Abstract
(−)-Cytoxazone, originally isolated from cultures of a Streptomyces species has an oxazolidin-2-one 4,5-disubstituted ring. It is known that this natural product presents a cytokine modulator effect through the signaling pathway of Th2 cells (type 2 cytokines), which are involved in the process of growth and differentiation of cells. From this, the interest in the development of research aimed at the total synthesis of this molecule and its analogs has remained high, which can be confirmed by the large number of publications on the topic, more than 30 to date. This review focuses on the various creative methods for the synthesis of (−)-cytoxazone and its congeners. The assessment of the preparation of this oxazolidinone and related structures serves as a treatise on the efforts made in the synthesis of this important class of compound from its first total synthesis in 1999.
1. Introduction
This review focuses on the various total synthesis strategies for the preparation of (−)-cytoxazone and its congeners. The assessment of the preparation of this oxazolidinone and related structures serves as a treatise on the efforts made in the synthesis of this class of compounds. Emphasis is placed on the literature since the first total synthesis in 1999 until mid-2016. Readers may also consult two excellent surveys of the building of oxazolidin-2-one rings by Rozwadowska et al. [1] and Zappia [2].
1.1. Compounds Containing 2-Oxazolidinone Structural Units and Biological Applications
New synthetic antimicrobial agents have been discovered from the preparation of a library of compounds of the oxazolidin-2-one family (Figure 1) by DuPont researchers [3,4], specifically exerting a bacteriostatic effect during in vitro and in vivo assays, using human pathogenic bacteria [3].
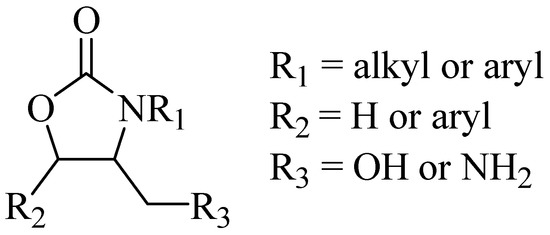
Figure 1.
Antimicrobial oxazolidin-2-ones.
The main biological activities exhibited by oxazolidin-2-ones include antiallergic, psychotropic, and neuroleptic activities. Moreover, oxazolidin-2-ones are also intermediates in the synthesis of renin inhibitors, β-lactams and macrolide antibiotics [5].
This class of compounds also exhibited remarkable activities against Gram-positive bacteria such as Staphylococcus aureus, Staphylococcus epidermidis, Streptococcus pneumoniae, Enterococcus faecalis, and Enterococcus faecium. They are also active against pathogens that are resistant to one or more antibiotics, as is the case with methicillin-resistant S. aureus, vancomycin-resistant enterococci, and penicillin-resistant pneumococci [4]. By contrast, the 5-substituted oxazolidinones exhibit an even more evident antibacterial activity. Linezolid (PNU-100766, Figure 2), for example, is recommended for the treatment of the infections caused by Gram-positive bacteria, such as those caused by vancomycin-resistant S. aureus [6], and was the first compound of this class to be licensed in the United States by the Food and Drug Administration (FDA) in 2000 [4].
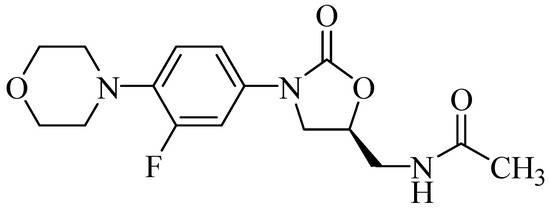
Figure 2.
Antibacterial linezolid (PNU-100766).
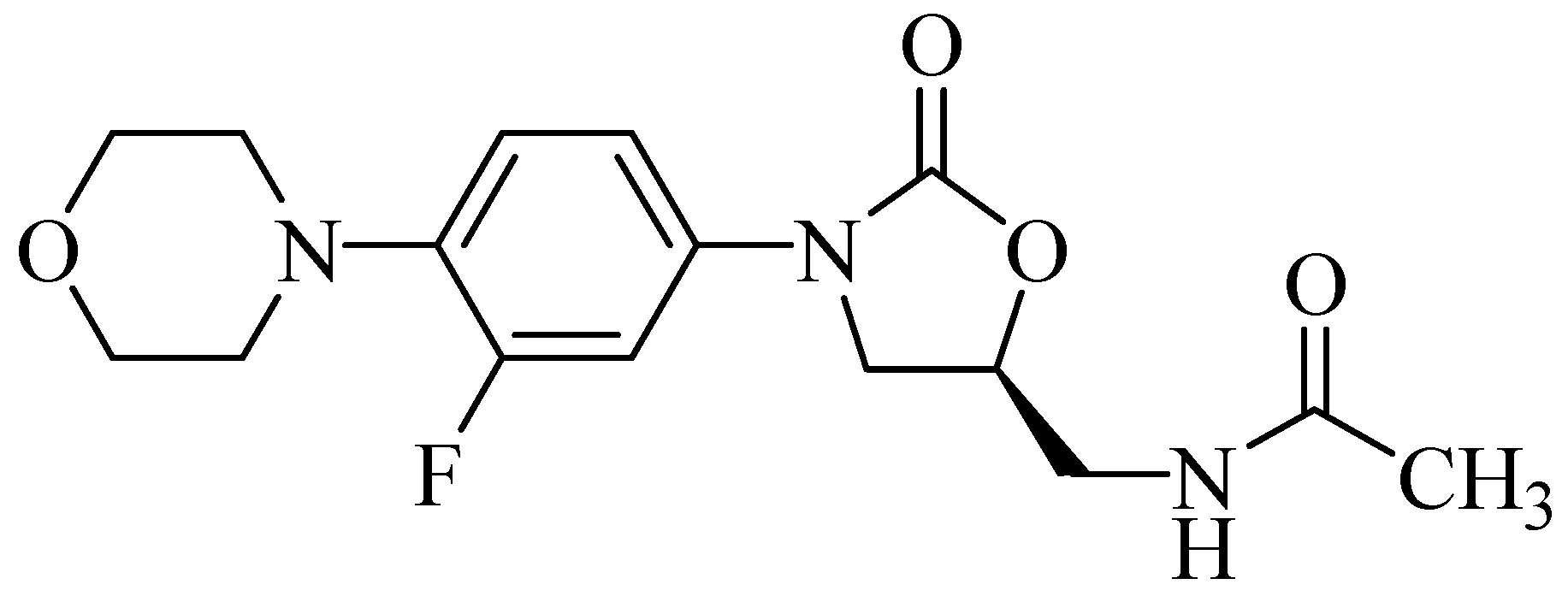
Furthermore, based on reports regarding new mechanisms of action against cancer, HIV, monoamine oxidase, glutamate receptor antagonists, and metabotropic receptors, Naresh et al. [7] synthesized a series of compounds that were linezolid analogs. Those, which had acrylic and ethyl substituents (Figure 3), exhibited excellent results against lung and prostate cancer cells, revealing a new option for the synthesis of drugs for the treatment of these types of cancer.
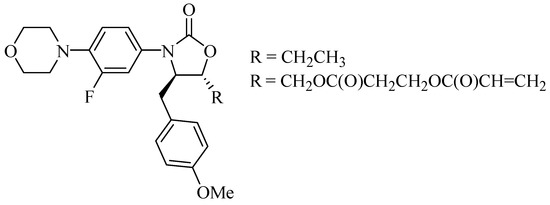
Figure 3.
Oxazolidinone compounds 4,5-di-substituted with anticancer activity.
In addition to the reported range of biological activities, after Evans reported their use as chiral auxiliaries in 1981, some of these oxazolidinones have been widely used in research involving asymmetric organic syntheses [2].
1.2. (−)-Cytoxazone
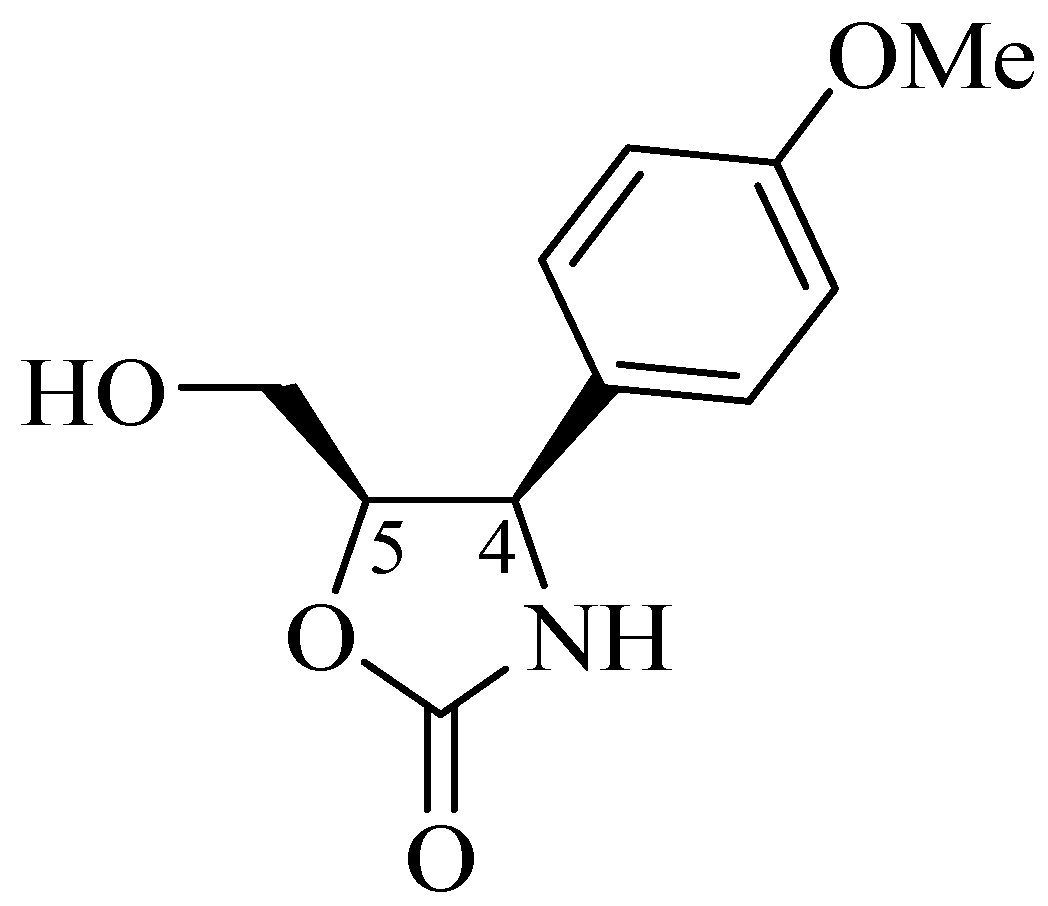
Although quite rare, a new type of oxazolidin-2-one [2], (−)-cytoxazone (Figure 4), was originally isolated by Kakeya et al. [8] from cultures of a Streptomyces species. (−)-Cytoxazone has an oxazolidin-2-one 4,5-disubstituted ring, and its absolute configuration, based on NMR data, CD, and X-ray crystallography, was defined as (4R,5R)-5-hydroxymethyl-4-p-methoxyphenyl-1,3-oxazolidin-2-one [8,9]. Nakata et al. [10] unequivocally confirmed the absolute configuration resulting from the first asymmetric total synthesis.
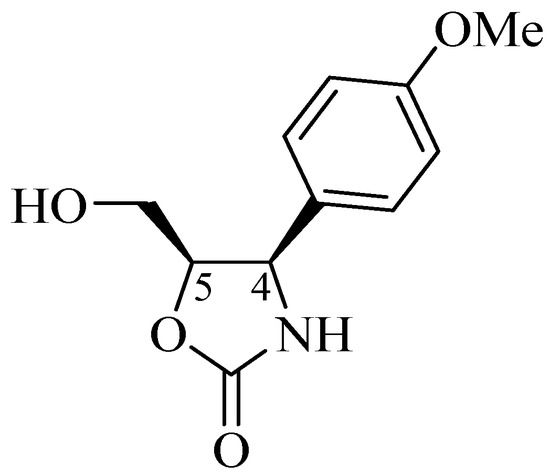
Figure 4.
Structure of (−)-cytoxazone.
It is known that the induction of humoral antibodies or cellular responses are affected by different sets of cells. The set of cells called Th1 (type 1 cytokines) are responsible for hypersensitivity reactions, however, the Th2 set (type 2 cytokines) are involved in the processes of cell growth and differentiation.
An imbalance in the production of these cytokines can result in immunological disorders such as allergies, progressive lymphoproliferation and severe immunodeficiency [9], since they are directly related to the production of antibodies. Looking for new immunomodulatory chemicals able to control the production of type 2 cytokines, Kakeya et al. [9] found that (−)-cytoxazone presented a cytokine modulator effect through the signaling pathway of Th2 cells, but not of Th1 cells. From this, the interest in the development of methods for the total synthesis of (−)-cytoxazone and its analogs has remained high, which can be confirmed by the large number of publications, more than 30 to date. In this context, the following review consists of a compilation of information about the synthesis of (−)-cytoxazone from its first approaches to the present day, as it is understood that this is a valuable contribution to the study of new synthetic strategies for this oxazolidinone, a very important molecule in the therapeutic arsenal today.
2. Total Synthesis Strategies of (−)-Cytoxazone and Congeners
The description of all publications on the preparation of (−)-cytoxazone and some congeners are presented in chronological order starting from 1999, when the first total syntheses of (−)-cytoxazone (1) were published, simultaneously, by Nakata [10] and Mori [11], respectively, both starting from 4-methoxycinnamate 2. Exhaustive coverage, however, cannot be guaranteed.
2.1. Stereoselective Syntheses of (−)-Cytoxazone, a Novel Cytokine Modulator
The route proposed by Nakata et al. [10] is summarized in Scheme 1. The key step of the synthesis of 1 consisted of a regio- and stereoselective introduction of an azide group into 4-methoxy-cinnamate 2. The sequence of Sharpless asymmetric dihydroxylation from 2, using AD-mix-α in t-BuOH/H2O, the reduction of the ester group with NaBH4, and the regioselective protection of the primary hydroxyl function with t-butyldiphenylsilyl chloride (TBDPSCl) afforded the (4S,5S)-diol 3 (cytoxazone numbering) in 65% yield (three steps). Treatment with SOCl2 and Et3N afforded the cyclic sulfite 5 in 99% yield. The regio- and stereoselective opening of sulfide was made by a nucleophilic reaction with LiN3 in DMF to generate azido alcohol 7 (74% yield) and desilylated azido-diol (24% yield) which was converted quantitatively to 7 by TBDPS/imidazole treatment. The azido alcohol 7 was treated with ClCO2Ph/pyridine, providing the phenyl carbonate 9 in 96% yield. The heterocyclization was performed in one pot from 9 by reduction of azide group to amine with Ph3P in THF/H2O and concomitant cyclization to produce oxazolidinone 10 (90% yield) as a properly controlled stereochemistry. Finally, the removal of the TBDPS group with TBAF, produced the desired (−)-cytoxazone (1) in 96% yield.
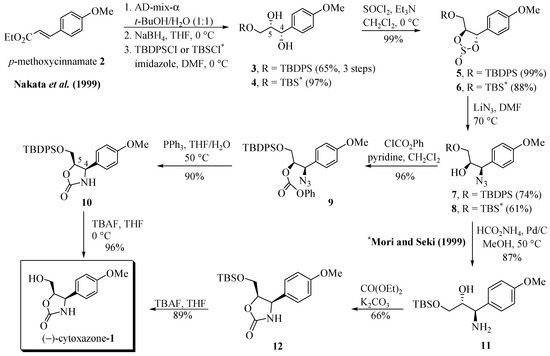
Scheme 1.
First total syntheses of (−)-cytoxazone reported by Nakata et al. [10] and Mori and Seki [11].
While Nakata et al. [10] converted the azido alcohol 7 into the corresponding phenyl carbonate 9, Mori and Seki [11] converted the azido alcohol 8 into the amino alcohol 11 in 87% yield, by reducing the azide group with ammonium formate and Pd/C (Scheme 1). N,O-Heterocyclization of 11 was accomplished by treatment with diethyl carbonate, providing 12 in 66% yield. The desilylation of 12 produced (−)-cytoxazone in 89% yield. These two studies established the absolute stereochemical configuration of (−)-cytoxazone.
