
Integrated Analysis of the Wood Oil from Xanthocyparis vietnamensis Farjon & Hiep. by Chromatographic and Spectroscopic Techniques



Abstract
:
1. Introduction
2. Results
2.1. Analysis of X. vietnamensis Wood Oil by GC(RI), GC-MS and 13C-NMR
- -
- Monoterpenes bearing the p-menthane skeleton: p-cymene (5), p-cymenene (7), p-cymen-8-ol (8), terpinen-4-ol (9), α-terpineol (10) and carvacrol (12);
- -
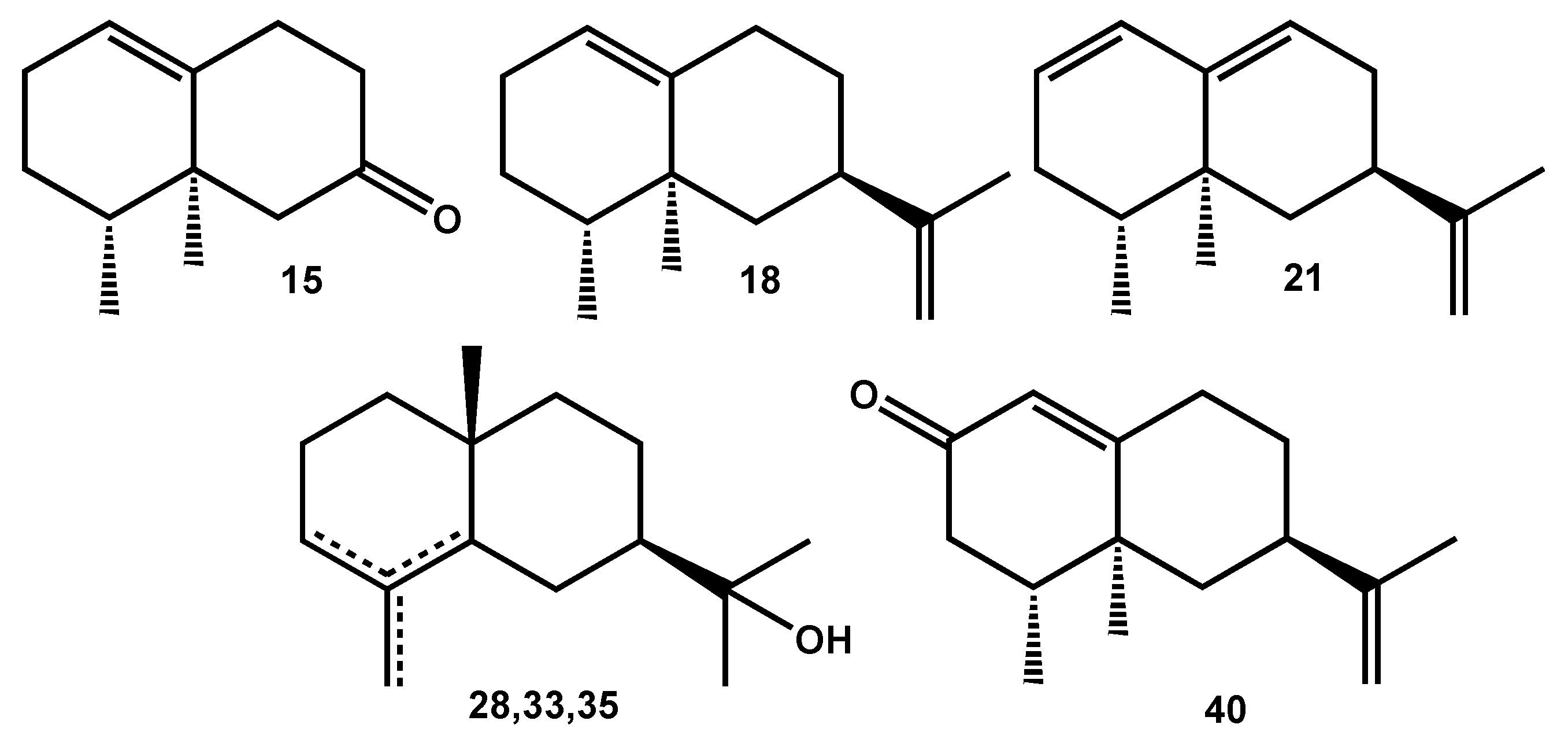
- Sesquiterpenes bearing the bicyclo[4.4.0]decane skeleton: β-selinene (17), valencene (18), δ-cadinene (22), γ-eudesmol (28), τ-muurolol (30), β-eudesmol (33), α-eudesmol (35);
- -
- Sesquiterpenes with substituted six-membered ring: β-elemene (14), β-elemol (23). It could be pointed out that β-elemene, being identified by NMR at room temperature, is a metabolite of the plant and it is not an artifact resulting from the thermal rearrangement of germacrene A in the GC injector [13];
- -
- Overall, the two major components of the EO—nootkatene (21, 20.7%) and 11,12,13-tri-nor-eremophil-1(10)-en-7-one (15, 17.2%) bear the eremophilane skeleton (Figure 2).
