
Binding Mode and Selectivity of Steroids towards Glucose-6-phosphate Dehydrogenase from the Pathogen Trypanosoma cruzi

 ,
,
Abstract
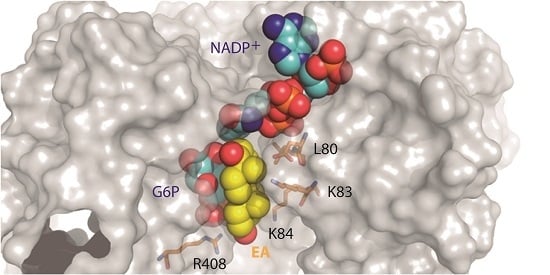
:
1. Introduction
2. Results and Discussion
2.1. EA Binding to the T. cruzi G6PDH-G6P-NADP+ Ternary Complex
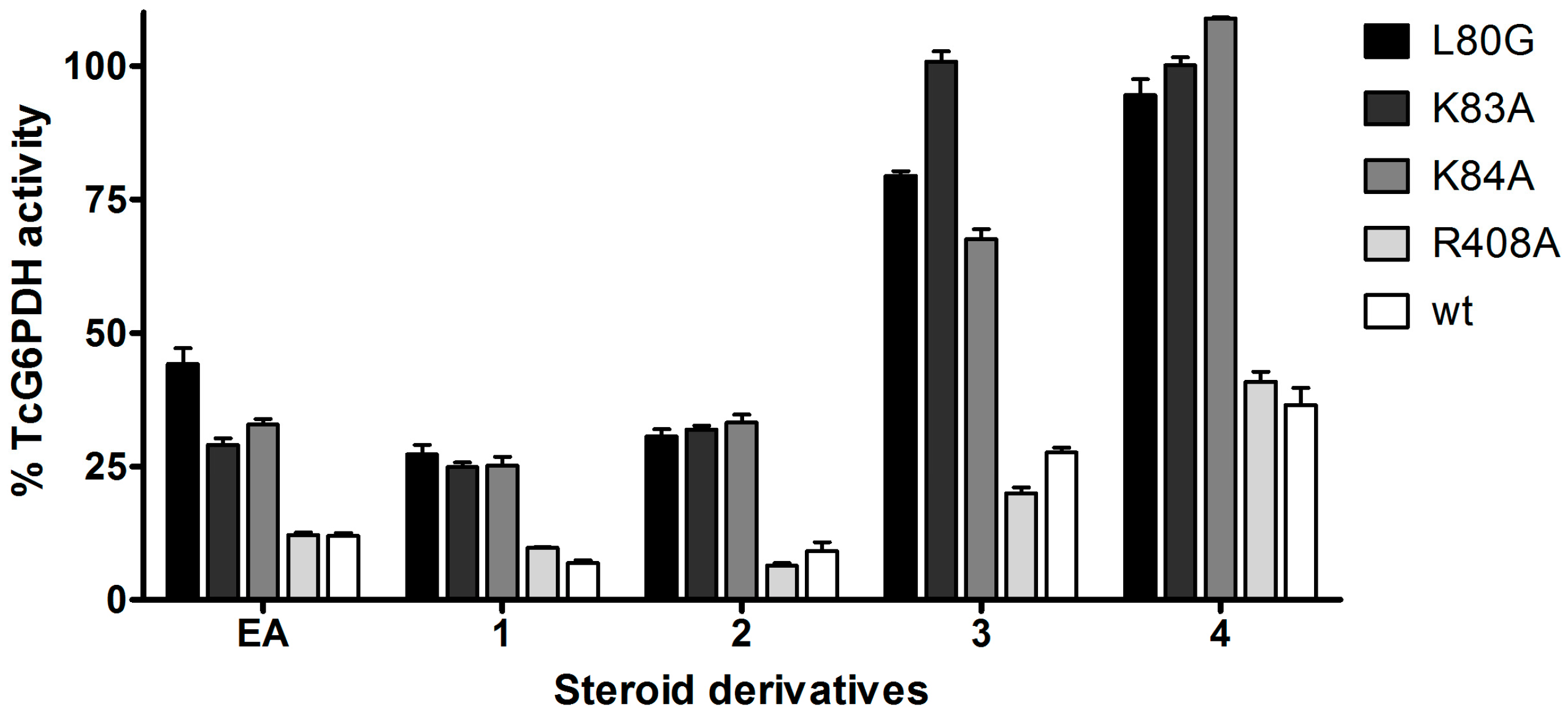
2.2. Biochemical Validation of EA Binding to TcG6PDH
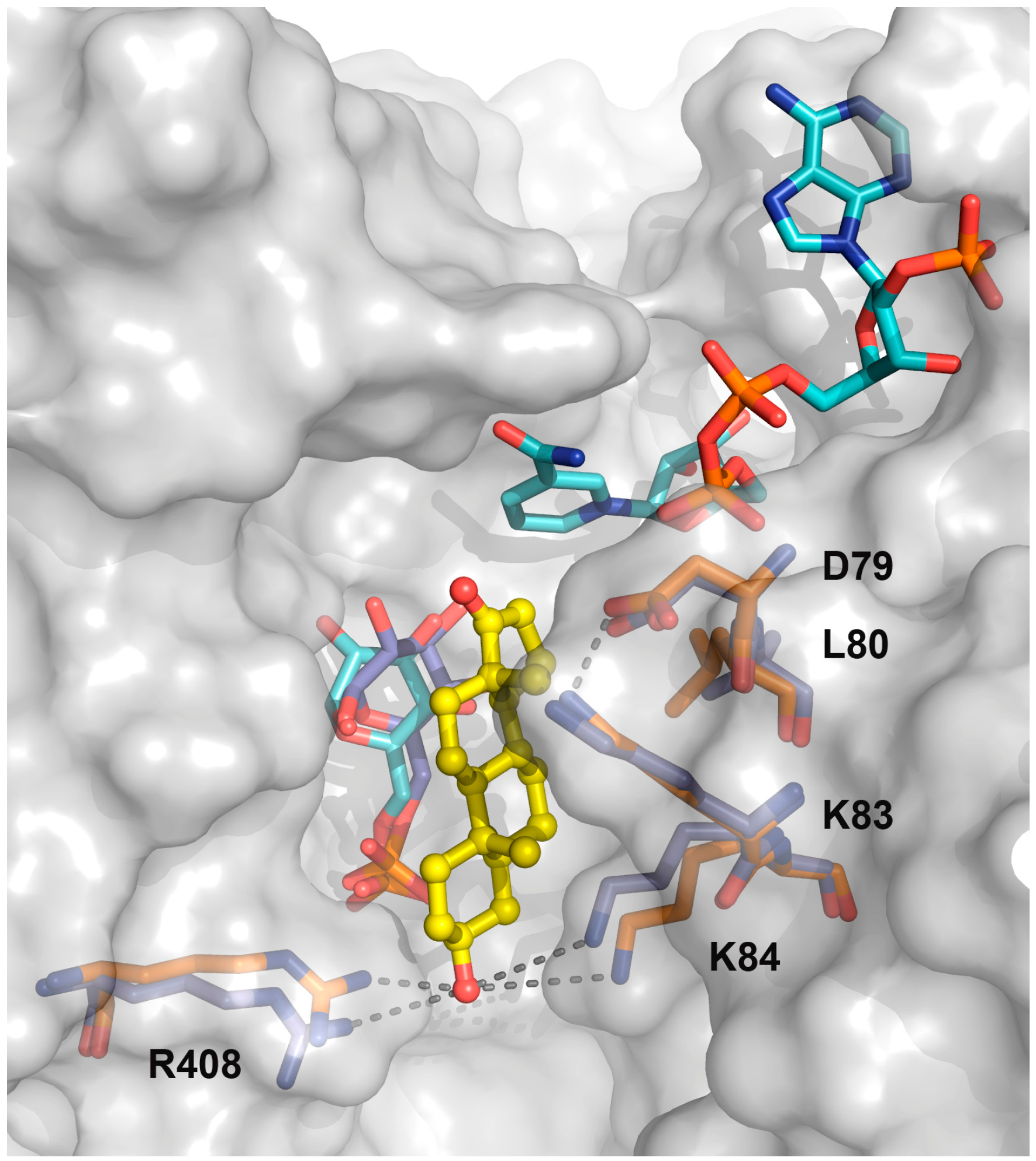
2.3. Activity and Binding Mode to TcG6PDH of Novel EA Derivatives
3. Experimental Section
3.1. Plasmids
3.2. Expression and Purification of Recombinant Proteins
3.3. Kinetic Assays
3.4. Inhibitor Assays with Wildtype or Mutants of TcG6PDH
3.5. Generation of TcG6PDH/G6P/NADP+ Ternary Complex and Steroids Docking
3.6. MD Simulation of TcG6PDH/G6P/NADP+/EA Complex
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hotez, P.J.; Bottazzi, M.E.; Franco-Paredes, C.; Ault, S.K.; Periago, M.R. The neglected tropical diseases of Latin America and the Caribbean: A review of disease burden and distribution and a roadmap for control and elimination. PLoS Negl. Trop. Dis. 2008, 2, e300. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.P.; Gilbert, I.H. Perspectives for new drugs against trypanosomiasis and leishmaniasis. Curr. Top. Med. Chem. 2002, 2, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Opperdoes, F.R.; Michels, P.A. Enzymes of carbohydrate metabolism as potential drug targets. Int. J. Parasitol. 2001, 31, 482–490. [Google Scholar] [CrossRef]
- Comini, M.A.; Ortíz, C.; Cazzulo, J.J. Drug targets in trypanosomal and leishmanial pentose phosphate pathway. In Trypanosomatid Diseases: Molecular Routes to Drug Discovery; Jäger, T., Koch, O., Flohé, L., Eds.; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2013; pp. 297–313. [Google Scholar]