2.2. Chemoenzymatic Synthesis of (−) and (+)-Cytoxazone Enantiomers
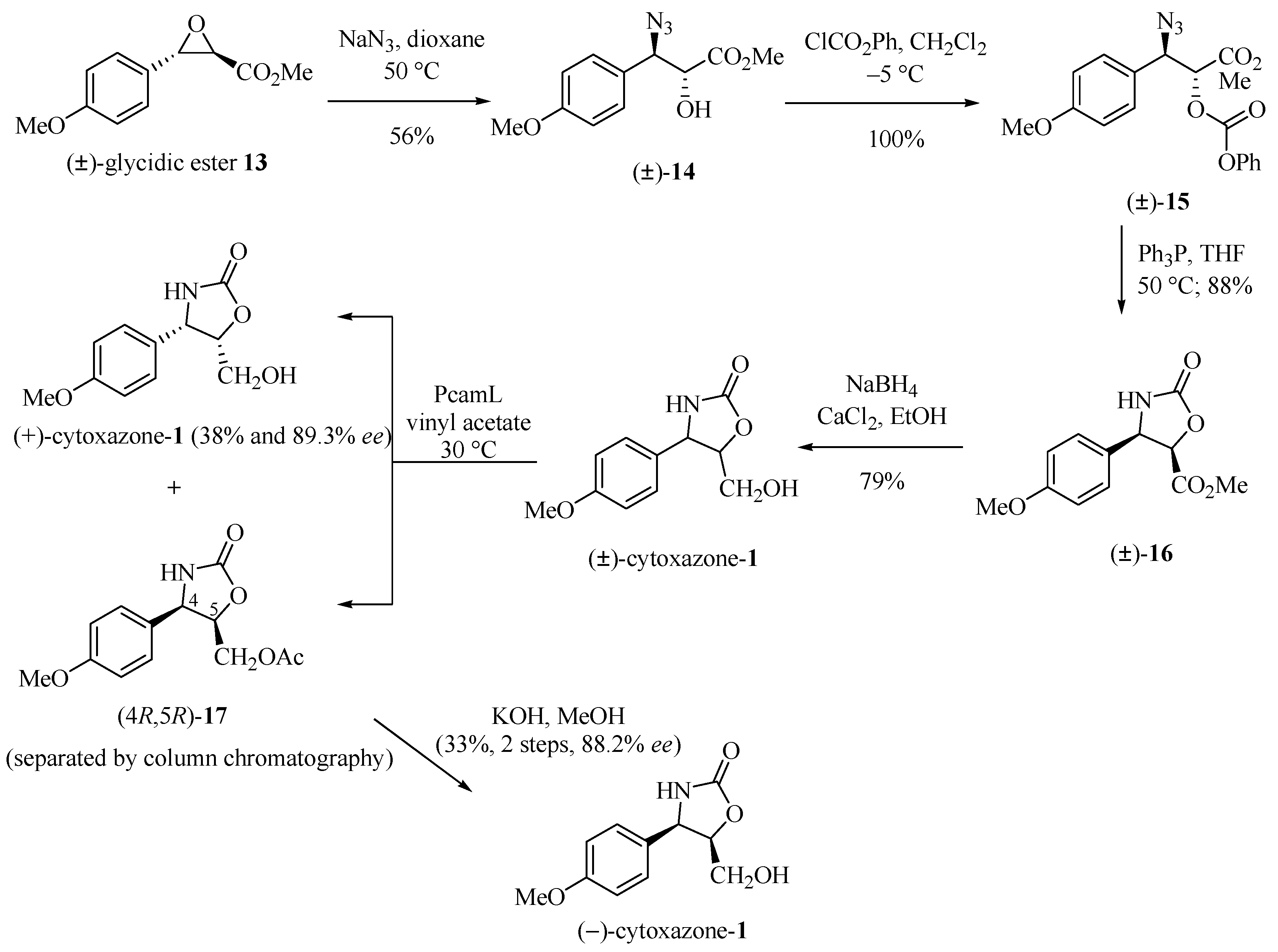
Sunjic and co-workers [12] described the chemoenzymatic synthesis of the enantiomers (−)- and (+)-cytoxazone whose resolution from the racemate (±)-1 was achieved by esterification catalyzed by Penicillium camambertii lipase (PcamL) (Scheme 2). Preparation of (±)-1 starts with the opening of the glycidic epoxide 13 by treatment with NaN3, producing azido alcohol 14 with complete regio- and stereoselectivity, in accordance with a well-stablished mechanism [13]. Treatment of this intermediate with ClCO2Ph produced the phenyl carbonate (±)-15. The reduction of the azide group and simultaneous cyclization generated (±)-oxazolidinone 16. Finally, the reduction of the ester group with sodium borohydride yielded (±)-cytoxazone (1) in 79% yield. Enzymatic kinetic resolution under acetylation of (±)-1 with vinyl acetate by PcamL-catalyzed and after their separation by chromatography column, produced the acetate (4R,5R)-17 and the (+)-cytoxazone (38% yield and 89.3% ee). Hydrolysis of ester 17 with KOH in MeOH produced (−)-cytoxazone (33% yield, two steps, and 88.2% ee).
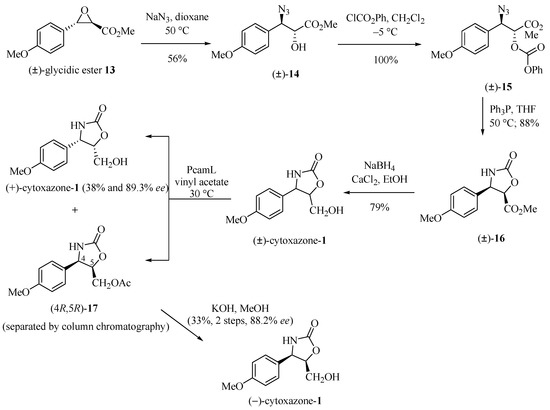
Scheme 2.
Chemoenzymatic synthesis of the (−)- and (+)-cytoxazones [12].
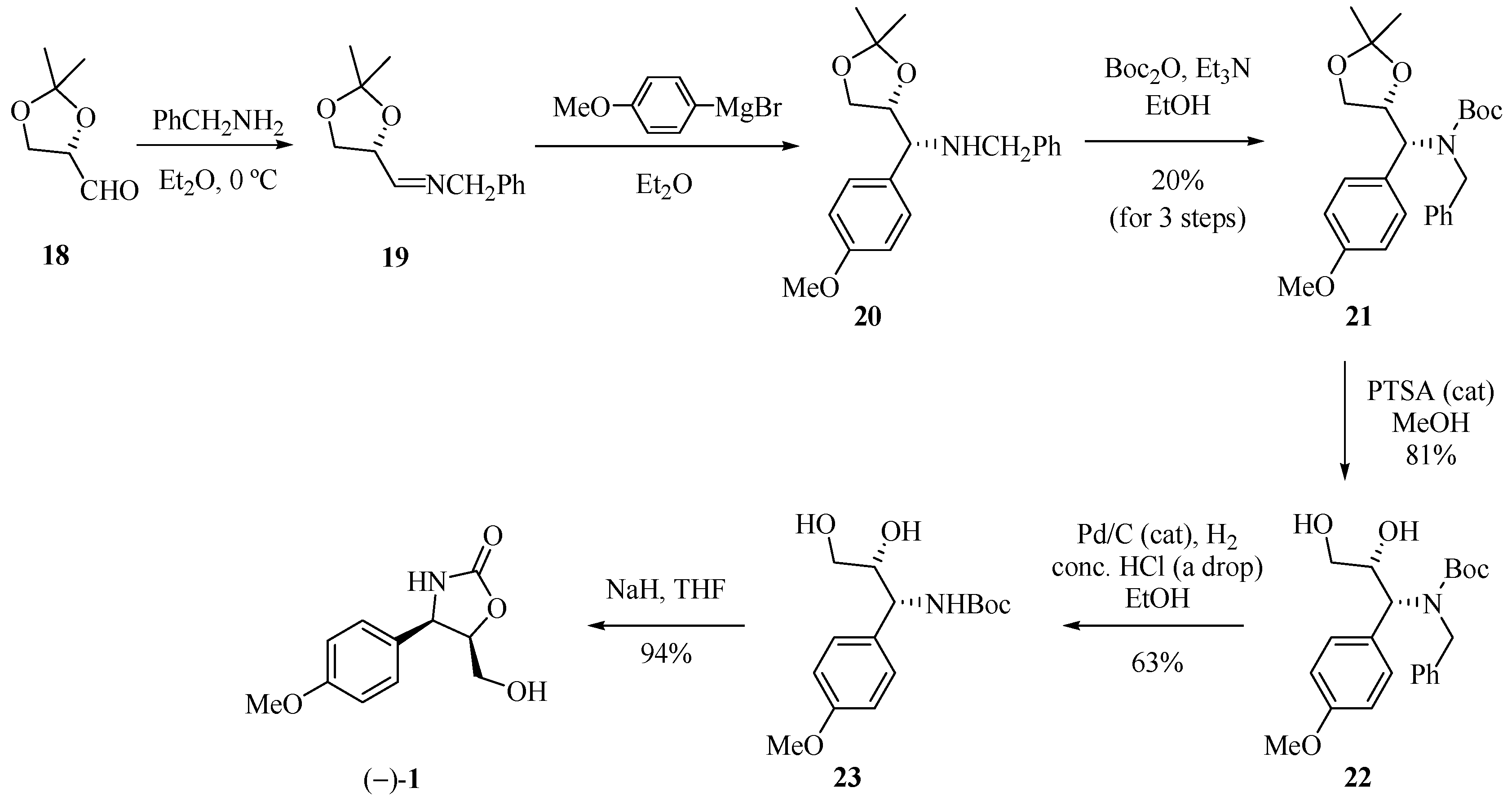
The synthetic approach reported by Rao and his group [14] to obtain (−)-cytoxazone started from (S)-2,3-O-isopropylidene glyceraldehyde (18, Scheme 3). Treatment of this compound with benzylamine produced imine 19, which when subjected to stereoselective addition of p-methoxyphenylmagnesium bromide led to amine 20. The amine function was protected with Boc2O producing the diprotected amine 21 (20% yield, three steps). Finally, the sequence of a chemoselective hydrolysis of the isopropylidene unit, debenzylation, and regioselective cyclization gave (−)-cytoxazone (1) from glyceraldehyde 18 in 9.6% overall yield.
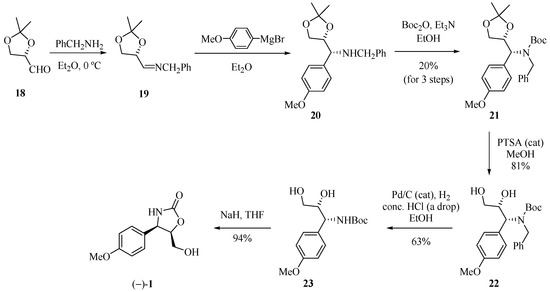
Scheme 3.
Total synthesis of (−)-cytoxazone proposed by Rao and co-workers [14].
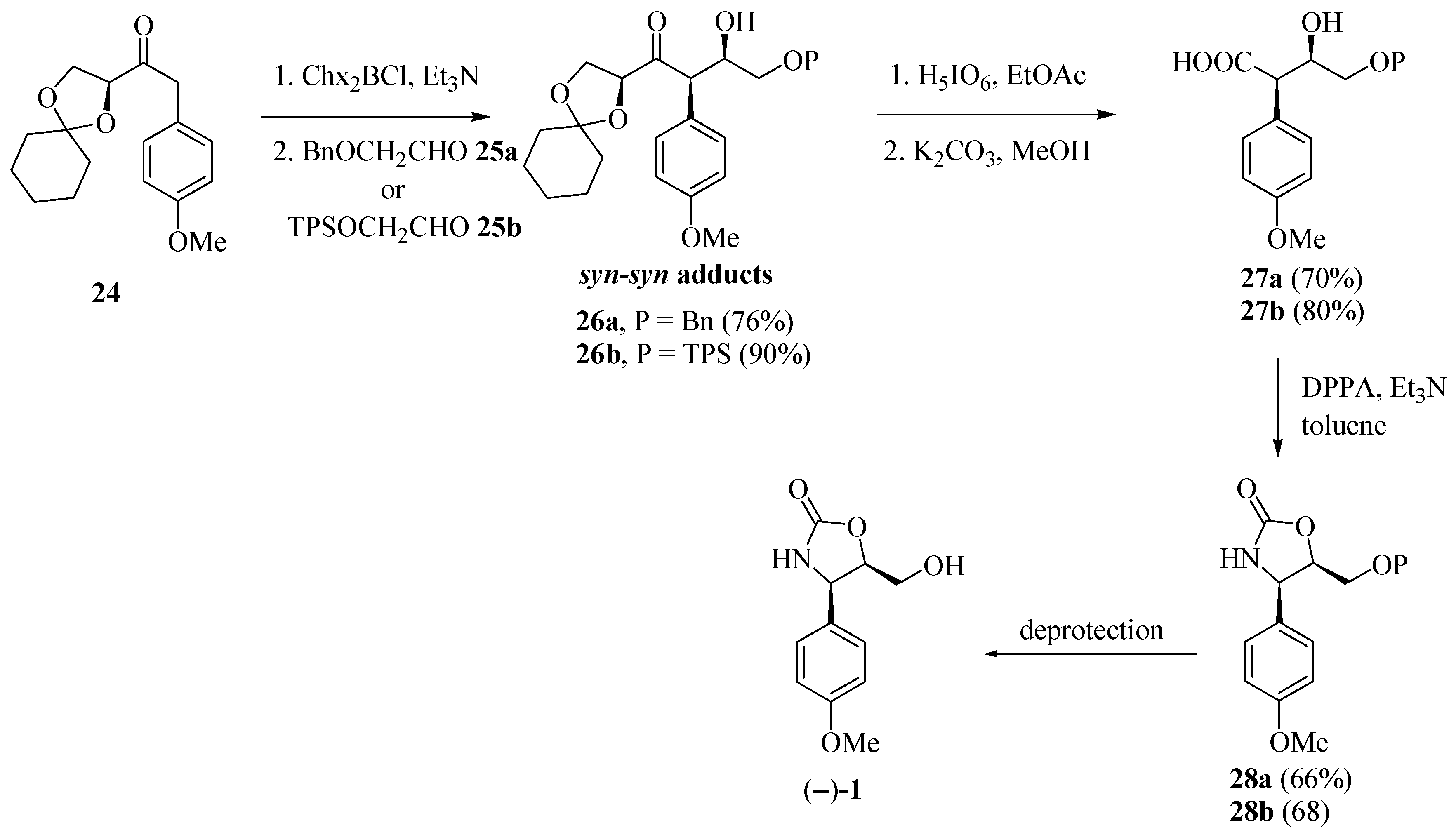
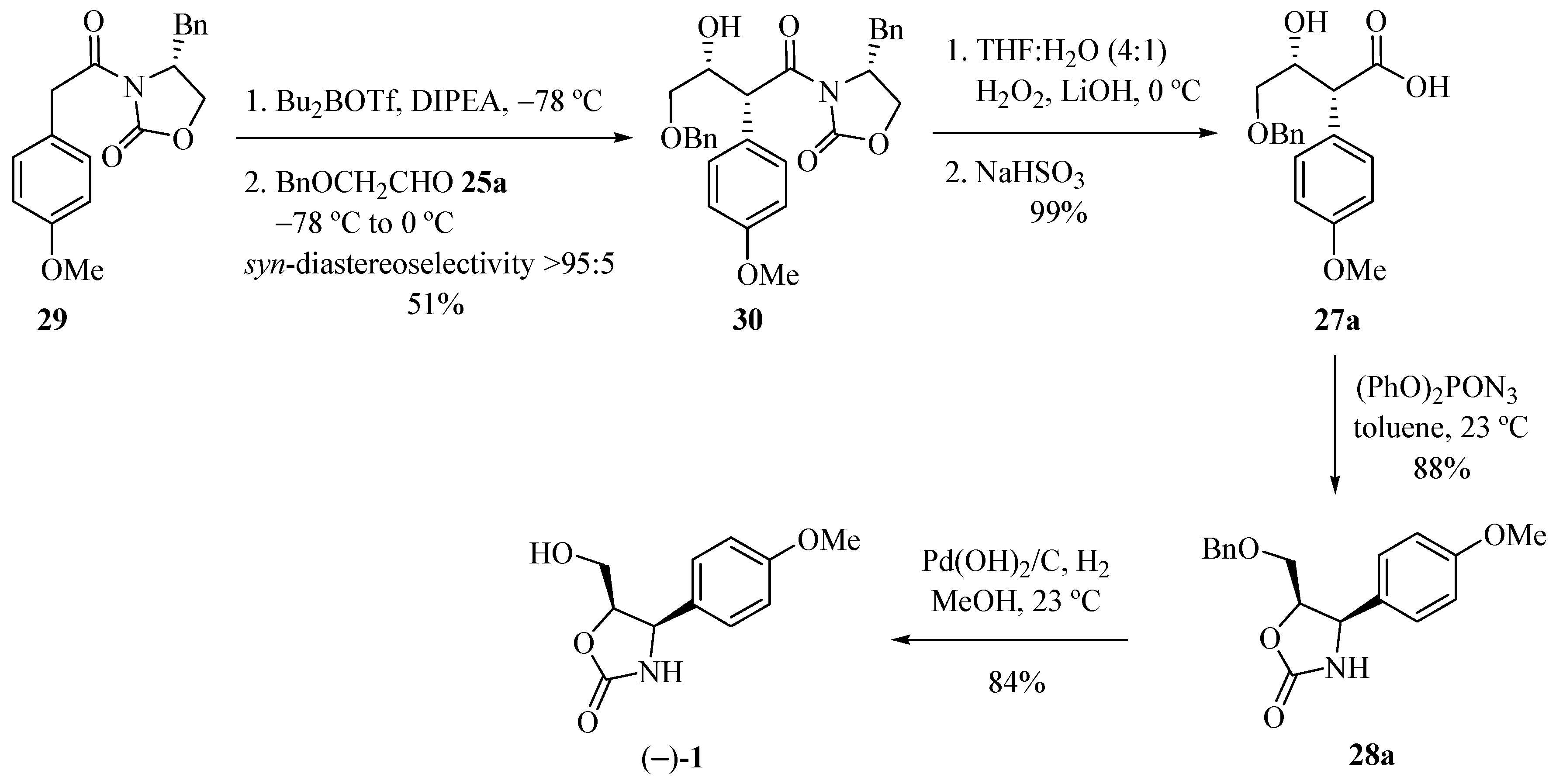
Marco and co-workers [15] obtained enantiomerically pure (−)-cytoxazone by a route that included a syn-stereoselective aldol addition by chlorodicyclohexylborane (Chx2BCl) mediation, with the unusual chiral ketone 24 and a Curtius rearrangement representing the key steps (Scheme 4). The aldol addition of ketone 24 to protected glycol aldehydes 25a–b, provided the expected syn-syn aldol adducts 26a–b. Oxidative cleavage of the acetonide rings by treatment with periodic acid provided the β-formyloxy acid intermediaries, which have been converted, subsequently, in basic conditions, into hydroxy acids 27a–b. Curtius rearrangement of these compounds was made by treatment with diphenyl phosphoryl azide (DPPA), whose isocyanate groups in the formed intermediates, were captured in situ by free hydroxyl groups, leading to the formation of oxazolidinone rings 28a–b, which after O-deprotection, produced (−)-cytoxazone.