2.2. Fractionation of X. vietnamensis Essential Oil
2.3. Structure Elucidation of New Natural Compounds
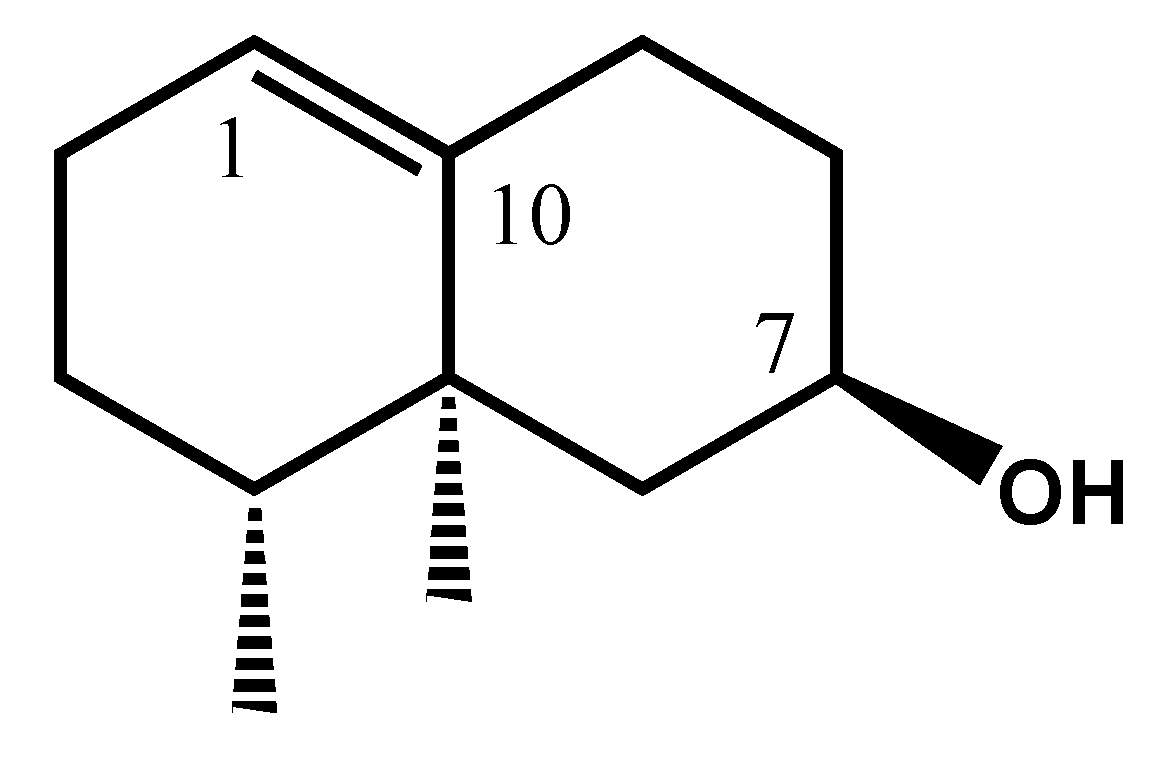
2.3.1. Identification of 11,12,13-Tri-nor-eremophil-1(10)-en-7-ol (16)
2.3.2. Identification of 10-epi-Nor-γ-eudesmen-11-one (24)
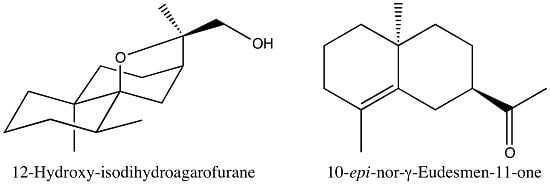
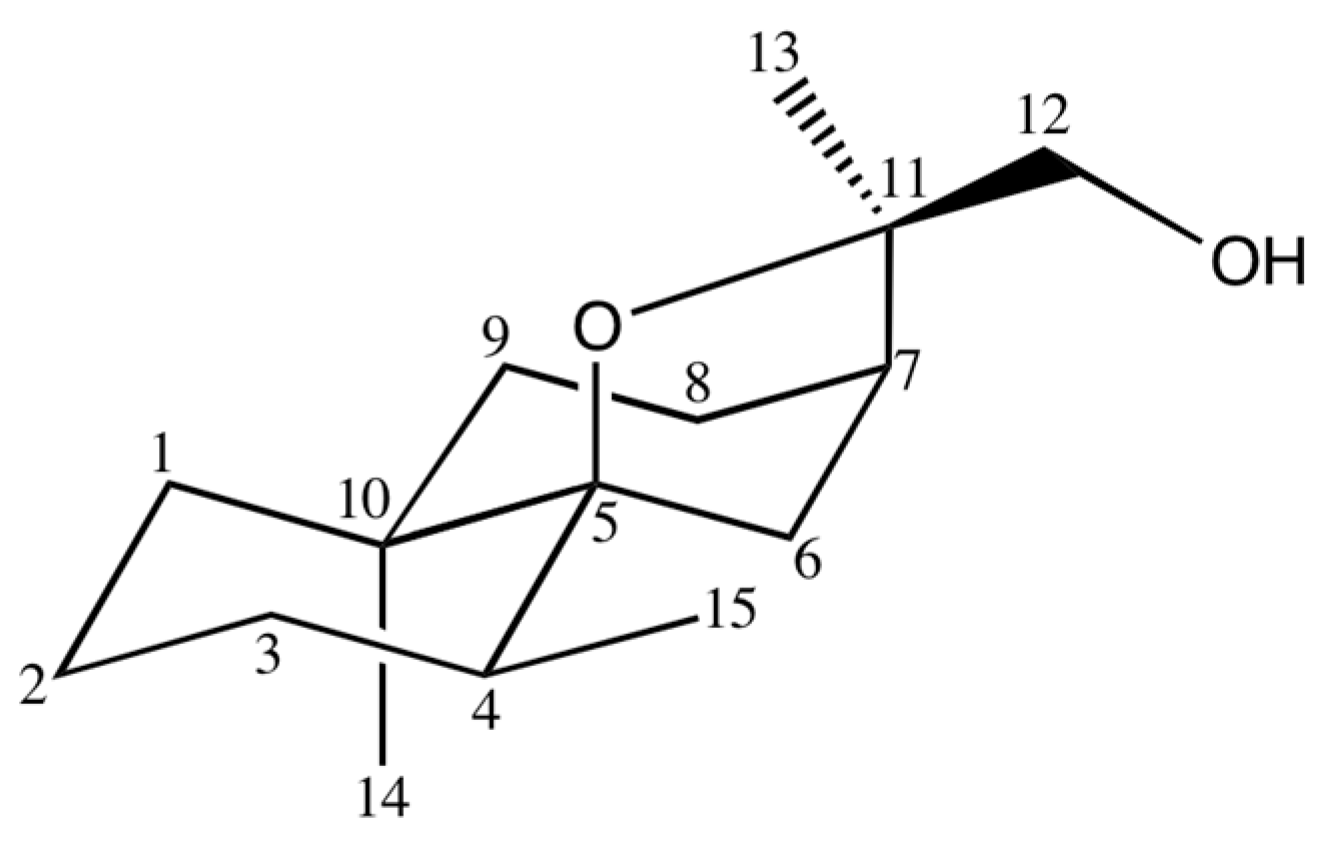
2.3.3. Identification of 12-Hydroxy-isodihydroagarofuran (39)
3. Discussion
4. Materials and Methods
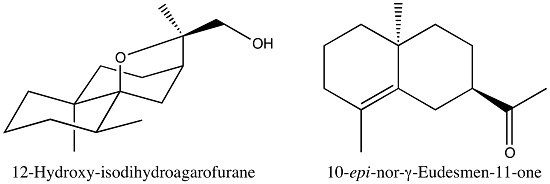
4.1. Plant Location and Essential Oil Isolation
4.2. Fractionation of Essential Oil
4.3. Gas Chromatography (GC) Analysis
4.4. Gas Chromatography-Mass Spectroscopy (GC/MS) Analysis
4.5. 13C-NMR Analysis
4.6. Identification of Individual Components
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Farjon, A. The discovery of a new genus of conifer in northern Vietnam. Dendrology 2002, 142–144. [Google Scholar]
- Thomas, P. IUCN Red List. Available online: http://threatenedconifers.rbge.org.uk/taxa/details/xanthocyparis-vietnamensis) (accessed on 7 July 2013).
- Missouri Botanical Garden—Plan Conservation. Available online: http://www.mobot.org/plantscience/ccsd/programs/cb/cbcloc/vietnam.shtml (accessed on 3 June 2013).
- Farjon, A. A new genus and species in Cupressaceae (Coniferales) from Northern Vietnam. Xanthocyparis vietnamensis. Novon 2002, 12, 179–189. [Google Scholar] [CrossRef]
- Little, D.P.; Schwarbach, A.E.; Adams, R.P.; Hsieh, C.F. The circumscription and phylogenetic relationships of Callitropsis and the newly described genus Xanthocyparis (Cupressaceae). Am. J. Bot. 2004, 91, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Russell, J. A new conifer species affects taxonomic classification in the Cupressaceae. TIC Talk. 2012, 11, 3–4. [Google Scholar]
