google Scholar] [CrossRef]
- ISO. Saffron (Crocus sativus Linneaus) Part 1: Specifications. In International Organization for Standardization; ISO 3632–1; ISO: Geneva, Switzerland, 2011. [Google Scholar]
- Del Campo, C.P.; Carmona, M.; Maggi, L.; Kanakis, C.D.; Anastasaki, E.G.; Tarantilis, P.A.; Polissiou, M.G.; Alonso, G.L. Picrocrocin content and quality categories in different (345) worldwide samples of saffron (Crocus sativus L.). J. Agric. Food Chem. 2010, 58, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Kyriakoudi, A.; Chrysanthou, A.; Mantzouridou, F.; Tsimidou, M.Z. Revisiting extraction of bioactive apocarotenoids from Crocus sativus L. dry stigmas (saffron). Anal.Chim. Acta 2012, 755, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, A.M.; Carmona, M.; Zalacain, A.; Carot, J.M.; Jabaloyes, J.M.; Alonso, G.L. Rapid determination of crocetin esters and picrocrocin from saffron spice (Crocus sativus L.) using UV-visible spectrophotometry for quality control. J. Agric. Food Chem. 2008, 56, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Kyriakoudi, A.; Ordoudi, S.A.; Roldan-Medina, M.; Tsimidou, M.Z. Saffron, A Functional Spice. Austin J. Nutr. Food Sci. 2015, 3, 1059–1063. [Google Scholar]
- Kyriakoudi, A.; Tsimidou, M.Z. A Food-grade approach to isolate crocetin from saffron (Crocus sativus L.) extracts. Food Anal. Methods 2015, 8, 2261–2272. [Google Scholar] [CrossRef]
- ISO. Saffron (Crocus sativus Linneaus) Part 2: Test methods. In International Organization for Standardization; ISO 3632-2; ISO: Geneva, Switzerland, 2010. [Google Scholar]
- Tarantilis, P.; Tsoupras, G.; Polissiou, M. Determination of saffron (Crocus sativus L.) components in crude plant extract using high-performance liquid chromatography-UV-visible photodiode-array detection mass spectrometry. J. Chromatogr. A 1995, 699, 107–118. [Google Scholar] [CrossRef]
- Carmona, M.; Zalacain, A.; Sánchez, A.M.; Novella, J.L.; Alonso, G.L. Crocetin esters, picrocrocin and its related compounds present in Crocus sativus stigmas and Gardenia jasminoides fruits. Tentative identification of seven new compounds by LC-ESI-MS. J. Agric. Food Chem. 2006, 54, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available by the authors.


{kind=link}
{kind=link}
| Quality Parameters | Samples per Commercial Category | ||
|---|---|---|---|
| I | II | III | |
| 1, 2, 3, 4, 6, 8, 9, 10, 17 | 7, 14, 16 | 5, 11, 12, 13, 15 | |
| E1%440nm | 199–275 (min 200) | 172–190 (min 170) | 141–162 (min 120) |
| E1%257nm | 83–100 (min 70) | 74–85 (min 55) | 69–79 (min 40) |
| E1%330nm | 35–49 (20–50) | 41–46 (20–50) | 39–49 (20–50) |
| Sample | Y Predicted FRESH (0–4 years) | Y Predicted NON-FRESH (5–14 years) |
|---|---|---|
| 1 * | 0.5 | 0.5 |
| 2 * | 0.5 | 0.5 |
| 3 | 0.6 | 0.4 |
| 4 * | 0.5 | 0.5 |
| 5 | 0.4 | 0.6 |
| 6 | 0.6 | 0.4 |
| 7 * | 0.5 | 0.5 |
| 8 | 0.7 | 0.3 |
| 9 | 0.7 | 0.3 |
| 10 * | 0.5 | 0.5 |
| 11 | 0.3 | 0.7 |
| 12 | 0.4 | 0.6 |
| 13 | 0.3 | 0.7 |
| 14 * | 0.5 | 0.5 |
| 15 | 0.4 | 0.6 |
| 16 | 0.4 | 0.6 |