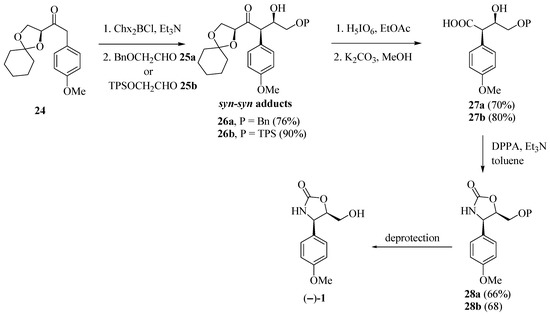
Scheme 4.
Total synthesis of (−)-cytoxazone proposed by Marco et al. [15].
2.3. A New Synthesis of (−)-Cytoxazone and Its Diastereomers
Carter and his team [16] explored a new (−)-cytoxazone synthesis route by means of an aldol reaction, starting from the chiral imide 29 with an aldehyde mediated by Bu2BOTf (Scheme 5). Subsequent reaction of the dibutylboron enolate of 29 with benzyloxyacetaldehyde 25a, according to the pioneering work of Evans [17], produced aldol 30 in good syn-diastereoselectivity (>95:5). Removal of the chiral auxiliary from 30 by treatment with lithium hydroxyperoxide led to the carboxylic acid 27a in 99% yield. The heterocyclization of the acid 27a was performed in a one pot (three-step) (acyl azide formation, Curtius rearrangement, isocyanate trapping) using diphenylphosphoryl azide producing the ether-oxazolidinone 28a. Deprotection using Pd(OH)2/C and hydrogen atmosphere produced enantiomerically pure (−)-cytoxazone in 84% yield. The enantiomer (+)-1 was similarly prepared to natural cytoxazone from (S)-4-phenylmethyl-oxazolidin-2-one.
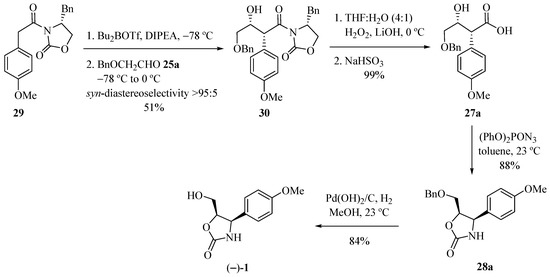
Scheme 5.
Total synthesis of (−)-cytoxazone reported by Carter and co-workers [16].
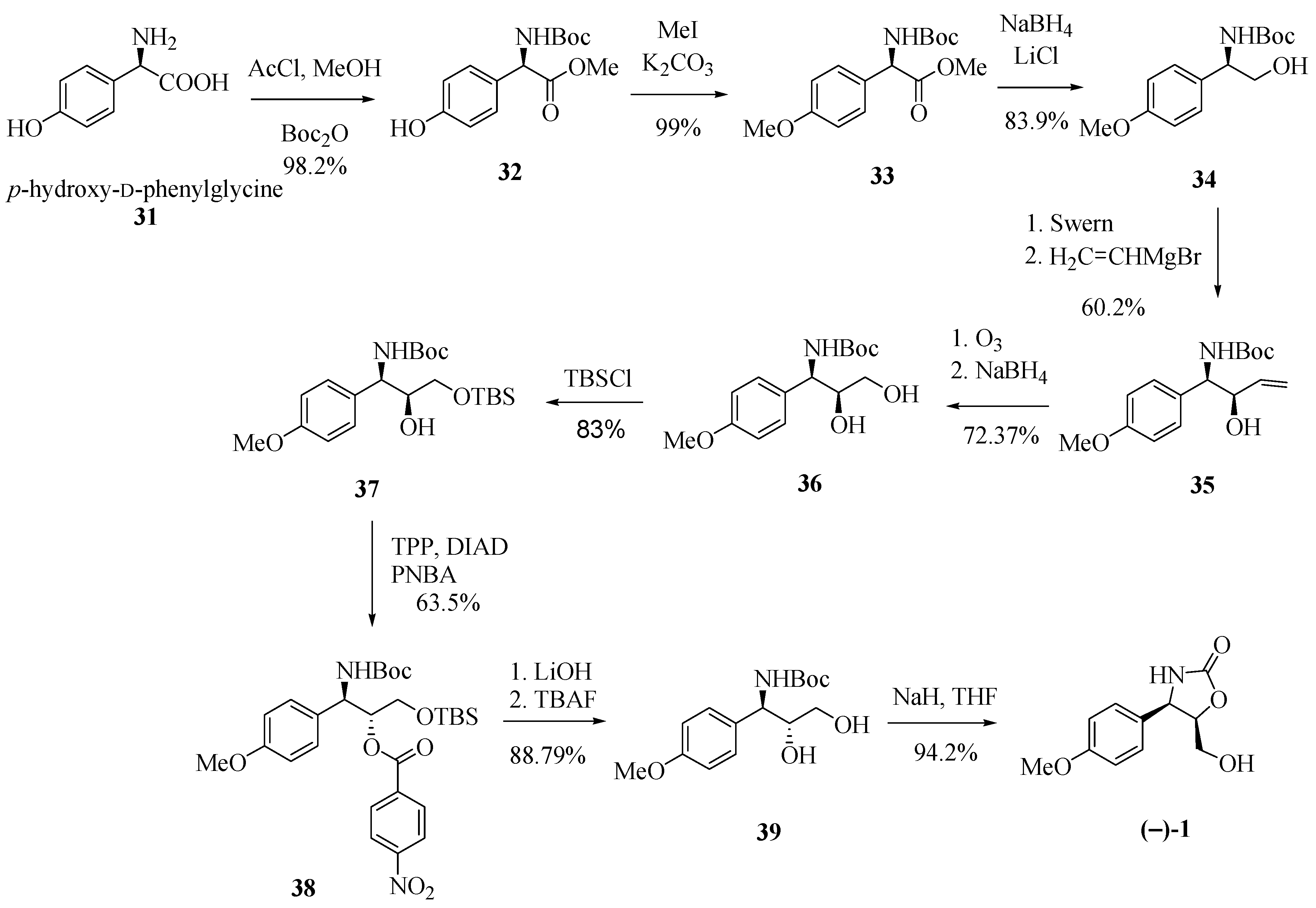
In another contribution to the preparation of (−)-cytoxazone, Rao and co-workers [18] proposed a stereoselective addition reaction of a vinyl Grignard at the N-Boc-aldehyde 34a, obtained from p-hydroxy-D-phenylglycine (31, Scheme 6). Starting from 31, the sequence of esterification, N-Boc-protection, phenol methylation, and the reduction of the ester function generated alcohol 34 in 81.6% yield (four steps). Swern oxidation of 34 followed by stereoselective addition of vinylmagnesium bromide to the resulting aldehyde led to the syn-1,2-amino alcohol 35 in 60.2% yield. Ozonolysis of the double bond and reduction of the formed aldehyde produced diol 36. Protection of the primary hydroxyl group, followed by an inversion of the configuration of the secondary hydroxyl by means of a Mitsunobu condition and subsequent desilylation led to trans-1,2-amino alcohol 39 in 46.79% overall yield from 36. Finally, the N,O-heterocyclization generated (−)-cytoxazone in 94.2% yield.
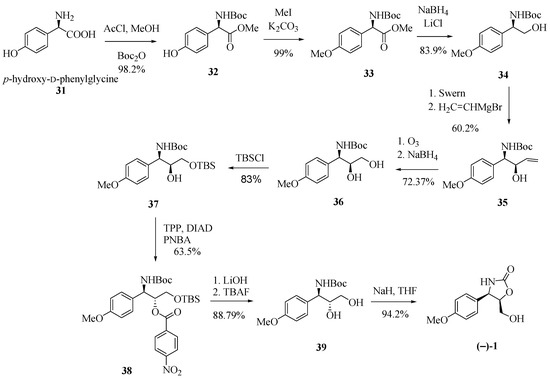
Scheme 6.
Synthesis of (−)-cytoxazone via stereoselective Grignard addition [18].
2.4. Asymmetric Synthesis of (4R,5R)-Cytoxazone
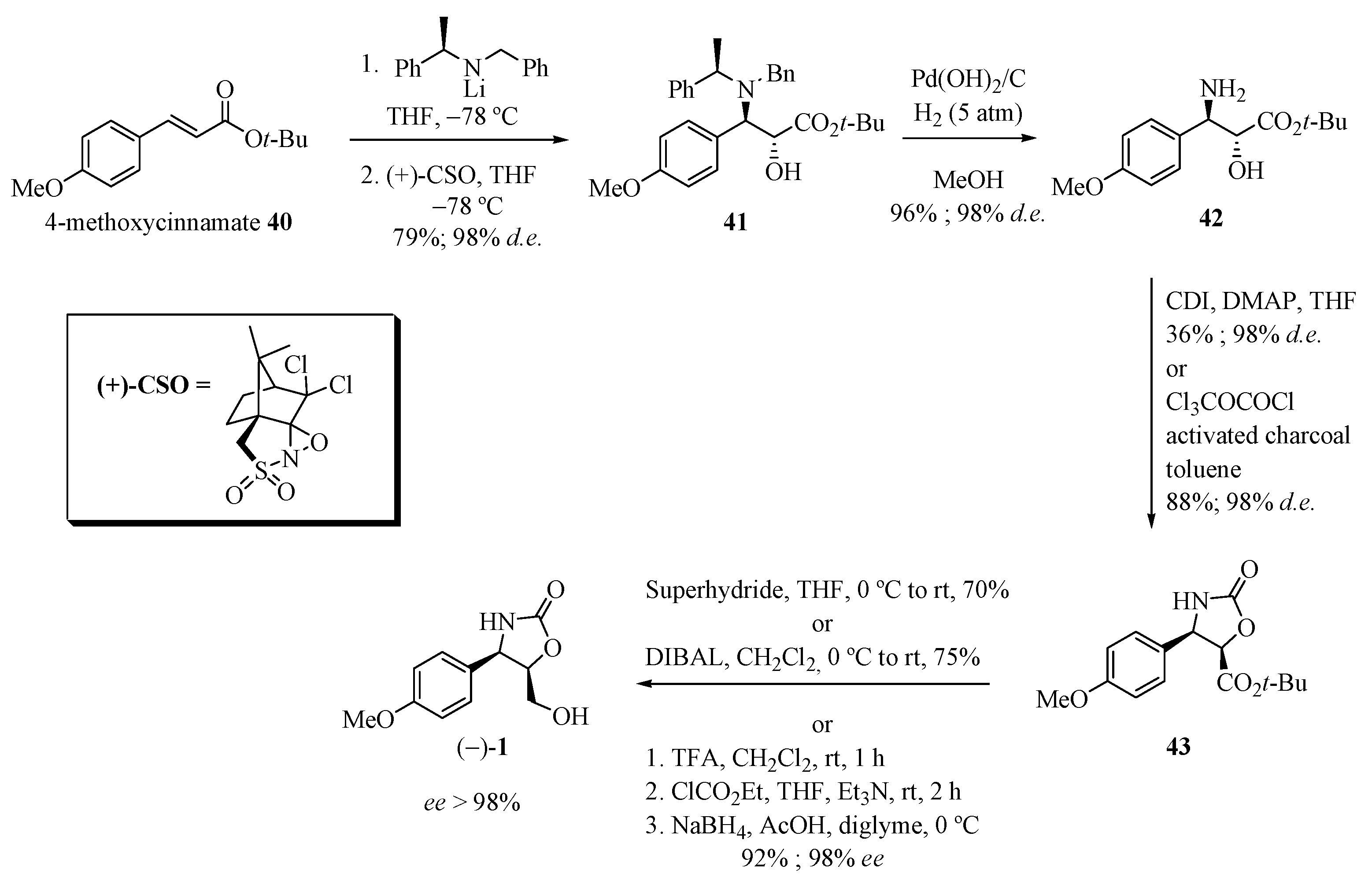
Davies and co-workers [19] reported the synthesis of (−)-cytoxazone in four steps from 4-methoxycinnamate (Scheme 7). 1,4-Addition of lithium (R)-N-benzyl-N-methylbenzylamide to cinnamate 40 and subsequent diastereoselective enolate oxidation using (+)-(camphorsulfonyl)-oxaziridine) [(+)-CSO] produced the α-hydroxy-β-amino ester 41. Hydrogenolysis of the N-benzyl group in the presence of Pearlman’s catalyst provided the free amino alcohol 42. Treatment with diphosgene generated 43 (88% yield and 99% de). Finally, the reduction of the tert-butyl ester group with superhydride or DIBAL-H produced (−)-cytoxazone in 70% or 75% yields, respectively, and with an enantiomeric excess of higher than 98%.
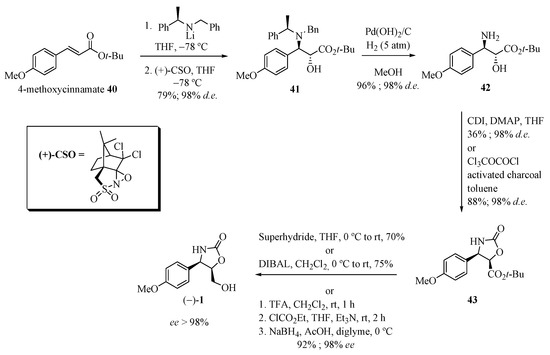
Scheme 7.
Total synthesis of (−)-cytoxazone reported by Davies and co-workers [19].