- Regalado, J.C., Jr.; Loc, P.K.; Hiep, N.T.; van Thao, T.; Averyanov, L.V. The Vietnamese Golden Cypress (Xanthocyparis vietnamensis) Conservation Status Assessment (CSA) and Conservation Action Plan (CAP). Fauna and Flora International. Hoang Lien Son Project 2006, 1–12. [Google Scholar]
- Adams, R.P.; Thomas, P.; Rushforth, K. The leaf essential oils of new conifer genus, Xanthocyparis: Xanthocyparis vietnamensis and X. nootkatensis. J. Essent. Oil Res. 2007, 19, 30–33. [Google Scholar] [CrossRef]
- Thai, T.H.; Hiep, N.T.; Hong, P.T.; Minh, D.T. Chemical composition of the essential oil of Xanthocyparis vietnamensis Farjon and Hiep from Vietnam. Vietnam J. Biol. 2007, 29, 92–94. [Google Scholar] [CrossRef]
- Tomi, F.; Bradesi, P.; Bighelli, A.; Casanova, J. Computer-aided identification of individual components of essential oils using carbon-13 NMR spectroscopy. J. Magn. Reson. Anal. 1995, 1, 25–34. [Google Scholar]
- Rezzi, S.; Bighelli, A.; Castola, V.; Casanova, J. Direct Identification and Quantitative Determination of Acidic and Neutral Diterpenes Using 13C-NMR Spectroscopy. J. Appl. Spectrosc. 2002, 56, 312–317. [Google Scholar] [CrossRef]
- Ouattara, Z.A.; Boti, J.B.; Ahibo, A.C.; Sutour, S.; Casanova, J.; Tomi, F.; Bighelli, A. The key role of 13C-NMR analysis in the identification of individual components of Polyalthia longifolia leaf oil. Flavour Fragr. J. 2014, 29, 371–379. [Google Scholar] [CrossRef]
- Ouattara, Z.A.; Boti, J.B.; Ahibo, A.C.; Casanova, J.; Tomi, F.; Bighelli, A. Analysis of Cleistopholis patens Leaf and Trunk Bark Oils Using Combined GC- Flame Ionisation Detection. GC-Retention Index. GC–MS and 13C-NMR. Phytochem. Anal. 2013, 24, 574–580. [Google Scholar] [CrossRef] [PubMed]
- König, W.A.; Hochmuth, D.H.; Joulain, D. Terpenoids and Related Constituents of Essential Oils; Library of MassFinder 2.1; Institute of Organic Chemistry: Hamburg, Germany, 2001. [Google Scholar]