- Maugeri, D.A.; Cazzulo, J.J. The pentose phosphate pathway in Trypanosoma cruzi. FEMS Microbiol. Lett. 2004, 234, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, D.A.; Cannata, J.J.; Cazzulo, J.J. Glucose metabolism in Trypanosoma cruzi. Essays Biochem. 2011, 51, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Igoillo-Esteve, M.; Cazzulo, J.J. The glucose-6-phosphate dehydrogenase from Trypanosoma cruzi: Its role in the defense of the parasite against oxidative stress. Mol. Biochem. Parasitol. 2006, 149, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, A.T.; Thiemann, O.H.; Michels, P.A. Inhibition of Trypanosoma brucei glucose-6-phosphate dehydrogenase by human steroids and their effects on the viability of cultured parasites. Bioorg. Med. Chem. 2009, 17, 2483–2489. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Banks, J. Inhibition of mammalian glucose-6-phosphate dehydrogenase by steroids. Proc. Natl. Acad. Sci. USA 1960, 46, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, A.T.; Thiemann, O.H. 16-Bromoepiandrosterone, an activator of the mammalian immune system, inhibits glucose 6-phosphate dehydrogenase from Trypanosoma cruzi and is toxic to these parasites grown in culture. Bioorg. Med. Chem. 2010, 18, 4762–4768. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Cordeiro, A.T.; Michels, P.A. Glucose-6-phosphate dehydrogenase is the target for the trypanocidal action of human steroids. Mol. Biochem. Parasitol. 