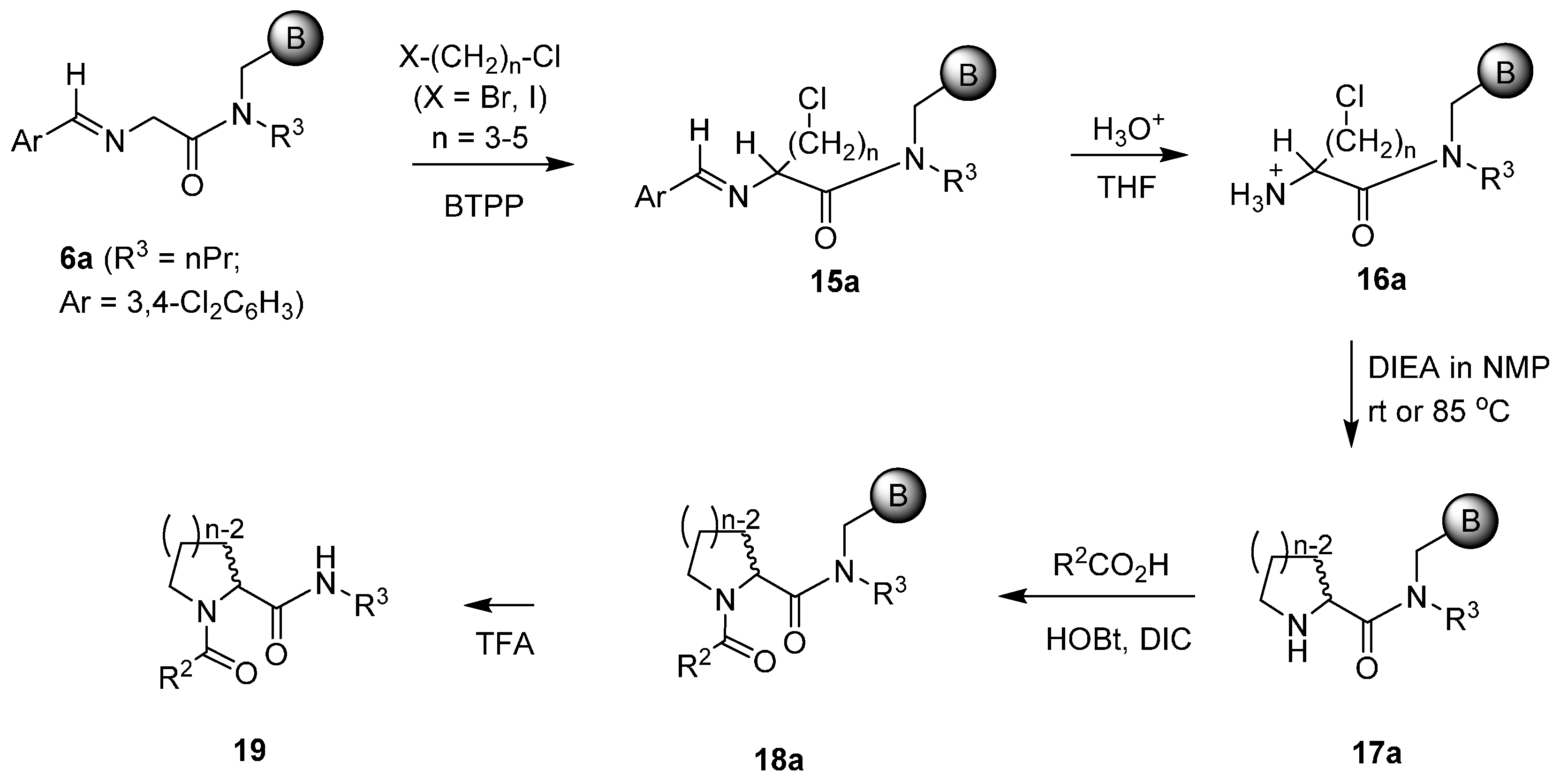
3.2.3. General Procedures for the Preparation of 19 from 17a
After the resin
17a was swelled in CH
2Cl
2 for 1 h and washed with DMF (4 × 1 mL), it was treated with a pre-mixed clear solution of carboxylic acid (0.25 mmol, 5 eq), HOBt (0.034 g, 0.25 mmol, 5 eq) and DIC (0.032 g, 0.25 mmol, 5 eq) in DMF. The completion of the reaction was indicated by a negative chloranil test [
14]. The resulting resin product
18a was then washed with DMF (4 × 1 mL), THF (3 × 1 mL), and CH
2Cl
2 (3 × 1 mL). Cleavage with 90% TFA/CH
2Cl
2 (1 mL) at room temperature over 1.5 h gave desired product
19a–
d. (
Scheme 3).