- Asakawa, Y.; Matsuda, R.; Tori, M.; Hashimoto, T. Preparation of biologically active substances and animal and microbial metabolites from menthols, cineoles and kauranes. Phytochemistry 1988, 27, 3861–3869. [Google Scholar] [CrossRef]
- Ishihara, M.; Tsuneya, T.; Uneyama, K. Fragrant sesquiterpenes from Agarwood. Phytochemistry 1993, 33, 1147–1155. [Google Scholar] [CrossRef]
- Tajuddin, S.N.; Yusoff, M.M. Chemical composition of volatile oils of Aquilaria malaccensis (Thymelaeaceae) from Malaysia. Nat. Prod. Commun. 2010, 5, 1965–1968. [Google Scholar] [PubMed]
- Gerhard, U.; Thomas, S.; Mortishire-Smith, R. Accelerated metabolite identification by “extraction NMR”. J. Pharm. Biomed. Anal. 2003, 32, 531–538. [Google Scholar] [CrossRef]
- Hubert, P.; Tomi, F.; Casanova, J.; Bighelli, A. Direct identification of two major components of an essential oil using “Extraction NMR”. Anal. Chem. Lett. 2011, 1, 115–122. [Google Scholar] [CrossRef]
- Revial, G.; Jabin, I.; Redolfi, M.; Pfau, M. Enantioselective imine Michael reaction for the preparation of the (8′R,8a′S)-8,8a′-dimethyl-1′,3′,4′,7′,8′,8a′-hexahydrospiro[1,3-dioxolane-2,2′(6′H)naphtalen]-6′-one building block. A formal synthesis of (+)-valencenol. Tetrahedron Asym. 2001, 12, 1683–1688. [Google Scholar] [CrossRef]
- Raharivelomanana, P.; Bianchini, J.P.; Cambon, A.; Azzaro, M.; Faure, R. Two-dimensional NMR of sesquiterpenes. Complete assignment of 1H and 13C-NMR spectra of seven sesquiterpene alcohols from Neocallitropsis pancheri. Magn. Reson. Chem. 1995, 33, 233–235. [Google Scholar] [CrossRef]
- Raharivelomanana, P.; Bianchini, J.P.; Ramanoelina, A.R.P.; Rasoarahona, J.R.E.; Faure, R.; Cambon, A. Eudesmane sesquiterpenes from Laggera alata. Phytochemistry 1998, 47, 1085–1088. [Google Scholar] [CrossRef]
- Marshall, J.A.; Pike, M.T. The stereoselective total synthesis of racemic γ-eudesmol. Tetrahedron Lett. 1966, 7, 4989–4992. [Google Scholar] [CrossRef]