2011, 176, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Mercaldi, G.F.; Ranzani, A.T.; Cordeiro, A.T. Discovery of new uncompetitive inhibitors of glucose-6-phosphate dehydrogenase. J. Biomol. Screen 2014, 19, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.; Mackow, M.C.; Levy, H.R. On the mechanism of interaction of steroids with human glucose 6-phosphate dehydrogenase. Arch. Biochem. Biophys. 1995, 318, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.B.; Liu, Y.; Yao, Y. Computational determination of binding structures and free energies of glucose 6-phosphate dehydrogenase with novel steroid inhibitors. J. Mol. Graph. Model. 2014, 51, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, C.; Larrieux, N.; Medeiros, A.; Botti, H.; Comini, M.; Buschiazzo, A. Expression, crystallization and preliminary X-ray crystallographic analysis of glucose-6-phosphate dehydrogenase from the human pathogen Trypanosoma cruzi in complex with substrate. Acta Cryst. Sect. F 2011, 67, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.M.; Dawson, M.; Fairweather, E.E.; Hamilton, N.S.; Hitchin, J.R.; James, D.I.; Jones, S.D.; Jordan, A.M.; Lyons, A.J.; Small, H.F.; et al. Novel steroid inhibitors of glucose 6-phosphate dehydrogenase. J. Med. Chem. 2012, 55, 4431–4445. [Google Scholar] [CrossRef] [PubMed]
- Au, S.W.; Gover, S.; Lam, V.M.; Adams, M.J. Human glucose-6-phosphate dehydrogenase: The crystal structure reveals a structural NADP+ molecule and provides insights into enzyme deficiency. Structure 2000, 8, 293–303. [Google Scholar] [CrossRef]
- Rowland, P.; Basak, A.K.; Gover, S.; Levy, H.R.; Adams, M.J. The three-dimensional structure of glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides refined at 2.0 Å resolution. Structure 1994, 2, 1073–1087. [Google Scholar] [CrossRef]