| 17 | 0.6 | 0.4 |
| Quality Parameters | Samples per Commercial Category 1,2 | ||
| I | II | III | |
| 3, 8 | 1, 4, 6, 17 | 2, 5, 7, 15 2 | |
| E1%440nm | 204–211 (min 200) | 169–195 (min 170) | 134–162 (min 120) |
| E1%257nm | 81 (min 70) | 70–81 (min 55) | 52–69 (min 40) |
| E1%330nm | 42–49 (20–50) | 45–50 (20–50) | 33–48 (20–50) |
| Sample Number | Labeling Information |
|---|---|
| 1 | Purchased from: Hypermarket (Qatar); Origin: Iran; Pack.: September 2013; Exp.: September 2015 |
| 2 | Purchased from: Open market (Qatar); Origin: Iran; Pack.: October 2013; Exp: October 2015 |
| 3 | Purchased from: Open market (Qatar); Origin: Iran; Pack.: December 2013; Exp.: December 2015 |
| 4 | Purchased from: Open market (Qatar); Origin: Iran; Pack.: March 2013; Exp.: March 2015 |
| 5 | Purchased from: Hypermarket (Qatar); Origin: Iran; Pack.: March 2013; Exp.: March 2015 |
| 6 | Purchased from: Hypermarket (Qatar); Product of Spain; Pack.: September 2013; Exp.: September 2015 |
| 7 | Purchased from: Open market (Qatar); Origin: -; Packed in Australia (NSW) Pack.: October 2012; Exp.: October 2014 |
| 8 | Purchased from: Open market (Qatar); Origin: -; Pack.: -; Exp.: - |
| 9 | Purchased from: Super market (Italy); Origin: -; Packed in Italy; Pack.: -; Exp.: 27 January 2017 |
| 10 | Purchased from: Super market (Italy); Origin: -; Packed in Italy; Pack.: -; Exp.: 2016 |
| 11 | Purchased from: Open market (Israel); Origin: Spain; Crop: 2011; Exp.: END 2017 |
| 12 | Purchased from: Open market (Israel); Origin: -; Pack.: -; Exp.: - |
| 13 | Purchased from: Herbal shop (KSA); Origin: Iran; Pack. :November 2012; Exp.: November 2015 |
| 14 | Purchased from: Herbal shop (KSA); Origin: Iran; Pack.: -; Exp.: - |
| 15 | Purchased from: Herbal shop (KSA); Origin: -; Pack.: -; Exp.: - |
| 16 | Purchased from: Retailer (KSA); Origin: Iran; Prod. Date 4/1434H; Exp. Date 4/1436H |
| 17 | Purchased from: Supermarket (Qatar); Origin: Iran; Pack.: November 2012; Exp.: November 2014 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Consonni, R.; Ordoudi, S.A.; Cagliani, L.R.; Tsiangali, M.; Tsimidou, M.Z. On the Traceability of Commercial Saffron Samples Using 1H-NMR and FT-IR Metabolomics. Molecules 2016, 21, 286. https://doi.org/10.3390/molecules21030286
Consonni R, Ordoudi SA, Cagliani LR, Tsiangali M, Tsimidou MZ. On the Traceability of Commercial Saffron Samples Using 1H-NMR and FT-IR Metabolomics. Molecules. 2016; 21(3):286. https://doi.org/10.3390/molecules21030286
Chicago/Turabian StyleConsonni, Roberto, Stella A. Ordoudi, Laura R. Cagliani, Maria Tsiangali, and Maria Z. Tsimidou. 2016. "On the Traceability of Commercial Saffron Samples Using 1H-NMR and FT-IR Metabolomics" Molecules 21, no. 3: 286. https://doi.org/10.3390/molecules21030286
APA StyleConsonni, R., Ordoudi, S. A., Cagliani, L. R., Tsiangali, M., & Tsimidou, M. Z. (2016). On the Traceability of Commercial Saffron Samples Using 1H-NMR and FT-IR Metabolomics. Molecules, 21(3), 286. https://doi.org/10.3390/molecules21030286