1-(4-Cyanobenzoyl)-N-propylpyrrolidine-2-carboxamide (19a): Product 19a was obtained from resin 6a, 1-chloro-3-iodopropane, and 4-cyanobenzoic acid as light yellow oil (4.0 mg, 28% isolated yield) following chromatographic purification over silica gel with CH2Cl2/MeOH (97:3). Initial LC/MS purity (for crude product) 43%, tR = 3.1 min. 1H-NMR (500 MHz, CDCl3): δ 0.93 (t, J = 7.3 Hz, 3H), 1.53 (tq, J = 7.3 Hz, J = 7.3 Hz, 2H), 1.82–1.89 (m, 1H), 1.96–2.09 (m, 3H), 2.43–2.52 (m, 1H), 3.20–3.28 (m, 2H), 3.34–3.44 (m, 1H), 3.49–3.58 (m, 1H), 4.68 (dd, J = 7.3 Hz, J = 3.2 Hz, 1H), 6.78 (brs, 1H), 7.62 (d, J = 8.5 Hz, 2H), 7.72 (d, J = 8.6 Hz, 2H). 13C-NMR (500 MHz, CDCl3): δ 11.4, 20.4, 22.7, 25.8, 41.4, 46.0, 52.7, 113.9, 117.9, 126.5, 132.5, 139.7, 170.3, 171.1.
1-(4-Cyanobenzoyl)-N-propylpiperidine-2-carboxamide (19b): Product 19b was obtained from resin 6a, 1-chloro-4-iodobutane, and 4-cyanobenzoic acid as light yellow oil (3.0 mg, 20% isolated yield) following chromatographic purification over silica gel with CH2Cl2/MeOH (98:2). Initial LC/MS purity (for crude product) 46%, tR = 5.9 min. 1H-NMR (500 MHz, CDCl3): δ 0.92 (t, J = 7.3 Hz, 3H), 1.46–1.58 (m, 4H), 1.60–1.76 (m, 2H), 1.83–1.93 (m, 1H), 2.22–2.32 (m, 1H), 3.15–3.35 (m, 3H), 3.46–3.55 (m, 1H), 5.20 (m, 1H), 6.32 (brs, 1H), 7.53 (d, J = 8.5 Hz, 2H), 7.72 (d, J = 8.5 Hz, 2H). 13C-NMR (500 MHz, CDCl3): δ 11.2, 21.0, 23.1, 23.8, 25.2, 42.5, 44.5, 62.3, 113.6, 118.1, 127.9, 132.1, 139.5, 170.5, 171.3.
1-(4-Cyanobenzoyl)-N-propylazepane-2-carboxamide (19c): Product 19c was obtained from resin 6a, 1-chloro-5-iodopentane, and 4-cyanobenzoic acid as light yellow oil (4.0 mg, 26% isolated yield) following chromatographic purification over silica gel with CH2Cl2/MeOH (98:2). Initial LC/MS purity (for crude product) 82%, tR = 6.1 min. 1H-NMR (500 MHz, CDCl3): δ 0.93 (t, J = 7.3 Hz, 3H), 1.34–1.40 (m, 4H), 1.49–1.60 (m, 2H), 1.97–2.21 (m, 4H), 3.18–3.29 (m, 2H), 3.37–3.45 (m, 2H), 4.85 (dd, J = 7.4 Hz, J = 3.0 Hz, 1H), 6.32 (brs, 1H), 7.47 (d, J = 8.5 Hz, 2H), 7.71 (d, J = 8.6 Hz, 2H). 13C-NMR (500 MHz, CDCl3): δ 11.5, 23.0, 25.2, 28.9, 29.3, 30.9, 41.4, 46.2, 58.5, 113.5, 118.3, 127.1, 132.7, 141.2, 171.5, 172.2.