- Thomas, A.F.; Ozainne, M. The stereochemistry of dihydroagarofurans. Tetrahedron Lett. 1976, 17, 1717–1718. [Google Scholar] [CrossRef]
- Itokawa, H.; Morita, H.; Watanabe, K.; Mihashi, S.; Iitaka, Y. Agarofurane. Eudesmane and Eremophilane-type Sesquiterpenoids from Alpinia japonica (Thunb.) Miq. Chem. Pharm. Bull. 1985, 33, 1148–1153. [Google Scholar] [CrossRef]
- Southwell, I.A.; Tucker, D.J. Cis-dihydroagarofuran from Prostanthera sp. aff. ovalifolia. Phytochemistry 1993, 33, 857–862. [Google Scholar] [CrossRef]
- Cavalli, J.F.; Tomi, F.; Bernardini, A.F.; Casanova, J. Dihydroagarofurans: The fourth isomer isolated from Cedrelopsis grevei bark oil. Magn. Reson. Chem. 2004, 42, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Khasawneh, M.A.; Xiong, Y.; Peralta-Cruz, J.; Karchesy, J.J. Biologically Important Eremophilane Sesquiterpenes from Alaska Cedar Heartwood Essential Oil and Their Semi-Synthetic Derivatives. Molecules 2011, 16, 4775–4785. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.P. Identification of Essential Oil Components by Gas Chromatography/Mass Spectrometry, 4th ed.; Allured Publishing Corporation Company: Carol Stream, IL, USA, 2007. [Google Scholar]
- US National Institute of Standards and Technology. PC Version 1.7 of the NIST/EPA/NIH Mass Spectra Library; US National Institute of Standards and Technology: Norwalk, CT, USA, 1999.
- Sample Availability: Samples of the compounds are not yet available from the authors.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Components | RI | RIa | RIp | % | Identification |
|---|---|---|---|---|---|---|
| 1 | α-Thujene | 932 | 922 | 1015 | 0.1 | RI, MS |
| 2 | Sabinene | 973 | 966 | 1124 | tr | RI, MS, 13C-NMR |
| 3 | Myrcene | 987 | 979 | 1161 | 0.2 | RI, MS, 13C-NMR |
| 4 | 1,4-Cineole | - | 1003 | 1178 | 0.6 | RI, MS, 13C-NMR |
| 5 | p-Cymene | 1015 | 1011 | 1271 | 1.3 | RI, MS, 13C-NMR |
| 6 | Limonene | 1025 | 1020 | 1202 | 0.2 | RI, MS, 13C-NMR |