- Kotaka, M.; Gover, S.; Vandeputte-Rutten, L.; Au, S.W.; Lam, V.M.; Adams, M.J. Structural studies of glucose-6-phosphate and NADP+ binding to human glucose-6-phosphate dehydrogenase. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-T.; Au, S.W.; Lam, V.M.; Engel, P.C. Recombinant human glucose-6-phosphate dehydrogenase. Eur. J. Biochem. 2002, 269, 3417–3424. [Google Scholar] [CrossRef] [PubMed]
- Cardi, M.; Chibani, K.; Castiglia, D.; Cafasso, D.; Pizzo, E.; Rouhier, N.; Jacquot, J.P.; Esposito, S. Overexpression, purification and enzymatic characterization of a recombinant plastidial glucose-6-phosphate dehydrogenase from barley (Hordeum vulgare cv. Nure) roots. Plant Physiol. Biochem. 2013, 73, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Glide; Version 6.5; Schrödinger, L.L.C.: New York, NY, USA, 2014.
- Raineri, R.R.; Levy, H.R. Specificity of steroid interaction with mammary glucose 6-phosphate dehydrogenase. Biochemistry 1970, 9, 2233–2243. [Google Scholar] [CrossRef] [PubMed]
- Cer, R.Z.; Mudunuri, U.; Stephens, R.; Lebeda, F.J. IC50-to-Ki: A web-based tool for converting IC50 to Ki values for inhibitors of enzyme activity and ligand binding. Nucleic Acids Res. 2009, 37, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Protein Preparation Wizard; Schrödinger, L.L.C.: New York, NY, USA, 2014.
- Prime; Version 3.8; Schrödinger, L.L.C.: New York, NY, USA, 2014.
- Epik; Version 3.0; Schrödinger, L.L.C.: New York, NY, USA, 2014.
- Olsson, M.H.M.; Søndergard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in Empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- LigPrep; Version 3.2; Schrödinger, L.L.C.: New York, NY, USA, 2014.
- Maestro; Version 9.2; Schrödinger, L.L.C.: New York, NY, USA, 2011.