2-(4-Cyanobenzoyl)-N-propyl-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (19d): Product 19d was obtained from resin 6a, α,α′-dichloro-o-xylene, and 4-cyanobenzoic acid as light yellow oil (9.0 mg, 52% isolated yield) following chromatographic purification over silica gel with CH2Cl2/MeOH (99:1). Initial LC/MS purity (for crude product) 75%, tR = 7.9 min. 1H-NMR (500 MHz, CDCl3): δ 0.91 (t, J = 7.3 Hz, 3H), 1.52 (m, 2H), 3.10–3.18 (m, 1H), 3.19–3.32 (m, 2H), 3.42–3.49 (m, 1H), 4.33–4.54 (m, 2H), 5.13 (m, 1H), 6.46 (brs, 1H), 7.16–7.28 (m, 4H), 7.54 (d, J = 8.5 Hz, 2H), 7.77 (d, J = 8.5 Hz, 2H). 13C-NMR (500 MHz, CDCl3): δ 11.3, 22.8, 29.7, 41.4, 49.6, 53.9, 114.1, 117.9, 125.2, 126.8, 127.5, 127.8, 128.2, 128.5, 133.2, 133.9, 139.7, 169.9, 170.1.
3.2.4. General Procedures for the Preparation of 3 and 22 from 17b
Synthesis of New Proline Amide and Homologue -based Piperazine Incorporated Products
3 and
22 through Acylation, Deprotection, Iodination,
N-Alkylation with Piperazines, and Cleavage: After the resin
17b was swelled in CH
2Cl
2 for 1 h and washed with DMF (4 × 1 mL), it was treated with a pre-mixed clear solution of carboxylic acid (0.25 mmol, 5 eq), HOBt (0.034 g, 0.25 mmol, 5 eq) and DIC (0.032 g, 0.25 mmol, 5 eq) in DMF. The completion of the reaction was indicated by a negative chloranil test [
14]. The resulting resin product
18b was then washed with DMF (4 × 1 mL), THF (3 × 1 mL), and CH
2Cl
2 (3 × 1 mL). After
18b was pre-swelled in CH
2Cl
2 for 40 min, it was washed with THF (3 × 1 mL), followed by treatment with 1M TBAF/THF (1 mL, 1 mmol, 20 eq) for removal of TBDPS group. After the reaction proceeded for 18 h, the drained resin product
20 was washed with THF (4 × 1 mL), and CH
2Cl
2 (3 × 1 mL). Iodination of alcohol resin
20 started with its pre-swelling in CH
2Cl
2 for 1 h, followed by washes with DMF (3 × 1 mL). The resin was then treated with a pre-mixed solution of iodine (0.063 g, 0.25 mmol, 5 eq), PPh
3 (0.066 g, 0.25 mmol, 5 eq), and imidazole (0.017 g, 0.25 mmol, 5 eq) in DMF (0.75 mL). The reaction was allowed to proceed at room temperature overnight. After 18 h of iodination reaction, the filtered iodinated resin product
21 was washed with DMF (3 × 1 mL), MeOH (3 × 1 mL), DMF (2 × 1 mL), and CH
2Cl
2 (3 × 1 mL). The air-dried resin was swollen in CH
2Cl
2 for 40 min, and to the resin was then added a solution of Piperazines in DMSO (1 M, 0.75 mL, 15 eq) except that 2,3-dichlorophenyl piperazine hydrochloride (0.201 g, 0.75 mmol, 15 eq) was added as the solid together with same equivalent of base DIEA (0.097 g, 0.75 mmol, 15 eq). The reaction was heated to 50 °C for 6 h with occasional agitation. The resulting resin product was drained, and sequentially washed with DMF (2 × 1 mL), MeOH (2 × 1 mL), DMF (3 × 1 mL), and CH
2Cl
2 (4 × 1 mL). Cleavage with 90% TFA/CH
2Cl
2 (1 mL) at room temperature over 1.5 h afforded target compound
3a–
3d,
22a and
22b.
1-(Cyclopropanecarbonyl)-N-(3-(4-(2-methoxyphenyl)piperazin-1-yl)propyl)pyrrolidine-2-carboxamide (3a): Product 3a was obtained from resin 6b, 1-chloro-3-iodopropane, cyclopropanecarboxylic acid, and 1-(2-methoxyphenyl)piperazine as light yellow oil (2.9 mg, 14% isolated yield) following chromatographic purification over silica gel with CH2Cl2/MeOH (95:5). Initial LC/MS purity (for crude product) 49%, tR = 3.3 min. 1H-NMR (500 MHz, CDCl3): δ 0.78–0.82 (m, 2H), 0.83–0.89 (m, 1H), 0.91–0.99 (m, 1H), 1.69–1.79 (m, 1H), 1.98–2.22 (m, 8H), 2.98–3.05 (m, 2H), 3.24–3.35 (m, 4H), 3.43–3.52 (m, 3H), 3.64–3.69 (m, 2H), 3.87 (s, 3H), 3.89–3.94 (m, 1H), 4.44 (dd, J = 7.6 Hz, J = 3.0 Hz, 1H), 6.89 (d, J = 7.9 Hz, 1H), 6.93–6.96 (m, 2H), 7.04–7.11 (m, 1H), 7.83 (m, 1H). 13C-NMR (500 MHz, CDCl3): δ 7.5, 8.0, 12.8, 23.0, 24.8, 28.9, 35.8, 47.3, 47.6, 51.8, 53.1, 54.2, 55.5, 60.9, 111.4, 118.9, 121.3, 124.5, 139.0, 152.2, 162.1, 173.0, 173.9.