| 7 | p-Cymenene | 1075 | 1072 | 1436 | 2.4 | RI, MS, 13C-NMR |
| 8 | p-Cymen-8-ol | 1169 | 1159 | 1842 | 4.1 | RI, MS, 13C-NMR |
| 9 | Terpinen-4-ol | 1164 | 1161 | 1597 | 3.0 | RI, MS, 13C-NMR |
| 10 | α-Terpineol | 1176 | 1171 | 1690 | 0.5 | RI, MS, 13C-NMR |
| 11 | Carvacryl methyl oxide | 1226 | 1223 | 1602 | 0.2 | RI, MS, 13C-NMR |
| 12 | Carvacrol | 1278 | 1275 | 2204 | 2.1 | RI, MS, 13C-NMR |
| 13 | α-Terpinyl acetate | 1335 | 1330 | 1685 | 0.1 | RI, 13C-NMR |
| 14 | β-Elemene | 1389 | 1386 | 1588 | 0.8 | RI, MS, 13C-NMR |
| 15 | 11,12,13-tri-nor-Eremophil-1(10)-en-7-one | - | 1438 | 1996 | 17.2 | RI, MS, 13C-NMR |
| 16 | 11,12,13-tri-nor-Eremophil-1(10)-en-7-ol (A) | - | 1458 | 2173 | 0.4 | RI, 2D-NMR |
| 17 | β-Selinene | 1486 | 1480 | 1720 | 0.8 | RI, MS, 13C-NMR |
| 18 | Valencene | 1494 | 1487 | 1715 | 3.5 | RI, MS, 13C-NMR |
| 19 | α-Selinene | 1494 | 1490 | 1723 | 0.3 | RI, 13C-NMR |
| 20 | γ-Cadinene | 1507 | 1492 | 1750 | 0.3 | RI, 13C-NMR |
| 21 | Nootkatene | 1512 | 1507 | 1811 | 20.7 | RI, MS, 13C-NMR |
| 22 | δ-Cadinene | 1520 | 1512 | 1753 | 1.2 | RI, MS, 13C-NMR |
| 23 | β-Elemol | 1541 | 1532 | 2070 | 0.9 | RI, MS, 13C-NMR |
| 24 | 10-epi-nor-γ-Eudesmen-11-one (B) | - | 1553 | 2039 | 0.9 | RI, 2D-NMR |
| 25 | 13-nor-Eremophil-1(10)-en-11-one | - | 1594 | 2133 | 2.6 | RI, 13C-NMR |
| 26 | Eremophil-9-en-11-ol (jinkoheremol) | - | 1613 | 2206 | tr | RI, 13C-NMR |
| 27 | Eremoligenol | - | 1616 | 2172 | tr | RI, 13C-NMR |
| 28 | γ-Eudesmol | 1618 | 1617 | 2159 | 5.1 | RI, MS, 13C-NMR |
| 29 | τ-Cadinol | 1633 | 1625 | 2159 | 0.7 | RI, 13C-NMR |
| 30 | τ-Muurolol | 1633 | 1627 | 2176 | 0.7 | RI, MS, 13C-NMR |
| 31 | δ-Cadinol (torreyol) | - | 1629 | 2193 | 0.3 | RI, 13C-NMR |
| 32 | Valerianol | 1647 | 1633 | 2207 | tr | RI, 13C-NMR |
| 33 | β-Eudesmol | 1641 | 1634 | 2220 | 3.7 | RI, MS, 13C-NMR |
| 34 | α-Cadinol | 1643 | 1636 | 2220 | 1.9 | RI, 13C-NMR |