2.5. Short Synthesis of Both Enantiomers of Cytoxazone using the Petasis Reaction
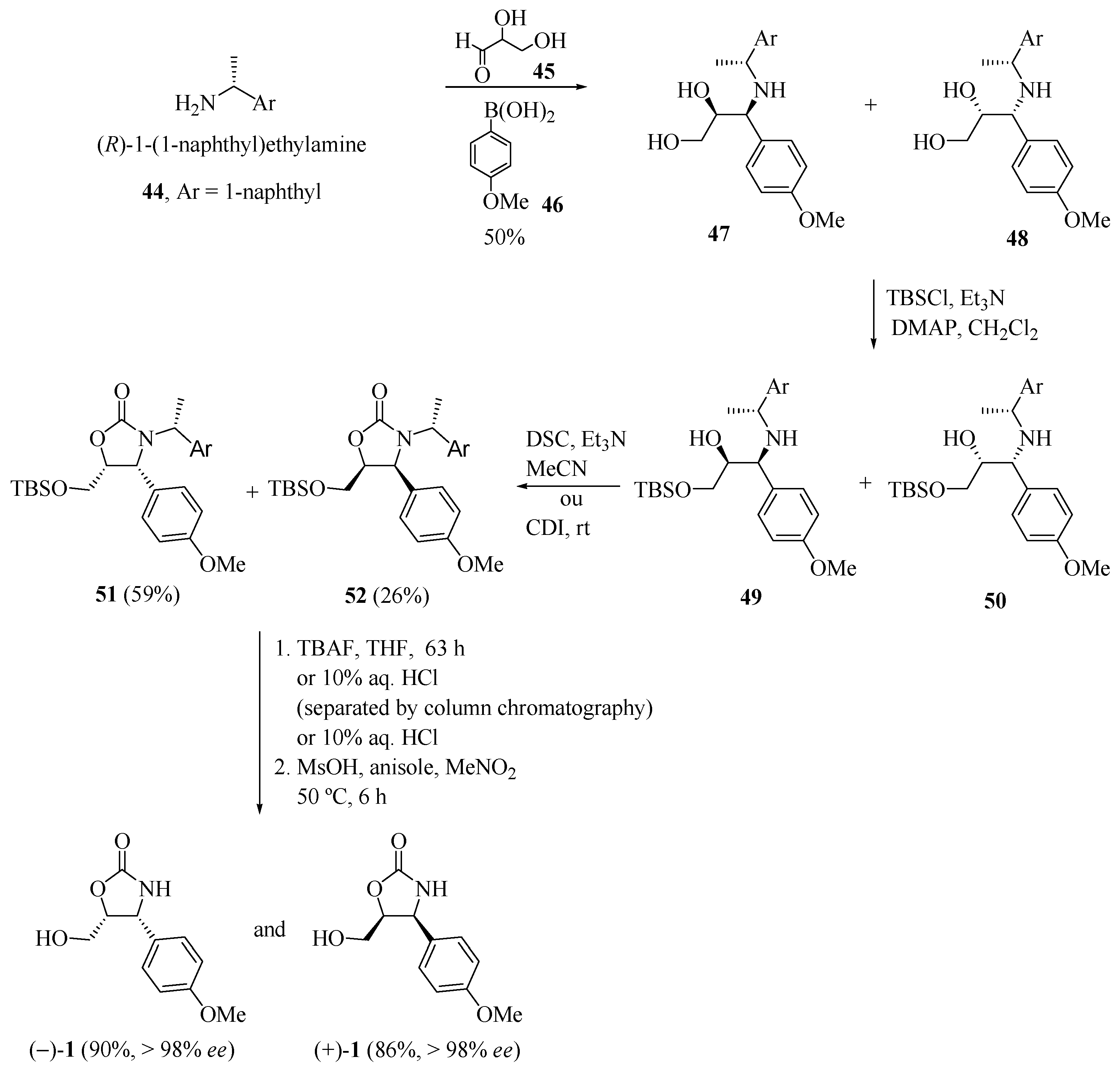
Using Petasis’s protocol [20], Sugiyama and co-workers [21] reported the synthesis of (−)-cytoxazone (Scheme 8). The coupling reaction between three components, (R)-1-(1-naphthyl)ethylamine (44), d,l-glyceraldehyde (45) and 4-methoxyphenylboronic acid (46) led to a diastereomeric mixture (1:1) of the aminodiols 47 and 48 in 50% yield. Protection of the primary hydroxyl of mixture led to their silyl ethers 49 and 50. Oxazolidin-2-one rings 51 and 52 were formed from 49 and 50 upon treatment with N,N'-succinimidyl carbonate (DSC) in acetonitrile. Finally, the sequence of desilylation, the separation of the diastereomeric mixture by column chromatography, and the removal of the 1-naphthylethyl group, generated both enantiomers of cytoxazone (−)-1 and (+)-1.
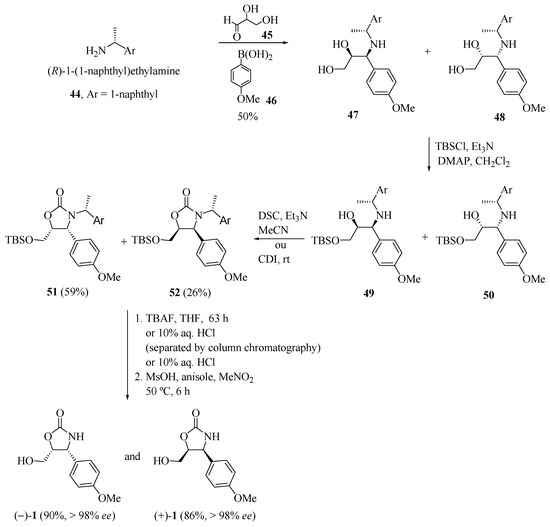
Scheme 8.
Synthesis of the enantiomers of cytoxazone through Petasis coupling [21].
Alternatively, the (−)- and (+)-cytoxazone enantiomers were also obtained in a one-pot process from the diastereomeric mixture of 47 and 48 (Scheme 8). The sequence of the selective protection of the primary hydroxylic groups of these diols, heterocyclization with 1,1-carbonyl bis-1H-imidazole (CDI), desilylation with 10% HCl, and the removal of the 1-naphthylethyl group led to (−)- and (+)-cytoxazone in 13% and 7% overall yields, respectively. In the one-pot process, CDI, rather than DSC, was used for the cyclization process due to its low solubility in dichloromethane, and 10% HCl, rather than TBAF, was used for desilylation to reduce the formation of unwanted products.
2.6. Imino 1,2-Wittig Rearrangement of Hydroximates and Its Application to the Synthesis of (−)-Cytoxazone
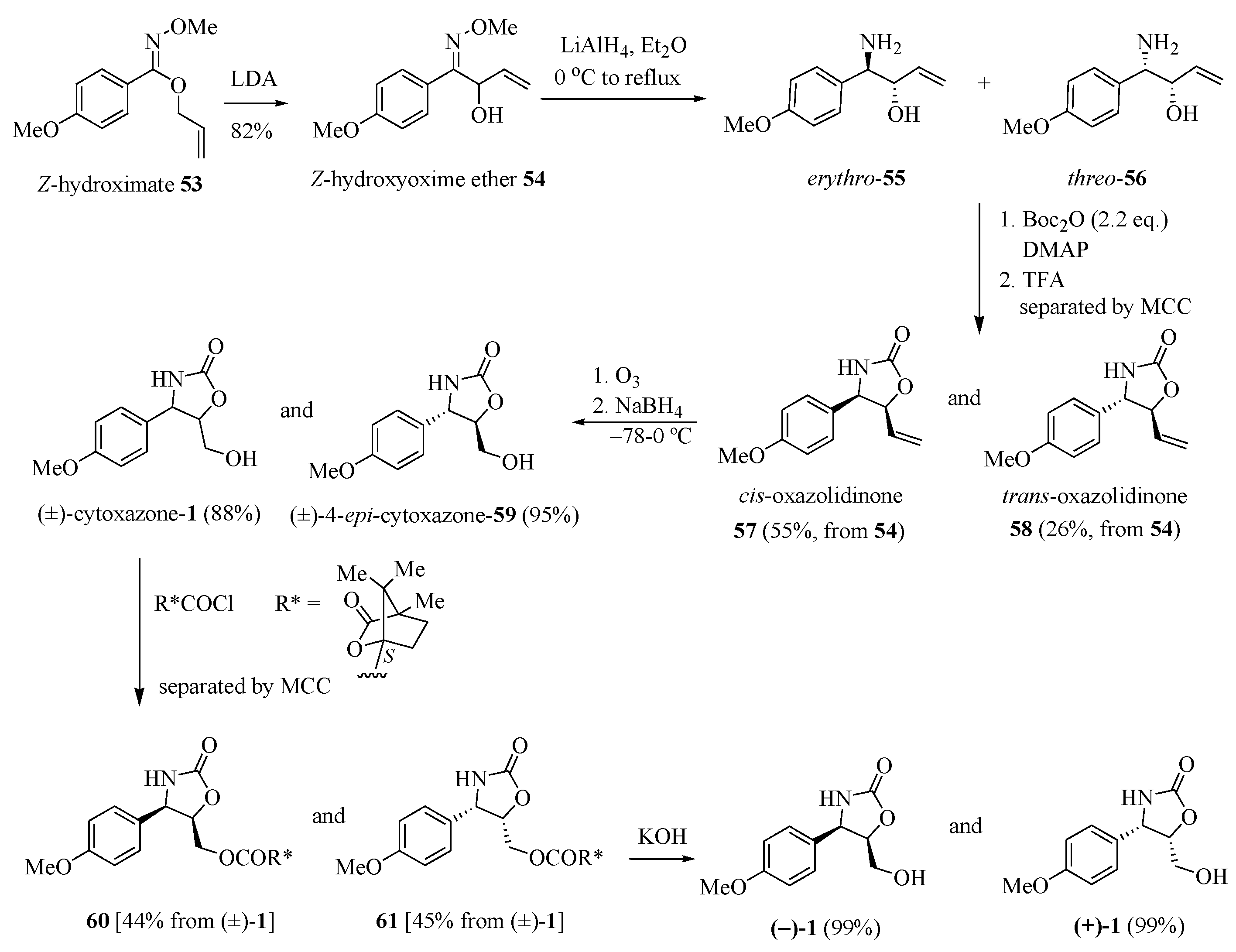
Naito and his group [22] reported a synthetic approach to prepare the (−)-cytoxazone based on the imino 1,2-Wittig rearrangement of hydroximates (Scheme 9). The Z-hydroximate 53, when treated with LDA, underwent 1,2-Wittig rearrangement to give Z-2-hydroxyoxime ether 54. Reduction and concomitant N-demethoxylation of 54 with LiAlH4 yielded a mixture of erythro- and threo-amino alcohols 55 and 56, respectively. Heterocyclization in situ of the mixture through the N-Boc di-protection by treatment with di-tert-butyl dicarbonate (Boc2O) (2.2 equiv.) in the presence of DMAP and subsequent N-Boc-deprotection with TFA led, after the separation of diastereomers, to cis- and trans-oxazolidin-2-one derivatives 57 and 58 in 55% and 26% yields, respectively (three steps). These oxazolidinone derivatives were separately converted into (±)-cytoxazone (1) and (±)-4-epi-cytoxazone (59) through the oxidative cleavage of terminal alkene with ozone and subsequent reduction with NaBH4 of the formed aldehyde. The chemical resolution of the racemate (±)-1 was carried out by acylation with (−)-camphanic chloride to give (4R,5R)- and (4S,5S)-oxazolidinones 60 and 61 in 44% and 45% yields, respectively, after the separation of isolated diastereomers by medium-pressure column chromatography (MCC). Hydrolysis of 60 and 61 led, respectively, to (−)- and (+)-1. Analogously, the enantiomers (−)- and (+)-4-epi-cytoxazone were also prepared from racemate (±)-59 (Scheme 9).
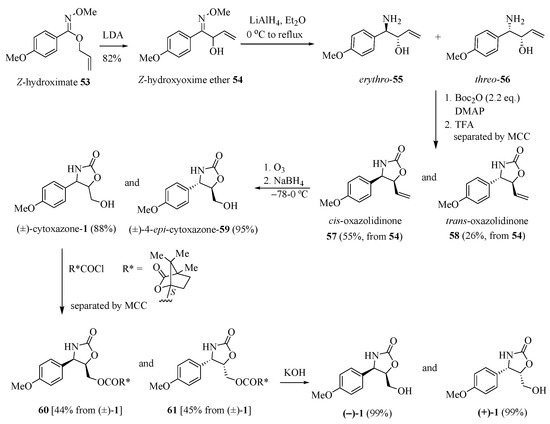
Scheme 9.
Synthetic approach of (−)- and (+)-cytoxazone reported by Naito’s group [22].
2.7. Highly Regioselective Ring Opening of Epoxides using NaN3: A Short and Efficient Synthesis of (−)-Cytoxazone
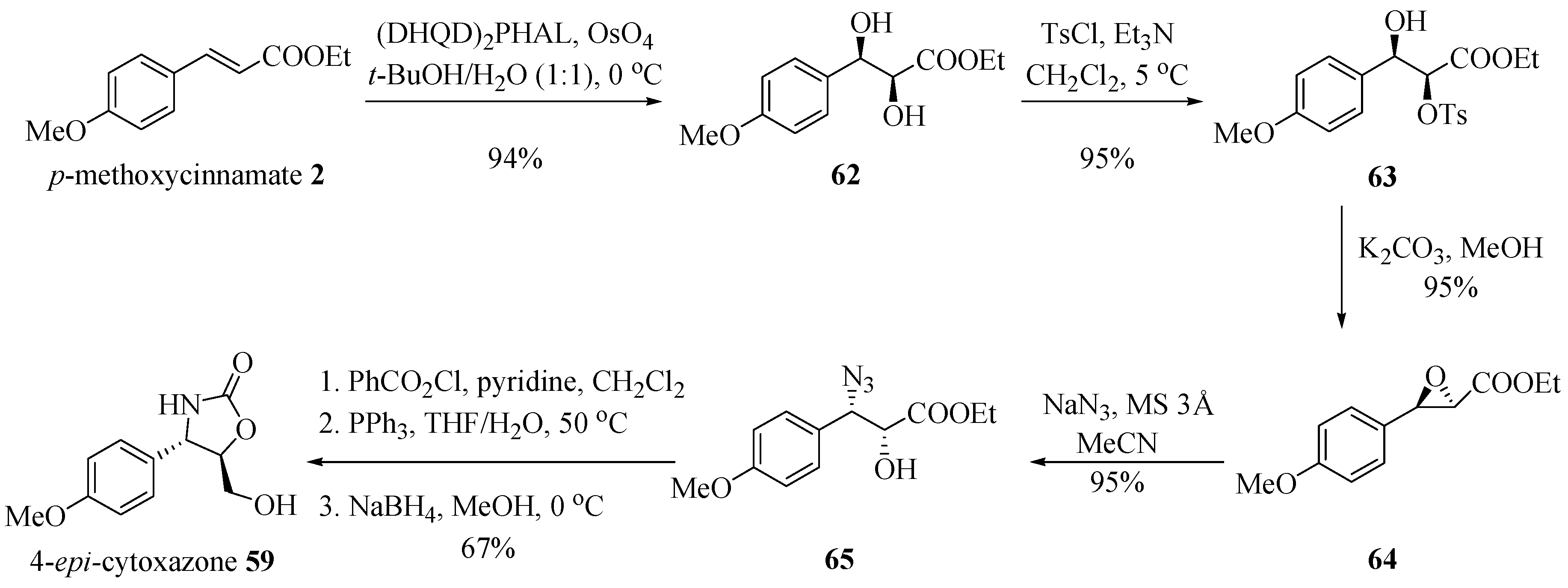
Barua et al. [23] applied a regioselective ring opening of epoxide, using NaN3 and molecular sieve as the catalyst to obtain (−)-cytoxazone from p-methoxycinnamate (Scheme 10). Asymmetric dihydroxylation of p-methoxycinnamate 2 by treatment with 1,4-bis(9-O-dihydroquinidine)-phthalazine [(DHQD) 2PHAL] under the so-called AD-mix-α® conditions [24], generated diol 62 in 94% yield and 99% ee. This diol was treated with p-toluenesulfonyl chloride and triethylamine, in turn leading to the monotosylated product 63, which in the presence of K2CO3 enabled the formation of epoxide 64 with no occurrence of epimerization. The regioselective azidolysis of 64 with NaN3 led to the azido alcohol 65, which was converted into epi-cytoxazone 59 in 57% overall yield from 62, following the protocol described by Nakata et al. (Scheme 1). NOTE: The synthetic strategy shown in Scheme 10 does not lead to the natural product structure (−)-(4R,5R)-cytoxazone, as indicated by the authors, but rather to the (4S,5R)-epi-cytoxazone (59), as corrected in the scheme below.
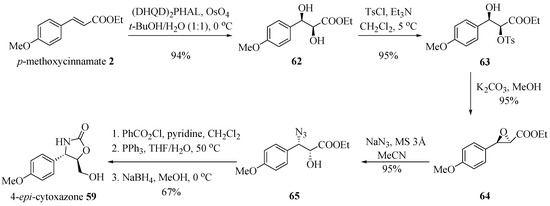
Scheme 10.
Total synthesis of (−)-cytoxazone proposed by Barua et al. [23].