N-(3-(4-(3-Chlorophenyl)piperazin-1-yl)propyl)-1-(cyclopropanecarbonyl)pyrrolidine-2-carboxamide (3b): Product 3b was obtained from resin 6b, 1-chloro-3-iodopropane, cyclopropanecarboxylic acid, and 1-(3-chlorophenyl)piperazine as light yellow oil (3.0 mg, 14% isolated yield) following chromatographic purification over silica gel with CH2Cl2/MeOH (92:8). Initial LC/MS purity (for crude product) 43%, tR = 5.1 min. 1H-NMR (500 MHz, CDCl3): δ 0.79–0.89 (m, 3H), 0.90–0.97 (m, 1H), 1.69–1.78 (m, 1H), 1.94–2.26 (m, 8H), 2.84–2.95 (m, 1H), 2.98–3.07 (m, 1H), 3.22–3.33 (m, 2 H), 3.38–3.49 (m, 3 H), 3.57–3.72 (m, 4H), 3.86–3.93 (m, 1H), 4.42 (dd, J = 7.5 Hz, J = 3.1 Hz), 6.79 (dd, J = 8.3 Hz, J = 2.2 Hz, 1H), 6.89 (m, 1H), 6.92–6.97 (m, 1H), 7.21 (t, J = 8.1 Hz, 1H), 7.73 (t, J = 5.2 Hz). 13C-NMR (500 MHz, CDCl3): δ 7.5, 8.1, 12.8, 23.1, 24.8, 29.0, 35.9, 46.3, 47.6, 54.3, 60.9, 114.9, 117.2, 121.5, 130.5, 135.3, 150.6, 173.0, 173.9. HRMS calcd for [M + H]+: C22H32N4O2Cl 419.2214, found 419.2229.
1-(Cyclopropanecarbonyl)-N-(3-(4-(2,3-dichlorophenyl)piperazin-1-yl)propyl)pyrrolidine-2-carboxamide (3c): Product 3c was obtained from resin 6b, 1-chloro-3-iodopropane, cyclopropanecarboxylic acid, and 1-(2,3-dichlorophenyl)-piperazine hydrochloride as light yellow oil (2.0 mg, 9% isolated yield) following chromatographic purification over silica gel with CH2Cl2/OH (95:5). Initial LC/MS purity (for crude product) 41%, tR = 6.1 min. 1H-NMR (500 MHz, CDCl3): δ 0.79–0.89 (m, 3H), 0.91–0.97 (m, 1H), 1.69–1.78 (m, 1H), 1.99–2.05 (m, 2H), 2.06–2.11 (m, 2H), 2.14–2.22 (m, 2H), 2.97–3.09 (m, 3H), 3.26–3.34 (m, 2H), 3.36–3.46 (m, 5H), 3.63–3.74 (m, 3H), 3.87–3.94 (m, 1H), 4.43 (dd, J = 7.6 Hz, J = 3.1 Hz, 1H), 7.02 (dd, J = 8.0 Hz, J = 1.4 Hz, 1H), 7.19 (t, J = 8.0 Hz, 1H), 7.25 (d, J = 1.3 Hz, 1H), 7.75 (t, J = 5.6 Hz, 1H). 13C-NMR (500 MHz, CDCl3): 7.5, 8.1, 12.8, 23.1, 24.8, 29.0, 35.8, 47.6, 47.9, 54.4, 60.9, 119.2, 126.2, 127.8, 127.9, 134.3, 149.1, 162.4, 173.0, 173.9. HRMS calcd for [M + H]+: C22H31N4O2Cl2 453.1824, found 453.1845 (RA 100%).