| 35 | α-Eudesmol | 1653 | 1639 | 2210 | 3.3 | RI, MS, 13C-NMR |
| 36 | Selin-11-en-4α-ol | - | 1643 | 2241 | 0.3 | RI, 13C-NMR |
| 37 | Cadalene | 1659 | 1653 | 2204 | 0.3 | RI, MS, 13C-NMR |
| 38 | Dehydro-jinkoheremol | - | 1671 | 2214 | 0.2 | RI, 13C-NMR |
| 39 | 12-Hydroxy-isodihydroagarofuran (C) | - | 1742 | 2245 | 2.3 | RI, 2D NMR |
| 40 | Nootkatone | 1782 | 1774 | 2501 | 4.7 | RI, 13C-NMR |
| Total | 87.9 |
| C | δ C (ppm) | DEPT | δ 1H (ppm) | Multiplicity (J, Hz) | HMBC | COSY | NOESY |
|---|---|---|---|---|---|---|---|
| 1 | 39.99 | CH2 | 1.42 (a) | m1 | - | - | - |
| 1.28 (b) | m | - | - | - | |||
| 2 | 18.88 | CH2 | 1.53 | m | - | - | - |
| 3 | 32.96 | CH2 | 1.98 (a) | m | - | - | - |
| 1.86 (b) | m | - | - | - | |||
| 4 | 126.29 | C | - | - | - | - | - |
| 5 | 131.73 | C | - | - | - | - | - |
| 6 | 25.89 | CH2 | 1.96 (b) | m | C12 | - | - |
| 3.01 (a) | dt (14.8;2.2) | C4; C5; C7; C8; C10 | H12 | H12; H13 | |||
| 7 | 49.02 | CH | 2.62 | m | C5; C6; C8; C9 | H12 | H12 |
| 8 | 22.7 | CH2 | 2.02 (a) | m | - | - | H8b |
| 1.84 (b) | m | C7; C9; C11/C5; C6; C10 | H9 | H8a | |||
| 9 | 38.29 | CH2 | 1.32 | s | C1; C5; C7; C8; C10; C10-Me | H8b | - |
| 10 | 34.57 | C | - | - | - | - | - |
| 11 | 211.19 | C | - | - | - | - | - |
| 12 | 28.06 | CH3 | 2.14 | s | C7; C11 | H6a; H7 | H7; H6a |
| C4-Me | 19.55 | CH3 | 1.67 | s | C3; C4; C5; C10-Me | - | H6a |
| C10-Me | 24.56 | CH3 | 1.06 | s | C1; C5; C9; C10 | - | - |
| C | δC (ppm) | DEPT | δH (ppm) | Multiplicity (J, Hz) | HMBC | COSY | NOESY |
|---|---|---|---|---|---|---|---|
| 1 | 37.90 | CH2 | 1.12 (a) | br s 1 | - | ||
| 1.55 (b) | m | - | |||||
| 2 | 21.33 | CH2 | 1.44 (a) | t (3.0) | - | ||
| 0.92 (b) | d (7.0) | C 15 | |||||
| 3 | 32.08 | CH2 | 1.44 (a) | t (3.0) | - | ||
| 1.35 (b) | m | - | |||||
| 4 | 32.23 | CH | 1.74 | m | C7 | H15 | |
| 5 | 88.21 | C | - | - | - | - | - |
| 6 | 33.39 | CH2 | 1.98 (a) | dd (11.7; 4.3) | C4; C5; C7; C8; C10 | H6b; H8 | H12a |