2.8. Stereoselective Synthesis of (−)-Cytoxazone and (+)-5-epi-Cytoxazone
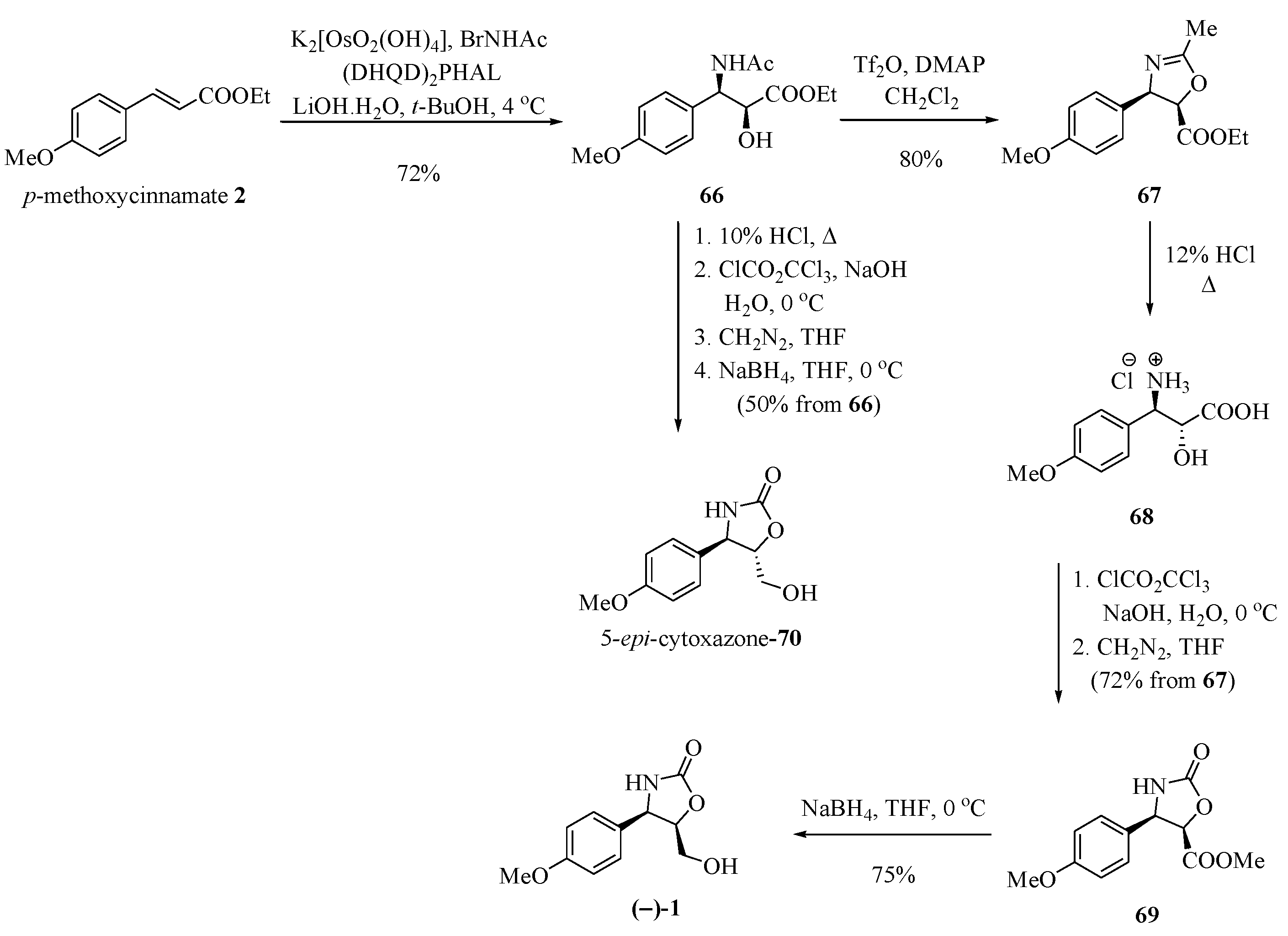
An asymmetric Sharpless aminohydroxylation reaction of p-methoxycinnamate with (DHQD)2PHAL produced amidoalcohol 66, which was used to syntheze (−)-cytoxazone, by Saicic and his group [25] (Scheme 11). The anti-aminoalcohol configuration required for the synthesis of (−)-1 was obtained by means of a configurational inversion of the amidoalcohol through an oxazoline. Thus, 66 was treated with Tf2O and DMAP, producing the cis-oxazoline 67 in an 80% yield. Acid hydrolysis led to hydroxyaminoacid 68, which was converted into the cyclic carbamate 69 upon treatment with diphosgene and esterification with diazomethane. Finally, the reduction of the ester function with sodium borohydride produced the optically pure (−)-cytoxazone. The 5-epi-cytoxazone (70) was also prepared from the common intermediate 66 without requiring the correction of stereochemistry at C-5 (Scheme 11).
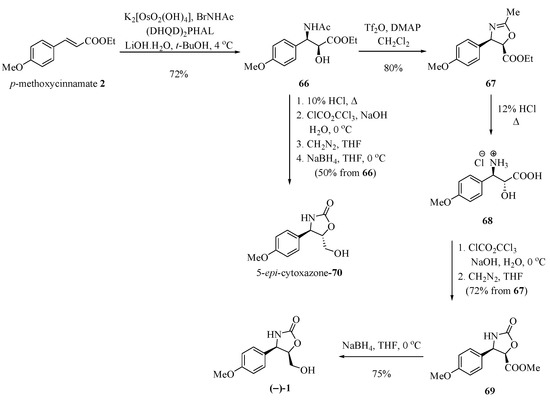
Scheme 11.
Total synthesis of (−)-cytoxazone by applying Sharpless asymmetric aminohydroxylation [25].
2.9. Regioselective and Diastereoselective Amination with the use of Chlorosulfonyl Isocyanate: A Short and Efficient Synthesis of (−)-Cytoxazone
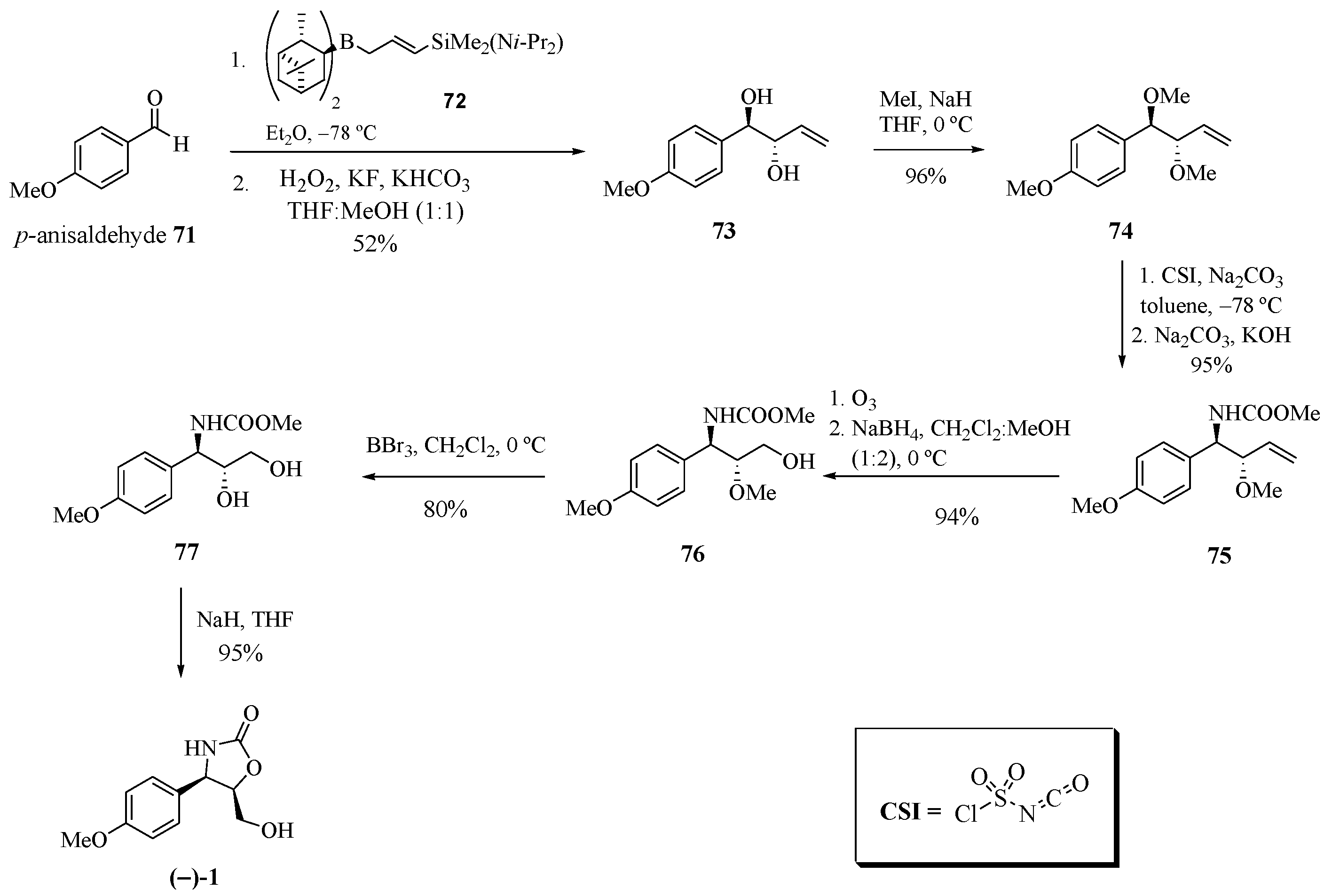
Jung et al. [26] developed a new route of regio- and diastereoselective amination by applying chlorosulfonyl isocyanate (CSI) for the synthesis of (−)-cytoxazone (Scheme 12). The treatment of p-anisaldehyde (71) with B-[3-((diisopropylamino)dimethylsilyl)allyl] diisopinocamphenylborane (72) produced the anti-diol 73. The dimethylation of compound 73 with NaH and iodomethane produced the anti-1,2-dimethyl ether 74. The regio- and diastereoselective amidation using CSI in the presence of sodium carbonate and the reduction of the N-chlorosulfonyl group with aqueous sodium thiosulfate and sodium hydroxide produced the desired anti-amino ether 75 in a high diastereoselectivity (27:1). The ozonolysis of the double bond and the reduction of generated aldehyde led to alcohol ether 76. Finally, demethylation with BBr3 and heterocyclization produced (−)-cytoxazone.
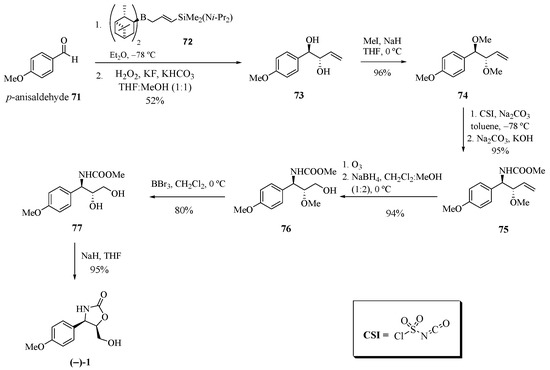
Scheme 12.
Total synthesis of (−)-cytoxazone from p-anisaldehyde [26].
2.10. Auxiliary Strategies to Prepare β-Amino Alcohols with Reductive Cross-coupling and the Synthesis of (−)-Cytoxazone
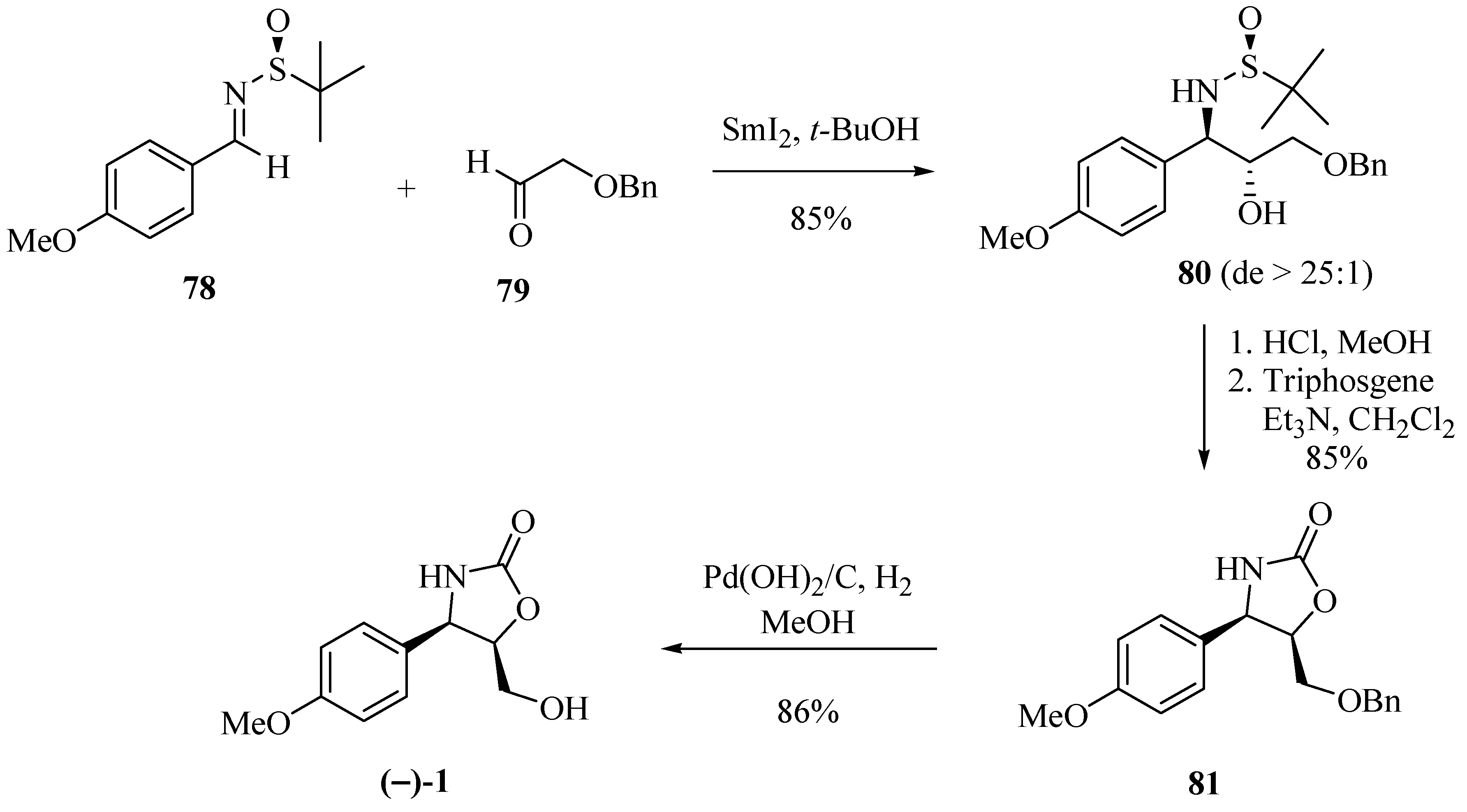
Ellman et al. [27] first used N-tert-butanesulfinyl chiral imines providing good results in stereocontrolled alkylation reactions. Based on this chiral auxiliary, Bentley et al. [28], seeking to synthesize (−)-cytoxazone, obtained the tert-butanesulfinyil amine derivative 80 with good yield and high diastereoselectivity (>25:1) through a reductive SmI2-mediated cross-coupling reaction of the (S)-N-tert-butanesulfinyl imine 78 and aldehyde 79 (Scheme 13). It is important to note that the presence of the p-methoxyl substituent on the aromatic ring in 78 led to an increase in diastereoselectivity coupling. Removal under acidic conditions of the tert-butanesulfinyl (t-BS) auxiliary, heterocyclization, and debenzylation produced (−)-cytoxazone from 78 in 62% yield.
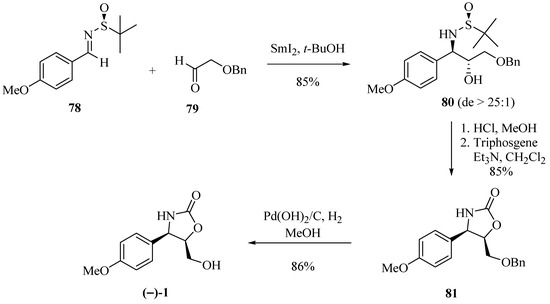
Scheme 13.
Total synthesis of (−)-cytoxazone using N-tert-butanesulfinyl imine as the chiral auxiliary [28].