1-(Adamantanecarbonyl)-N-(3-(4-(2-methoxyphenyl)piperazin-1-yl)propyl)pyrrolidine-2-carboxamide (3d): This product was obtained from resin 6b, 1-chloro-3-iodopropane, 1-adamantanecarboxylic acid, and 1-(2-methoxyphenyl)piperazine as light yellow oil (5.7 mg, 22% isolated yield) following chromatographic purification over silica gel with CH2Cl2/MeOH (92:8). Initial LC/MS purity (for crude product) 44%, tR = 9.3 min. 1H-NMR (500 MHz, CDCl3): δ 1.71 (m, 6H), 1.85–1.95 (m, 3H), 1.96–1.99 (m, 5H), 2.00–2.09 (m, 7H), 3.05 (t, J = 10.8 Hz, 2H), 3.13–3.18 (m, 1H), 3.20–3.32 (m, 4H), 3.41–3.47 (m, 1H), 3.48–3.54 (m, 2H), 3.66 (d, J = 11.8 Hz, 1H), 3.76–3.84 (m, 3H), 3.87 (s, 3H), 4.43 (dd, J = 7.4 Hz, J = 5.3 Hz, 1H), 6.89 (d, J = 8.1 Hz, 1H), 6.92–6.95 (m, 2H), 7.03–7.12 (m, 1H), 7.41 (m, 1H). 13C-NMR (500 MHz, CDCl3): δ 23.7, 28.3, 35.9, 36.6, 38.2, 41.8, 47.5, 48.6, 52.4, 52.6, 54.6, 55.5, 62.9, 111.4, 118.9, 121.2, 124.4, 139.0, 152.2, 162.5, 173.9, 176.9. HRMS calcd for [M + H]+: C30H45N4O3 509.3492, found 509.3474.
N-(3-(4-(3-Chlorophenyl)piperazin-1-yl)propyl)-1-(adamantanecarbonyl)pyrrolidine-2-carboxamide (3e): This product was obtained from resin 6b, 1-chloro-3-iodopropane, 1-adamantanecarboxylic acid, and 1-(3-chlorophenyl)piperazine as light yellow oil (2.0 mg, 8% isolated yield) following chromatographic purification over silica gel with CH2Cl2/MeOH (92:8). Initial LC/MS purity (for crude product) 39%, tR = 10.8 min. 1H-NMR (500 MHz, CDCl3): δ 1.63–1.78 (m, 6H), 1.89–1.98 (m, 7H), 1.99–2.12 (m, 8H), 2.95 (m, 2H), 3.12–3.20 (m, 1H), 3.21–3.31 (m, 2H), 3.36–3.52 (m, 3H), 3.56–3.71 (m, 3H), 3.78–3.89 (m, 3H), 4.38 (dd, J = 7.7 Hz, J = 5.2 Hz, 1H), 6.78 (dd, J = 8.2 Hz, J = 2.1 Hz, 1H), 6.86–6.90 (m, 1H), 6.91–6.95 (m, 1H), 7.16–7.24 (m, 2H). 13C-NMR (500 MHz, CDCl3): δ 23.8, 28.3, 36.1, 36.6, 38.2, 41.8, 46.5, 48.6, 54.5, 62.9, 114.9, 117.2, 121.4, 130.4, 135.3, 150.6, 173.9, 176.9. HRMS calcd for [M + H]+: C29H42N4O2Cl 513.2996, found 513. 2987.
1-(Adamantanecarbonyl)-N-(3-(4-(2,3-dichlorophenyl)piperazin-1-yl)propyl)pyrrolidine-2-carboxamide (3f): This product was obtained from resin 6b, 1-chloro-3-iodopropane, 1-adamantanecarboxylic acid, and 1-(2,3-dichlorophenyl)-piperazine hydrochloride as light yellow oil 2.0 mg (7% isolated yield) following chromatographic purification (CH2Cl2/MeOH (92:8)): initial LC/MS purity (for crude product) 39%, tR = 11.5 min. 1H-NMR (500 MHz, CDCl3): δ 1.66–1.77 (m, 6H), 1.91–1.99 (m, 7H), 2.00–2.13 (m, 8H), 3.00–3.12 (m, 2H), 3.14–3.23 (m, 1H), 3.23–3.31 (m, 2H), 3.35–3.42 (m, 4H), 3.45–3.55 (m, 1H), 3.65–3.73 (m, 1H), 3.78–3.89 (m, 3H), 4.39 (dd, J = 7.7 Hz, J = 5.3 Hz, 1H), 7.01 (dd, J = 7.9 Hz, J = 1.1 Hz, 1H), 7.19 (t, J = 7.9 Hz, 2H), 7.24 (m, 1H). 13C-NMR (500 MHz, CDCl3): δ 23.9, 28.3, 35.9, 36.6, 38.2, 41.8, 48.2, 48.7, 54.7, 62.9, 119.2, 126.2, 127.8, 127.9, 134.3, 149.1, 173.8, 176.9. HRMS calcd for [M + H]+: C29H41N4O2Cl2 547.2607, found 547.2627.