| 1.55 (b) | br s | C7; C8; C10; C11 | H6a | ||||
| 7 | 39.99 | CH | 1.87 | m | C6; C8; C9; C12 | ||
| 8 | 24.84 | CH2 | 1.70 | m | C9 | ||
| 9 | 36.12 | CH2 | 1.67 (a) | m | C14 | ||
| 1.20 (b) | m | C8 | |||||
| 10 | 38.83 | C | - | - | - | - | - |
| 11 | 82.91 | C | - | - | - | - | - |
| 12 | 69.29 | CH2 | 3.40 (a) | d (10.4) | C7; C11; C13 | H12a; H13 | H6a; H15 |
| 3.24 (b) | d (10.4) | C13 | H12b; H13 | ||||
| 13 | 17.76 | CH3 | 1.41 | s | C7; C11; C12 | ||
| 14 | 23.69 | CH3 | 1.01 | s | C1; C5; C9; C10 | ||
| 15 | 15.52 | CH3 | 0.85 | d (7.0) | C3; C4; C5 | H4 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bazzali, O.; Thai, T.H.; Hoi, T.M.; Khang, N.S.; Hien, N.T.; Casanova, J.; Bighelli, A.; Tomi, F. Integrated Analysis of the Wood Oil from Xanthocyparis vietnamensis Farjon & Hiep. by Chromatographic and Spectroscopic Techniques. Molecules 2016, 21, 840. https://doi.org/10.3390/molecules21070840
Bazzali O, Thai TH, Hoi TM, Khang NS, Hien NT, Casanova J, Bighelli A, Tomi F. Integrated Analysis of the Wood Oil from Xanthocyparis vietnamensis Farjon & Hiep. by Chromatographic and Spectroscopic Techniques. Molecules. 2016; 21(7):840. https://doi.org/10.3390/molecules21070840
Chicago/Turabian StyleBazzali, Ophélie, Tran Huy Thai, Tran Minh Hoi, Nguyen Sinh Khang, Nguyen Thi Hien, Joseph Casanova, Ange Bighelli, and Félix Tomi. 2016. "Integrated Analysis of the Wood Oil from Xanthocyparis vietnamensis Farjon & Hiep. by Chromatographic and Spectroscopic Techniques" Molecules 21, no. 7: 840. https://doi.org/10.3390/molecules21070840
APA StyleBazzali, O., Thai, T. H., Hoi, T. M., Khang, N. S., Hien, N. T., Casanova, J., Bighelli, A., & Tomi, F. (2016). Integrated Analysis of the Wood Oil from Xanthocyparis vietnamensis Farjon & Hiep. by Chromatographic and Spectroscopic Techniques. Molecules, 21(7), 840. https://doi.org/10.3390/molecules21070840