2.11. Synthesis of (−)-Cytoxazone and (+)-epi-Cytoxazone: The Chiral Pool Approach
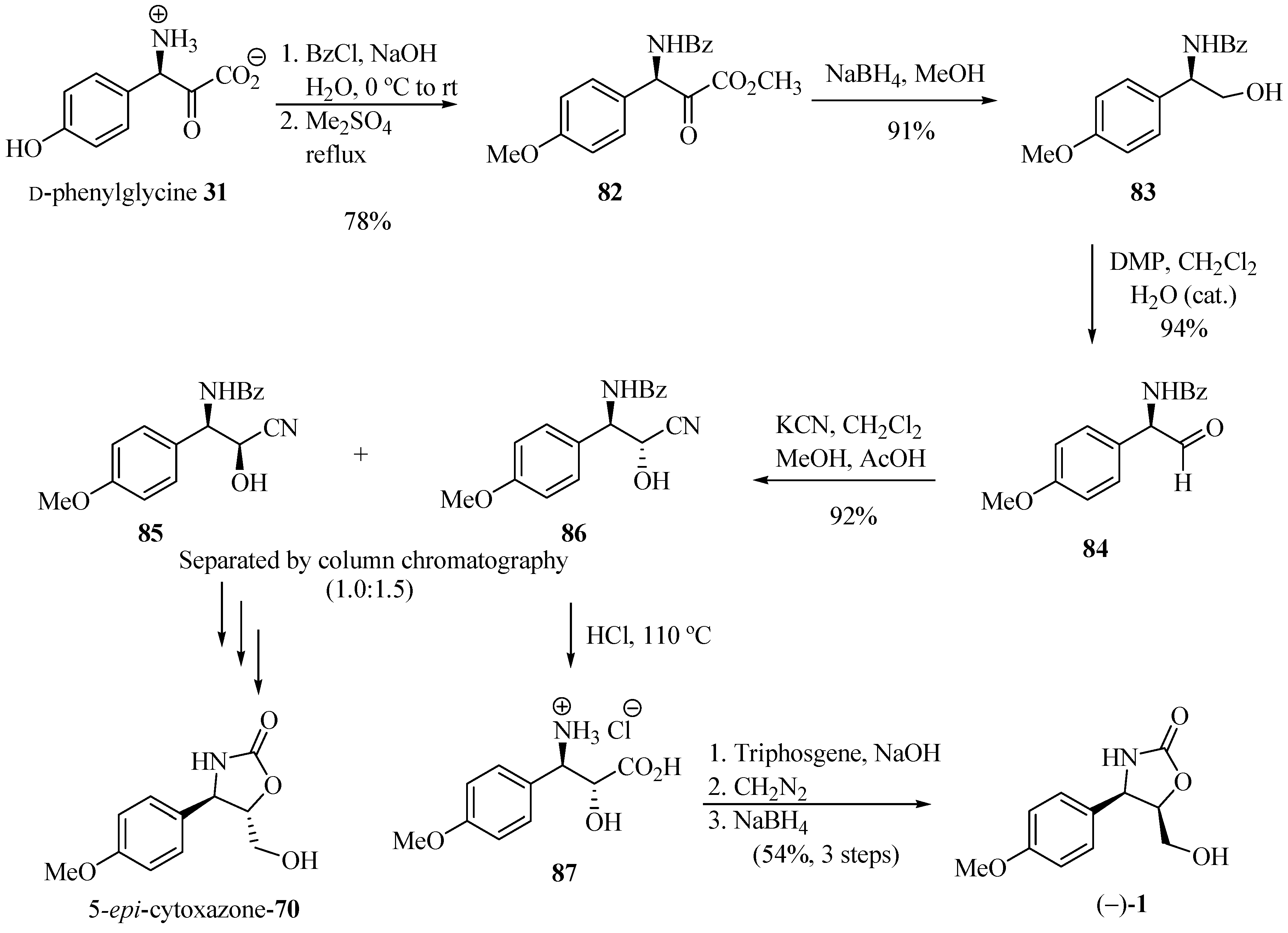
Saici et al. [29] reported the total synthesis of (−)-cytoxazone from p-hydroxy-D-phenylglycine 31 (Scheme 14). This amino acid was converted into its amino alcohol 83 after N-benzoylation, simultaneous esterification and methyl etherification, and the reduction of the ester group. Oxidation with water-catalyzed Dess-Martin periodinane led to aldehyde 84 in 94% yield and 98% ee, with no noticeable racemization. Homologation of the amino acid through the addition of KCN to the aldehyde 84 yielded a mixture of cyanohydrin diastereomers 85 and 86. Separation of diastereomers by column chromatography produced the optically pure cyanohydrins, which were individually subjected to acid hydrolysis, cyclization, esterification, and reduction, leading to (−)-cytoxazone and (+)-5-epi-cytoxazone (70).
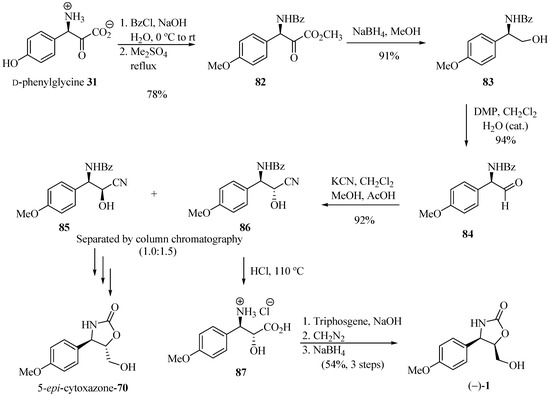
Scheme 14.
Total synthesis of (−)-cytoxazone and (+)-5-epi-cytoxazone from the (D)-(−)-4-hidroxyphenylglycine [29].
2.12. Short-step and Scalable Synthesis of (±)-Cytoxazone
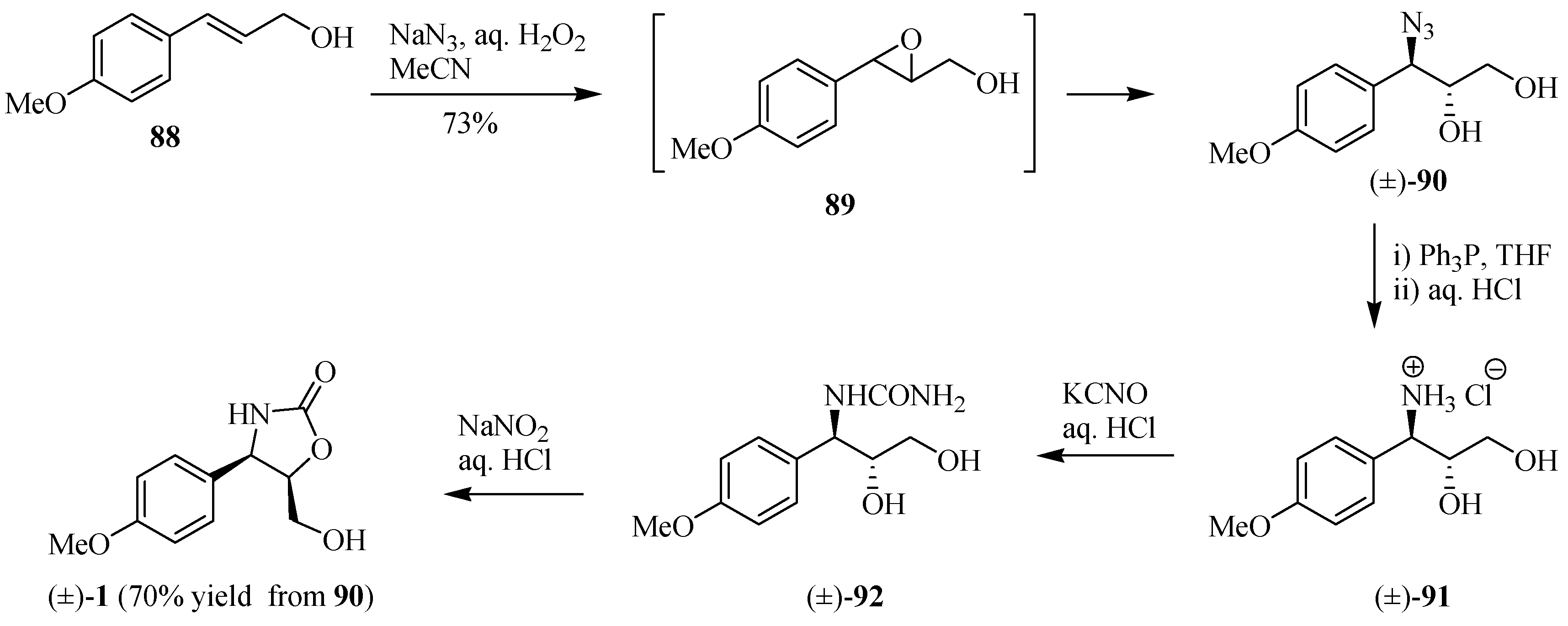
Sugai and co-workers [30] introduced a new one-pot azidohydroxylation procedure from 88 by combining the mixture of NaN3-H2O2-CH3CN to prepare (±)-cytoxazone by means of an epoxy-generated intermediate 89 (Scheme 15). The opening of epoxide 89 happened immediately after its formation through the presence of NaN3 in the medium reaction without any further addition of a Lewis acid to give azidoalcohol (±)-90. Reduction of the azide group led to amine salt 91, after acidification with aqueous HCl, which was treatment with aqueous KCNO and an additional 0.2 eq. of aqueous HCl to produce N-carbamylamino alcohol (±)-92. Treatment with NaNO2 led to a nitrosation-deaminocyclization cascading sequence, yielding (±)-cytoxazone in 70% yield (three steps). Note that this strategy does not require any protection of the hydroxyl groups.
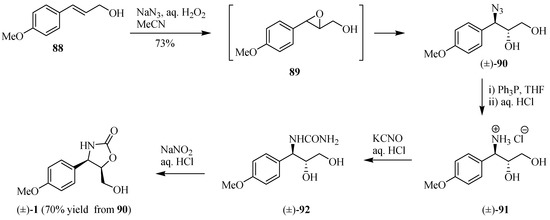
Scheme 15.
Synthesis of (±)-cytoxazone reported by Sugai and co-workers [30].
2.13. One-pot Sequential Catalytic Asymmetric Epoxidation-regioselective Epoxide-opening Process. Application for the Synthesis of (−)-Cytoxazone
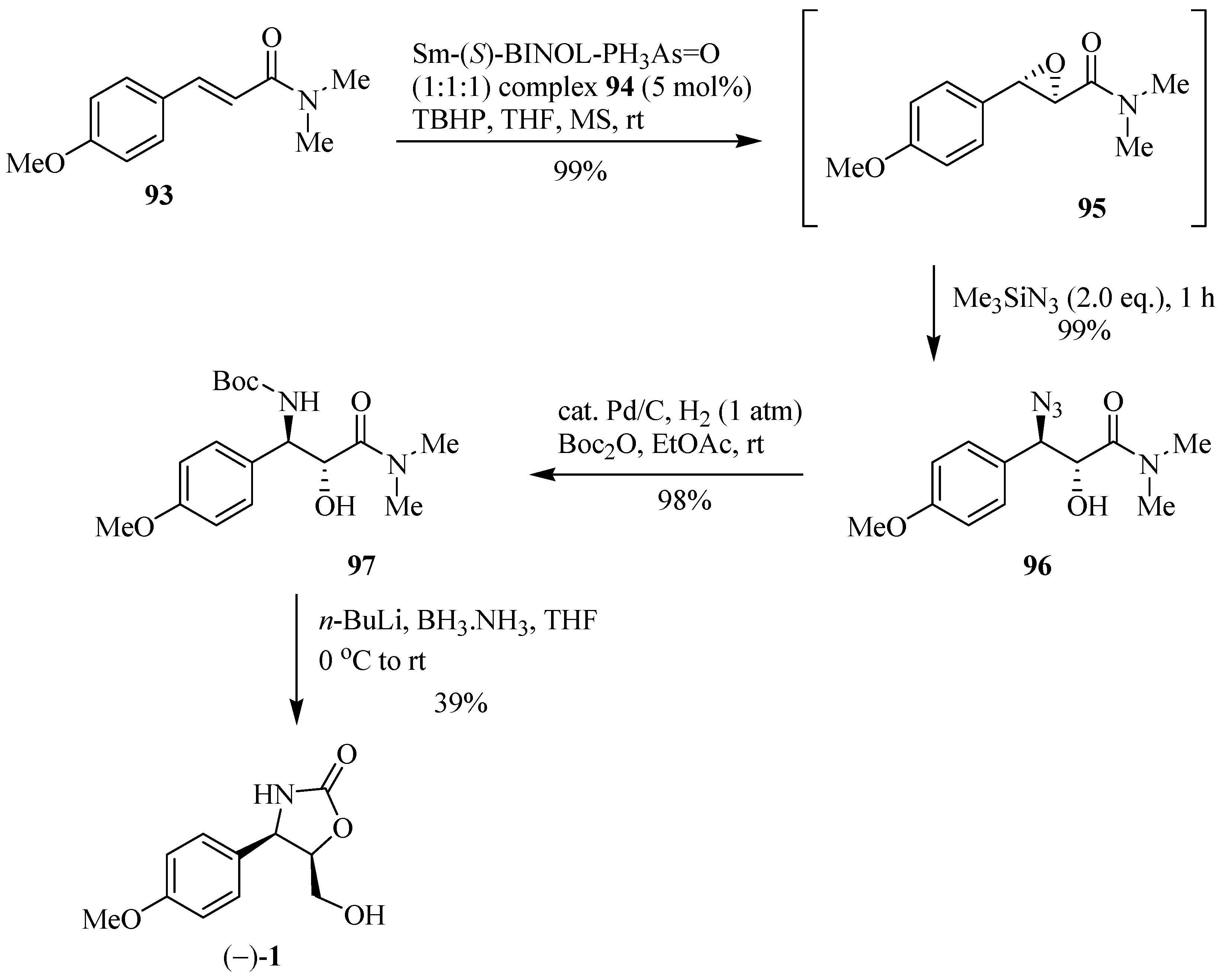
The application of a study executed by Shibasaki et al. [31] on the asymmetric epoxidation of α,β-unsaturated amide systems, in the presence of the Sm-(S)-BINOL-Ph3-As=O complex 94 and the immediate epoxide-opening process leading to the corresponding anti-β-azido-α-hydroxyamide, with complete regioselectivity and excellent ee (>99%) allows for the synthesis of (−)-cytoxazone (Scheme 16). Thus, the formation of the α,β-epoxy amide 95 and its immediate opening with Me3SiN3 provided efficient access to the β-azido alcohol 96. Reduction of the azide group and subsequent N-Boc protection led to intermediate 97. Finally, the reduction of the tertiary amide with n-BuLi and BH3.NH3 and immediate cyclization produced (−)-cytozaxone in 39% yield.
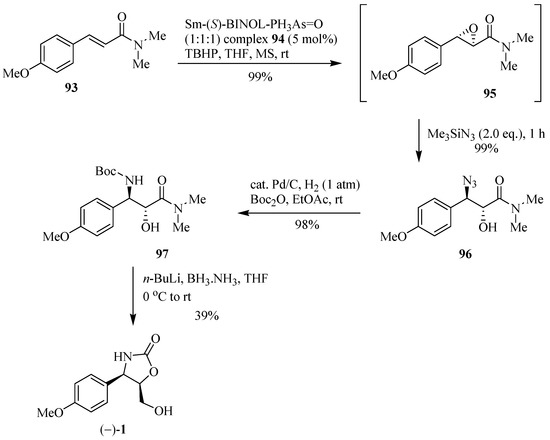
Scheme 16.
Synthesis of (±)-cytoxazone reported by Shibasaki and co-workers [31].
2.14. Enantioselective Synthesis of (−)-Cytoxazone and (+)-epi-Cytoxazone, Novel Cytokine Modulators via Sharpless Asymmetric Epoxidation and L-Proline Catalyzed Mannich Reaction
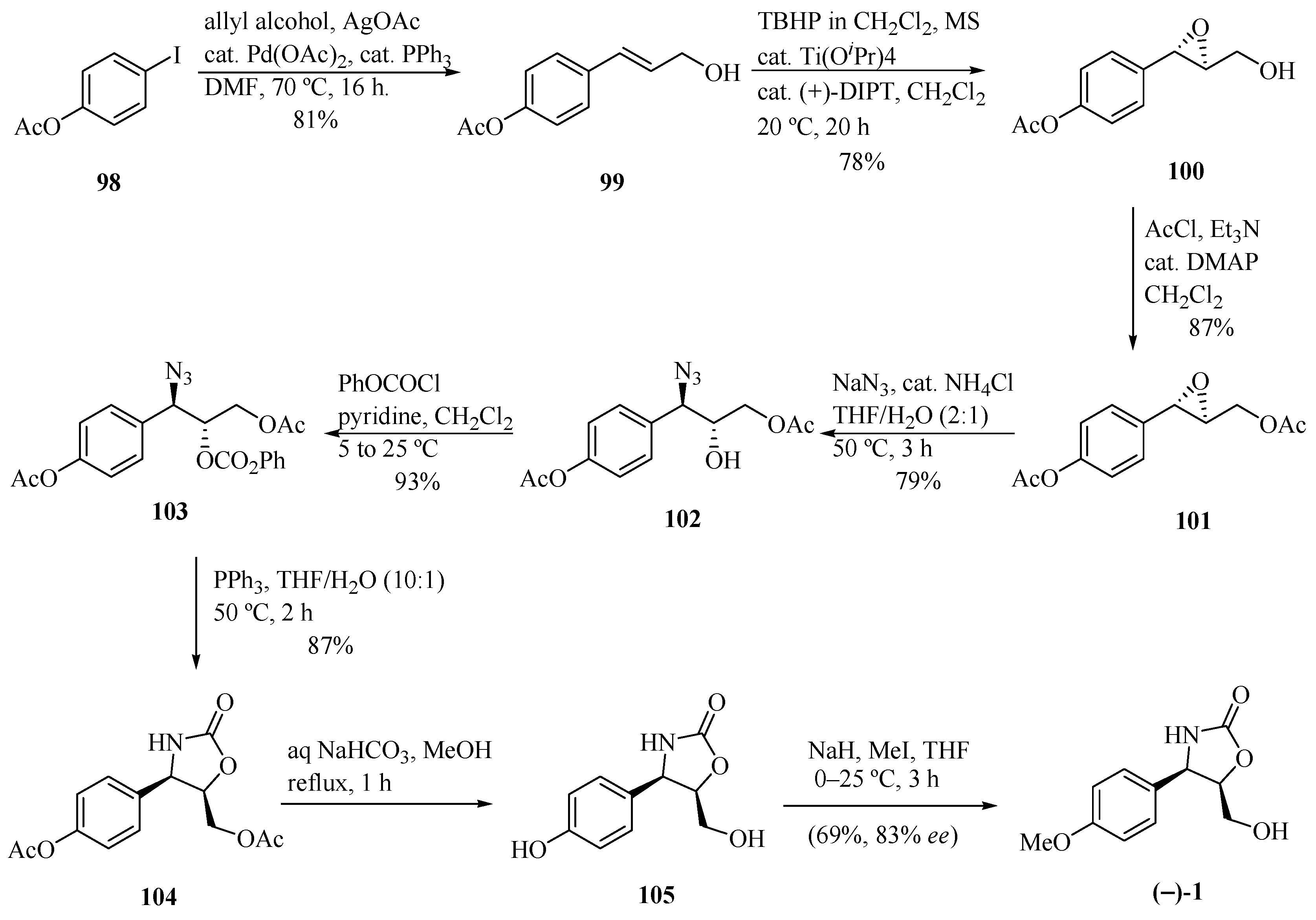
Sudalai and Paraskar [32] developed interesting routes for the synthesis of (−)-cytoxazone and (+)-epi-cytoxazone by means of the Ti-catalyzed Sharpless asymmetric epoxidation of allylic alcohol and L-proline catalyzed Mannich reaction, respectively, as key steps to introducing the stereocenters into the molecule (Scheme 17). The Pd-catalyzed arylation of allylic alcohol with iodine 98 produced the trans-allylic alcohol 99, which was converted into the chiral epoxide 100 via Sharpless asymmetric epoxidation [(+)-diisopropyl tartrate ((+)-DIPT), Ti(PriO)4, anhyd. tert-butyl hydroperoxide (TBHP)]. Acetylation of the hydroxyl led to epoxyacetate 101. Regioselective opening, using NaN3 and NH4Cl (cat.), yielded the trans-azido alcohol 102. Subsequently, successive reactions of alcohol protection, reductive cyclization using PPh3, basic hydrolysis of both acetate groups, and methylation led to (−)-cytoxazone in 56% yield from 102 and 83% ee.