2-(Cyclopropanecarbonyl)-N-(3-(4-(2-methoxyphenyl)piperazin-1-yl)propyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (22a): Product 22a was obtained from resin 6b, α,α′-dichloro-o-xylene, cyclopropanecarboxylic acid, and 1-(2-methoxyphenyl)piperazine as amorphous white solid (6.0 mg, 25% isolated yield) following chromatographic purification over silica gel with CH2Cl2/MeOH (92:8). Initial LC/MS purity (for crude product) 65%, tR = 6.2 min. 1H-NMR (500 MHz, CDCl3, 1:1 mixture of two rotamers): δ 0.78–0.91 (m, 2H), 0.93–1.05 (m, 1H), 1.05–1.19 (m, 1H), 1.38–1.49 (m, 1H), 1.56–1.61 (m, 1H), 1.69–1.83 (m, 2H), 1.90–2.05 (m, 1H), 2.07–2.23 (m, 1H), 2.47–2.59 (m, 2H), 2.63–2.75 (m, 2H), 2.97–3.05 (m, 1H), 3.07–3.16 (m, 3H), 3.17–3.26 (m, 2H), 3.35–3.45 (m, 1H), 3.86 (s, 3H), 4.56 (d, J = 15.9 Hz, 0.5 H), 4.81 (d, J = 15.9 Hz, 1H), 4.89 (m, 0.5H), 4.94 (d, J = 15.0 Hz, 0.5H), 5.16 (m, 0.5H), 6.87 (d, J = 7.8 Hz, 1H), 6.90–6.96 (m, 2H), 6.96–7.03 (m, 2H), 7.13–7.25 (m, 4H). 13C-NMR (500 MHz, CDCl3): δ 8.2, 12.4, 23.9, 29.7, 30.9, 32.9, 44.5, 48.5, 50.5, 53.4, 55.4, 111.2, 118.3, 121.0, 125.8, 126.4, 126.6, 127.3, 127.6, 127.9, 128.5, 133.9, 152.3, 173.3, 175.5. HRMS calcd for [M + H]+: C28H37N4O3 477.2866, found 477.2879.
N-(3-(4-(3-Chlorophenyl)piperazin-1-yl)propyl)-2-(cyclopropanecarbonyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxamide (22b): Product 22b was obtained from resin 6b, α,α′-dichloro-o-xylene, cyclopropanecarboxylic acid, and 1-(3-chlorophenyl)piperazine as amorphous white solid (6.0 mg, 25% isolated yield) following chromatographic purification over silica gel with CH2Cl2/MeOH (92:8). Initial LC/MS purity (for crude product) 64%, tR = 7.8 min. 1H-NMR (500 MHz, CD3OD/CDCl3, 1 : 1 mixture of two rotamers): δ 0.87–0.91 (m, 2H), 1.06–1.12 (m, 1H), 1.39–1.44 (m, 2H), 1.59–1.66 (m, 2H), 1.85–1.93 (m, 1H), 1.97–2.05 (m, 2H), 2.68–2.76 (m, 1H), 3.02–3.09 (m, 1H), 3.22–3.28 (m, 2H), 3.32–3.40 (m, 2H), 3.42–2.59 (m, 4H), 3.63–3.71 (m, 1H), 4.86 (d, J = 15.0 Hz, 1H), 4.91–4.94 (m, 0.5H), 4.99–5.05 (m, 1H), 5.05–5.11 (m, 0.5H), 6.78 (dd, J = 8.2 Hz, J = 2.4 Hz, 1H), 6.86–6.89 (m, 1H), 6.91–6.95 (m, 1H), 7.14–7.25 (m, 5H). 13C-NMR (500 MHz, CD3OD/CDCl3): δ 12.2, 13.5, 19.7, 21.4, 23.9, 29.8, 46.5, 46.7, 51.8, 58.8, 114.9, 116.9, 126.3, 127.1, 127.8, 130.5, 133.4, 133.7, 134.0, 135.3, 151.0, 175.3, 176.4. HRMS calcd for [M + H]+: C27H34N4O2Cl 481.2370, found 481.2393.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