- Wlodek, S.; Skillman, A.G.; Nicholls, A.J. Ligand entropy in gas-phase, upon solvation and protein complexation. Fast estimation with Quasi-Newton Hessian. J. Chem. Theory Comput. 2010, 6, 2140–2152. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 1.7.1.3; Schrödinger, L.L.C.: New York, NY, USA, 2014.
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 2, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.C. Rattle: A velocity version of the shake algorithm for molecular-dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1, 2, 3 and 4 are available from the authors.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G6PDH | KM G6P (µM) | kcat (s−1) | kcat/KM G6P (s−1·µM−1) | KM NADP+ (µM) | kcat (s−1) | kcat/KM NADP+ (s−1·µM−1) | Ki EA b (µM) |
|---|---|---|---|---|---|---|---|
| WT | 77 ± 20 | 62 ± 3 | 0.8 | 16 ± 3 | 52 ± 2 | 3.2 | 2.5 c |
| L80G | 74 ± 17 | 2.0 ± 0.1 | 0.027 | 75 ± 9 | 2.7 ± 0.1 | 0.036 | 31 d |
| K83A | 537 ± 99 | 2.8 ± 0.2 | 0.005 | 21 ± 3 | 3.0 ± 0.1 | 0.14 | 11 e |
| K84A | 618 ± 78 | 24.6 ± 0.9 | 0.04 | 17 ± 4 | 58 ± 2 | 3.4 | 8 f |
| R408A | 168 ± 7 | 15.0 ± 0.3 | 0.089 | 17 ± 3 | 19 ± 1 | 1.1 | 3 g |
| Compound | Substituent | IC50 (µM) a | G6PDH Activity [%] ± SD at 30 µM Compound b | |||
|---|---|---|---|---|---|---|
| 3-α | 3-β | 17- | 16-α | |||
| EA | H | OH | O | H | 3.0 ± 0.4 c | 12 ± 2 |
| 16Br-EA | H | OH | O | Br | 0.015 (13.3 to 16.6) d | ND e |
| 1 | H | NHCONHEt | O | H | 1.5 (1.0 to 2.0) f | 7 ± 1 |
| 2 | H | NHSO2NH2 | O | H | 2.2 (1.8 to 2.9) f | 9 ± 1 |
| 3 | H | NHSO2NH2 | α-H, β-COCH2OH | H | ND e | 30 ± 2 |
| 4 | H | OH | α-H, β-COCH2OH | H | ND e | 43 ± 2 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz, C.; Moraca, F.; Medeiros, A.; Botta, M.; Hamilton, N.; Comini, M.A. Binding Mode and Selectivity of Steroids towards Glucose-6-phosphate Dehydrogenase from the Pathogen Trypanosoma cruzi. Molecules 2016, 21, 368. https://doi.org/10.3390/molecules21030368
Ortiz C, Moraca F, Medeiros A, Botta M, Hamilton N, Comini MA. Binding Mode and Selectivity of Steroids towards Glucose-6-phosphate Dehydrogenase from the Pathogen Trypanosoma cruzi. Molecules. 2016; 21(3):368. https://doi.org/10.3390/molecules21030368
Chicago/Turabian StyleOrtiz, Cecilia, Francesca Moraca, Andrea Medeiros, Maurizio Botta, Niall Hamilton, and Marcelo A. Comini. 2016. "Binding Mode and Selectivity of Steroids towards Glucose-6-phosphate Dehydrogenase from the Pathogen Trypanosoma cruzi" Molecules 21, no. 3: 368. https://doi.org/10.3390/molecules21030368
APA StyleOrtiz, C., Moraca, F., Medeiros, A., Botta, M., Hamilton, N., & Comini, M. A. (2016). Binding Mode and Selectivity of Steroids towards Glucose-6-phosphate Dehydrogenase from the Pathogen Trypanosoma cruzi. Molecules, 21(3), 368. https://doi.org/10.3390/molecules21030368