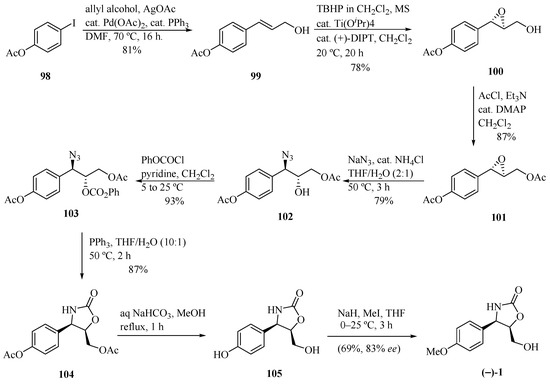
Scheme 17.
Total synthesis of (−)-cytoxazone reported by Sudalai and Paraskar [32].
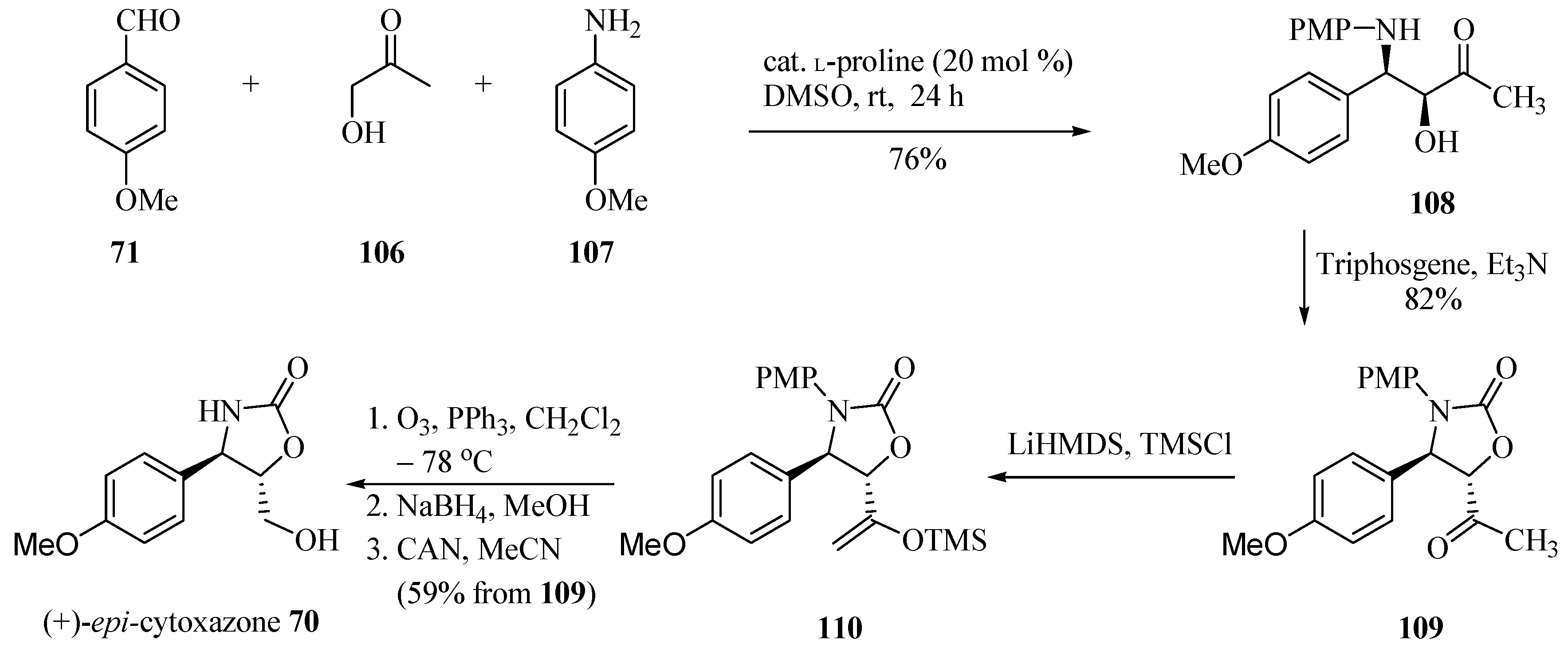
By contrast, the same authors [32] proposed another approach for the synthesis of (+)-epi-cytoxazone (70, Scheme 18). Thus, 4-methoxybenzaldehyde (71) was condensed with hydroxyacetone (106) and p-anisidine (107) in the presence of L-proline (30 mol %) to prepare chiral amino alcohol 108 in 76% and a (2:1) syn/anti ratio. The diastereomers were separated by column chromatography. The cyclization with triphosgene generated oxazolidinone 109 in 82% yield. Finally, silyl enol ether 110 was prepared from 109 by treatment with LiHMDS and TMSCl, which was subjected to ozonolysis without further purification. The reduction of generated aldehyde and N-PMP deprotection with CAN yielded (+)-epi-cytoxazone (70) in 59% yield from 109 and 81% ee.
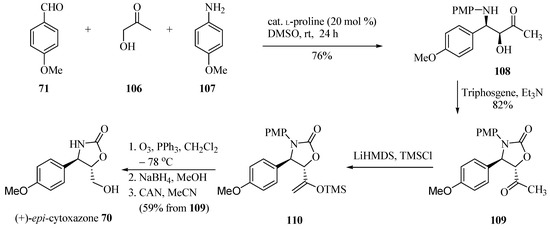
Scheme 18.
Total synthesis of (+)-epi-cytoxazone reported by Sudalai and Paraskar [32].
2.15. A Concise Synthesis of (−)-Cytoxazone and (−)-4-epi-Cytoxazone using Chlorosulfonyl Isocyanate
Jung et al. [33] reported on the synthesis of (−)-cytoxazone and (−)-4-epi-cytoxazone from p-anisaldehyde (71) conducted in six steps, resulting in good diastereoselectivity. The key step in the synthesis includes the regio- and diastereoselective amination of the anti- and syn-1,2-dimethyl ethers with chlorosulfonyl isocyanate and the subsequent regioselective cyclization of the diols to build of the oxazolidin-2-one cores. This strategy proved to be equal to that previously published by the same authors in 2005 (see Scheme 12).
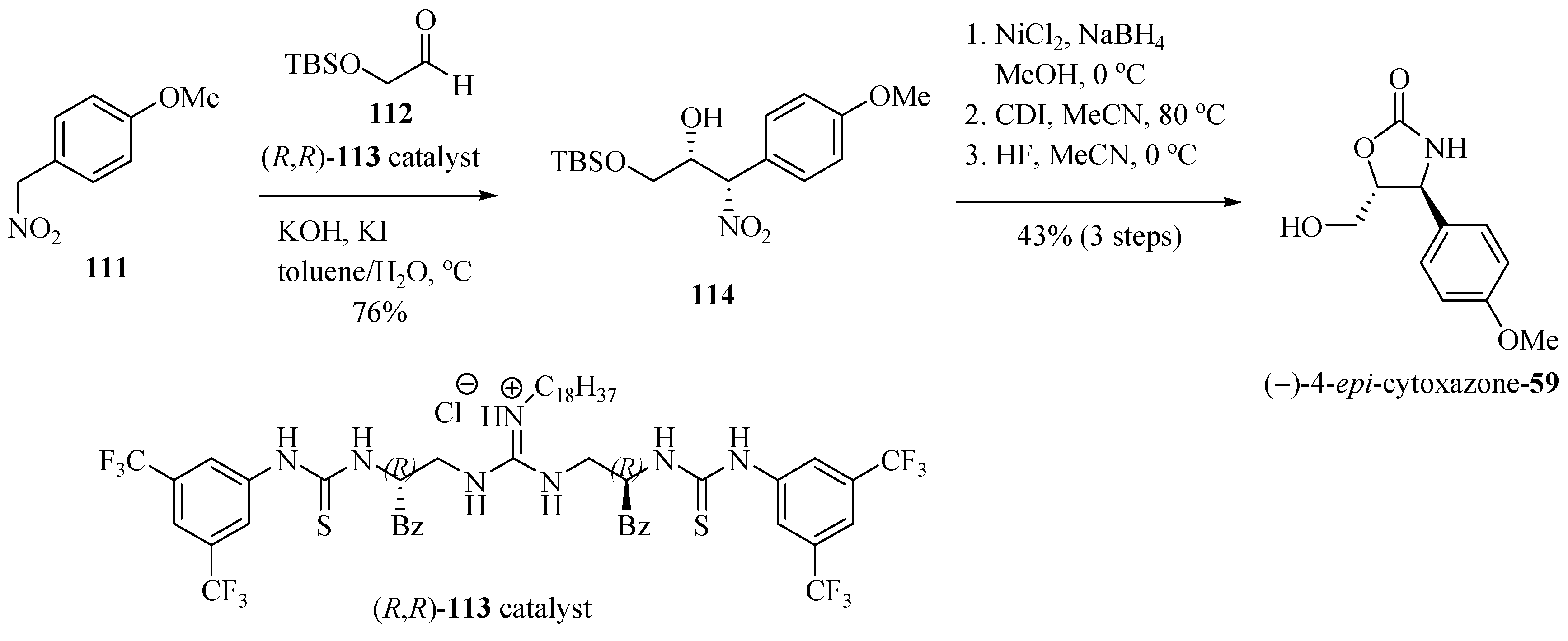
2.16. Diastereoselective and Enantioselective Henry (Nitroaldol) Reaction using a Guanidine-thiourea Bifunctional Organocatalyst. Application to the Synthesis of (4S,5R)-epi-Cytoxazone
Nagasawa and his team [34] reported the application of highly diastereoselective and enantioselective Henry reaction (nitroadol), between the aldehyde 112 and 1-methoxy-4-nitromethylbenzene (111), using the bifunctional guanidine-thiourea 113 as a chiral catalyst, producing 1,2-syn-nitro alcohol 114 used to prepare (−)-epi-cytoxazone (Scheme 19). The formation of the oxazolidinone ring came through a sequence of reduction of the nitro group, cyclization, and desilylation steps, which produced (−)-4-epi-cytoxazone (59) in 33% overall yield and 95% ee from 111.
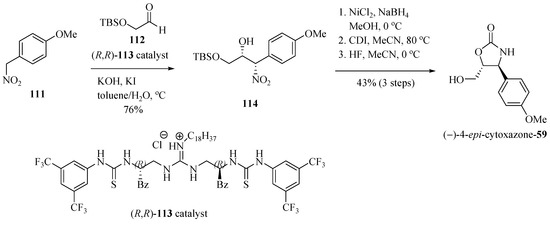
Scheme 19.
Synthesis of (4S,5R)-epi-cytoxazone (59) reported by Nagasawa et al. [34].
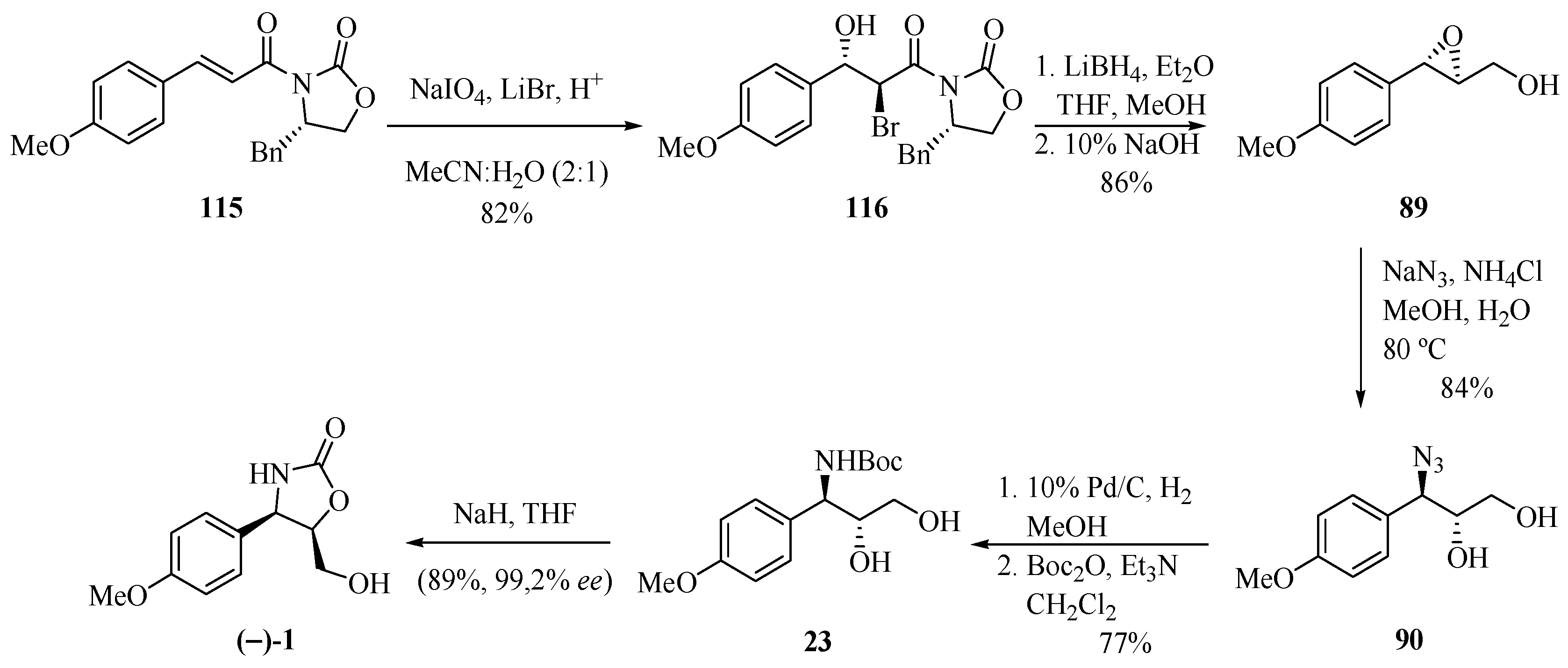
2.17. NaIO4-mediated Asymmetric Bromohydroxylation of α,β-Unsaturated Carboxamides with High Diastereoselectivity: A Short Route to (−)-Cytoxazone
Following the same line of research, Sudalai and co-workers [35] reported another contribution to the preparation of (−)-cytoxazone (Scheme 20), in addition to other previously published by the main author [32]. The diastereoselective bromohydroxylation reaction of the α,β-unsaturated carboxamide 115, when in the presence of NaIO4 and LiBr, generated bromohydrin 116. The chiral residue was removed when in presence of LiBH4, followed by treatment with 10% NaOH, in turn leading to epoxyalcohol 89, whose regioselective opening with NaN3 yielded the respective azido alcohol 90. The latter was converted into a product of interest after the reduction of the azide group, N-Boc protection, and cyclization.
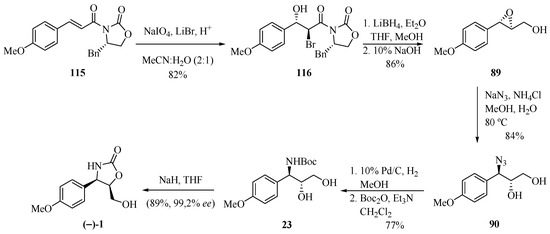
Scheme 20.
Total synthesis of (−)-cytoxazone proposed by Sudalai et al. [35].
2.18. Enantioselective Synthesis of (−)-Cytoxazone and (+)-epi-Cytoxazone via Rh-catalyzed Diastereoselective Oxidative C–H Aminations
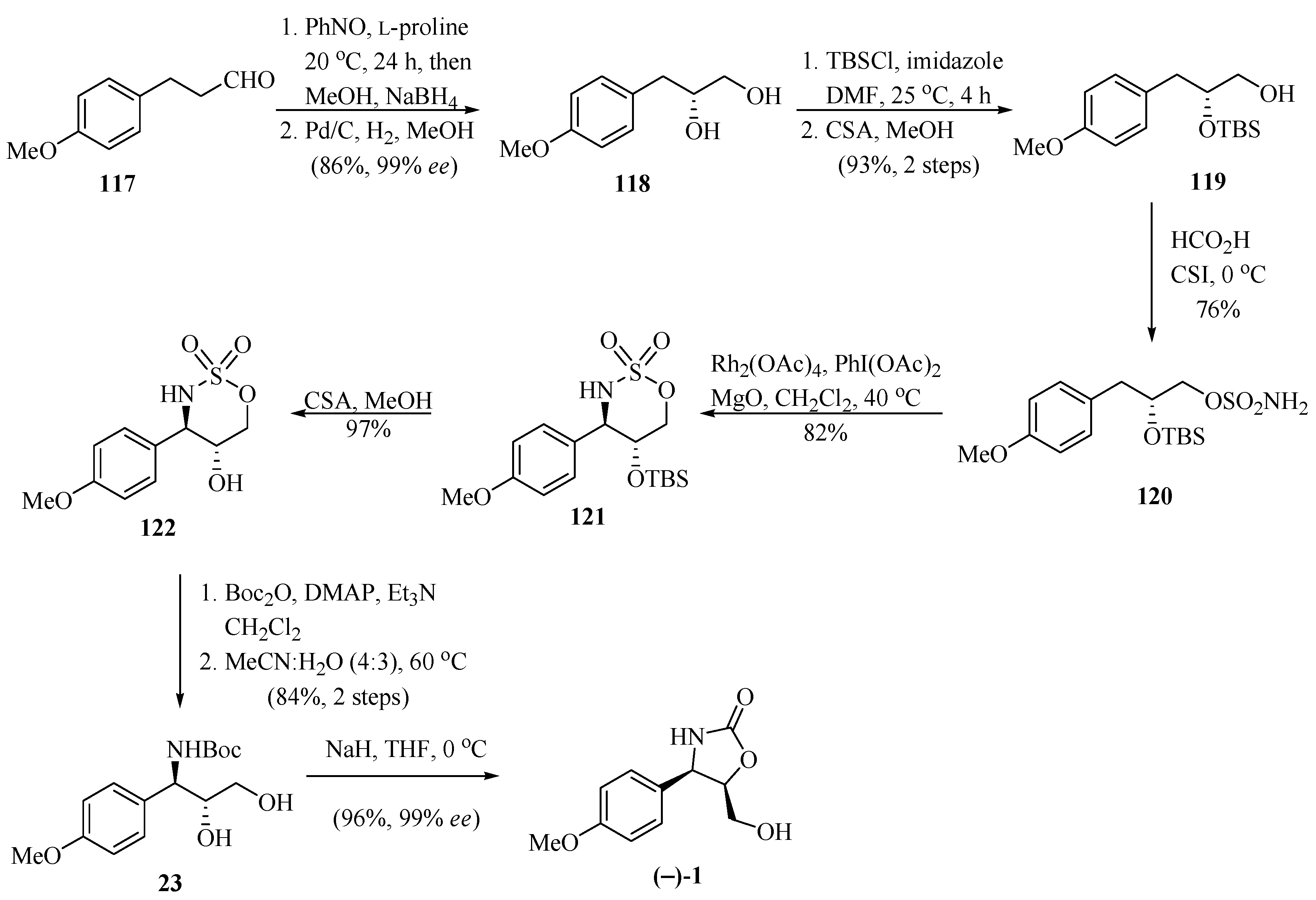
In another study, following the same line of research on the construction of oxazolidinone rings, Sudalai and his group [36], reported on an elegant enantioselective synthetic route for the preparation of (−)-cytoxazone (Scheme 21). The reaction of aldehyde 117 with nitrosobenzene, as the oxygen source, in the presence of L-proline, followed by its reduction with NaBH4 led to the crude aminoxy alcohol. The subsequent reduction of the crude aminoxyalcohol with Pd/C and an atmosphere of H2 produced chiral diol 118 in 86% yield and 99% ee. The bis-TBS-protection and selective deprotection of the primary hydroxyl from 118 generated the corresponding silyl ether 119. This alcohol was converted into the sulfamate ester 120 using known conditions (HCO2H and CSI). Selective γ-C-H insertion of the sulfamate ester 120 was carried out in the presence of a catalytic amount of Rh2(OAc)4 and PhI(OAc)2 as the oxidizing agent with anti (10:1) diastereoselectivity to produce the corresponding six-membered ring insertion product 121. The anti-diastereomer 121 was readily separated by column chromatography. The sequence of desilylation, carbamoylation with Boc2O of the -NH moiety of oxathiazinane 122, followed by ring opening of the crude N-Boc protected oxathiazinane with aqueous CH3CN and regioselective intramolecular cyclization, generated (−)-cytoxazone. The preparation of (+)-epi-cytoxazone was performed from aldehyde 117, similarly to (−)-cytoxazone, by exchanging the L-proline for D-proline. It is important to mention that the second stereocenter in diol 118 can be generated with excellent distereoselectivity by Rh-catalyzed intramolecular amidation of the γ-C-H bonds of carbamate or sulfamate esters.
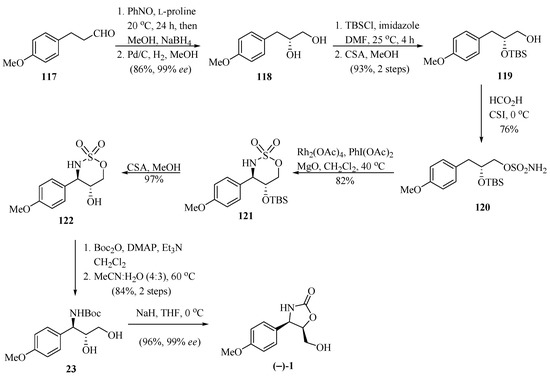
Scheme 21.
Synthetic approach of (−)-cytoxazone reported by Sudalai and his group [36].
2.19. Iridium(I)-catalyzed Regio- and Enantioselective Allylic Amidation
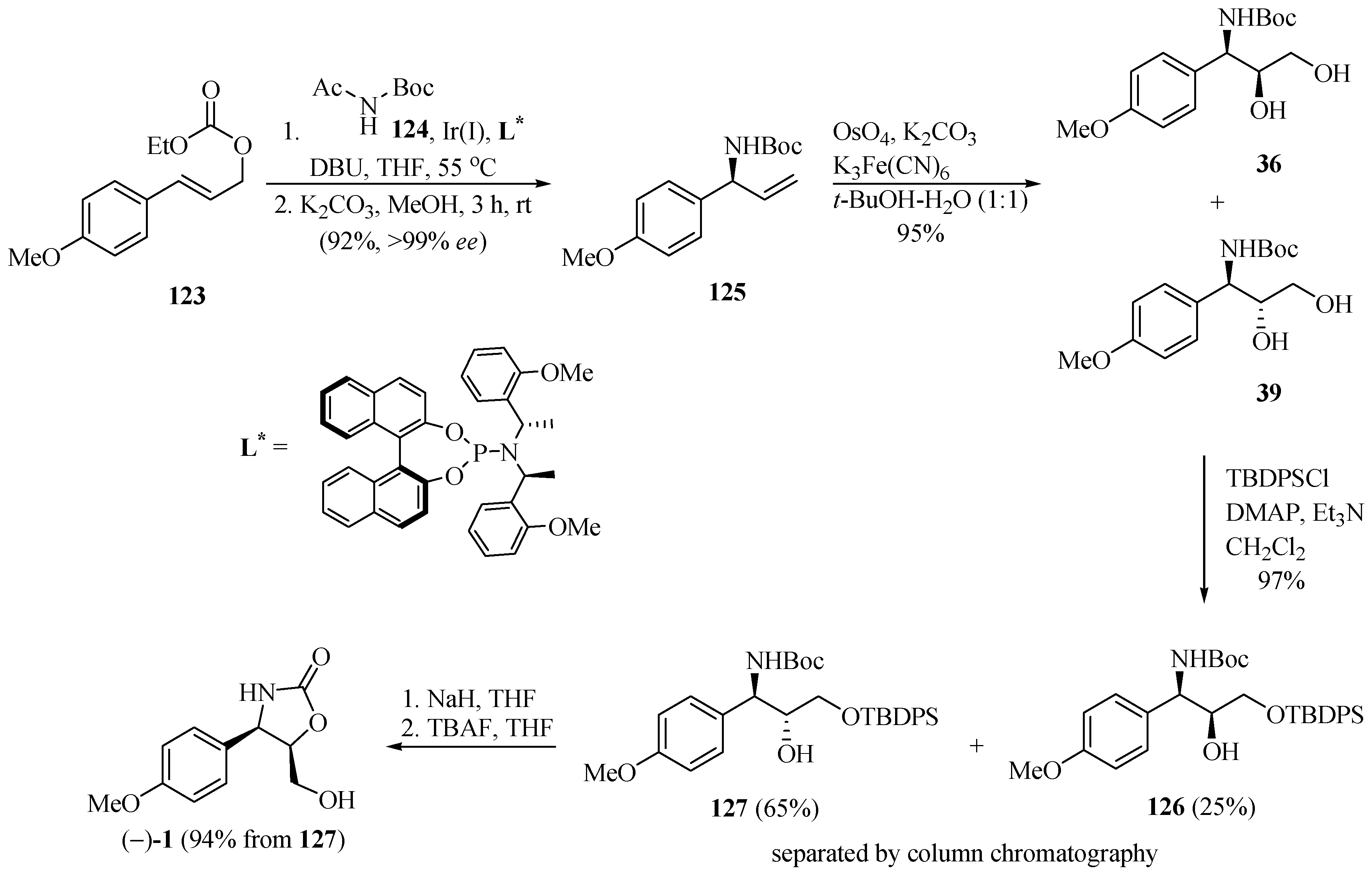
Han and Singh [37] developed a new route for the asymmetric synthesis of (−)-cytoxazone by applying an Ir(I)-catalyzed intermolecular allylic amidation reaction of aryl allyl carbonate with soft nitrogen nucleophiles and a chiral phosphoramidite ligand (L*) combined with the Sharpless dihydroxylation reaction (Scheme 22). The sequence of a one-pot process of Ir(I)-catalyzed allylic amidation of aryl allyl carbonate 123 with N-acetyl-N-tert-butyl carbamate 124 and chiral phosphoramidite ligand (L*), followed by N-deacetylation, led to the corresponding N-Boc protected allyl amine 125 in 99% ee. The Sharpless dihydroxylation reaction provided a mixture of syn-36/anti-39 diols in a 3:7 ratio. After converting the diols into their silylated ethers 126 and 127, these were separated by column chromatography. Finally, the cyclization of 127, performed in five steps starting from 123, by treatment with NaH and desilylation, produced (−)-cytoxazone in 52% yield.
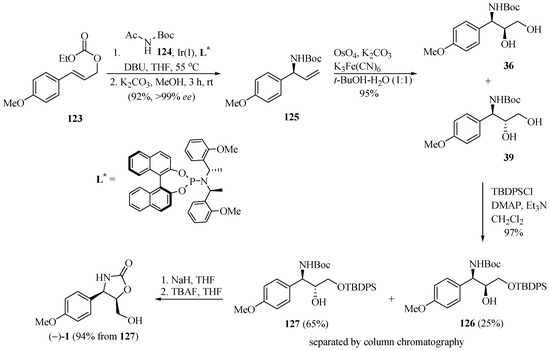
Scheme 22.
Total synthesis of (−)-cytoxazone reported by Han and Singh [37].
2.20. An Advance on Exploring N-tert-Butanesulfinyl Imines in Asymmetric Synthesis of Chiral Amines
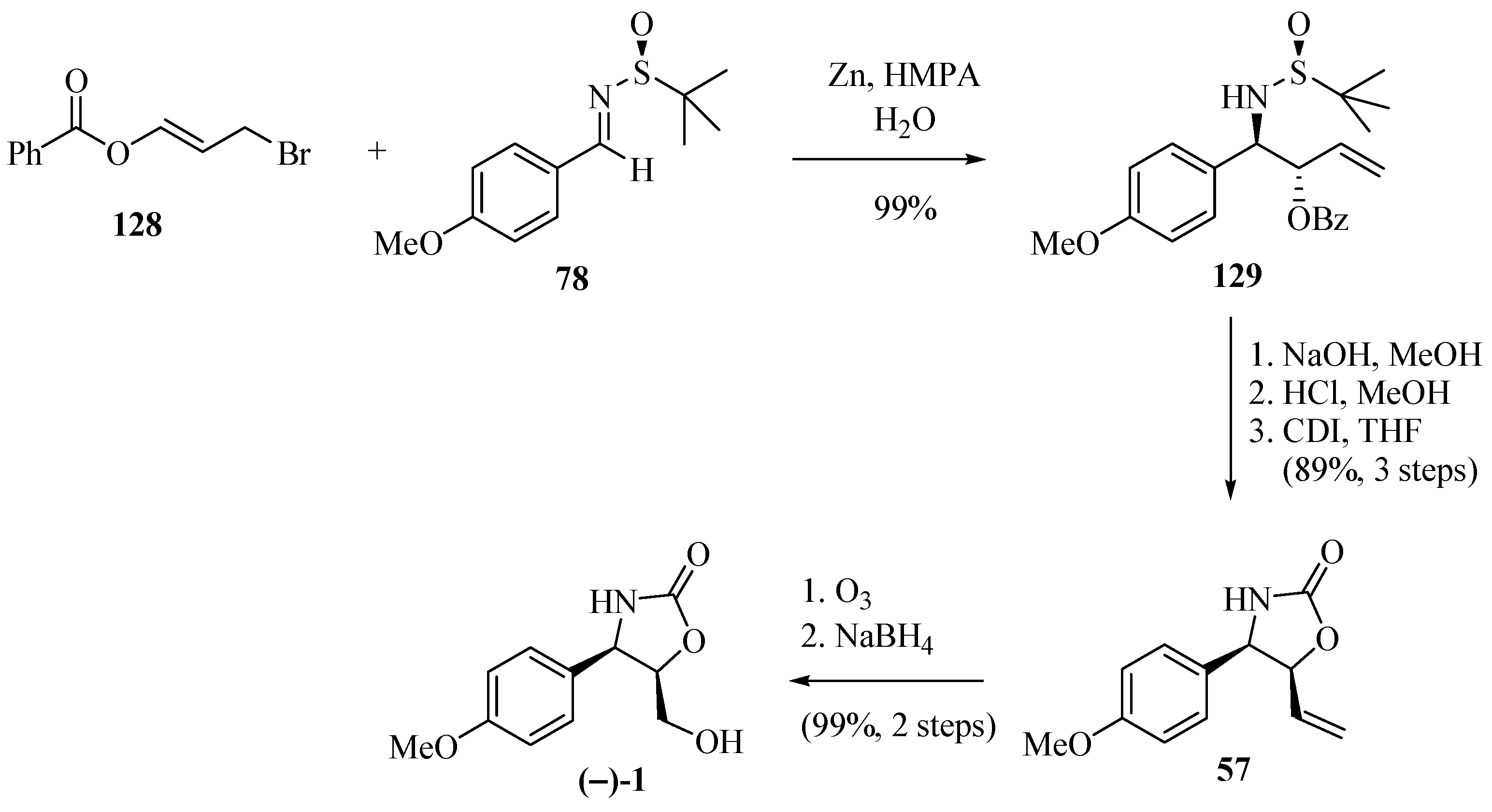
The application of the allylation of N-tert-butanesulfyl imine by the benzoyl-substituted allyl bromide 128 was illustrated by the synthesis of (−)-cytoxazone and described by Lin et al. [38] (Scheme 23). The allylation of imine 78 with 128 in the presence of Zn/HMPA produced 129 (99% yield, 96:4 rd). The routine removal of the benzoyl group and the chiral auxiliary, followed by ring closure in the presence of carbonyldiimidazole (CDI), produced 57. The ozonolysis of the double bond in 57, followed by the reduction of the generated aldehyde, completed the synthesis of the desired (−)-cytoxazone.
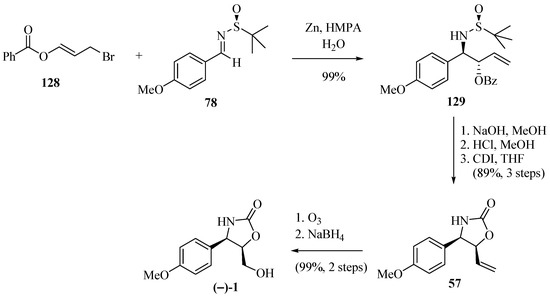
Scheme 23.
Synthesis of (−)-cytoxazone described by Lin and co-workers [38].
2.21. Asymmetric Synthesis of Chiral Amines by Highly Diastereoselective 1,2-Additions of Organometallic Reagents to N-tert-Butanesulfinyl Imines
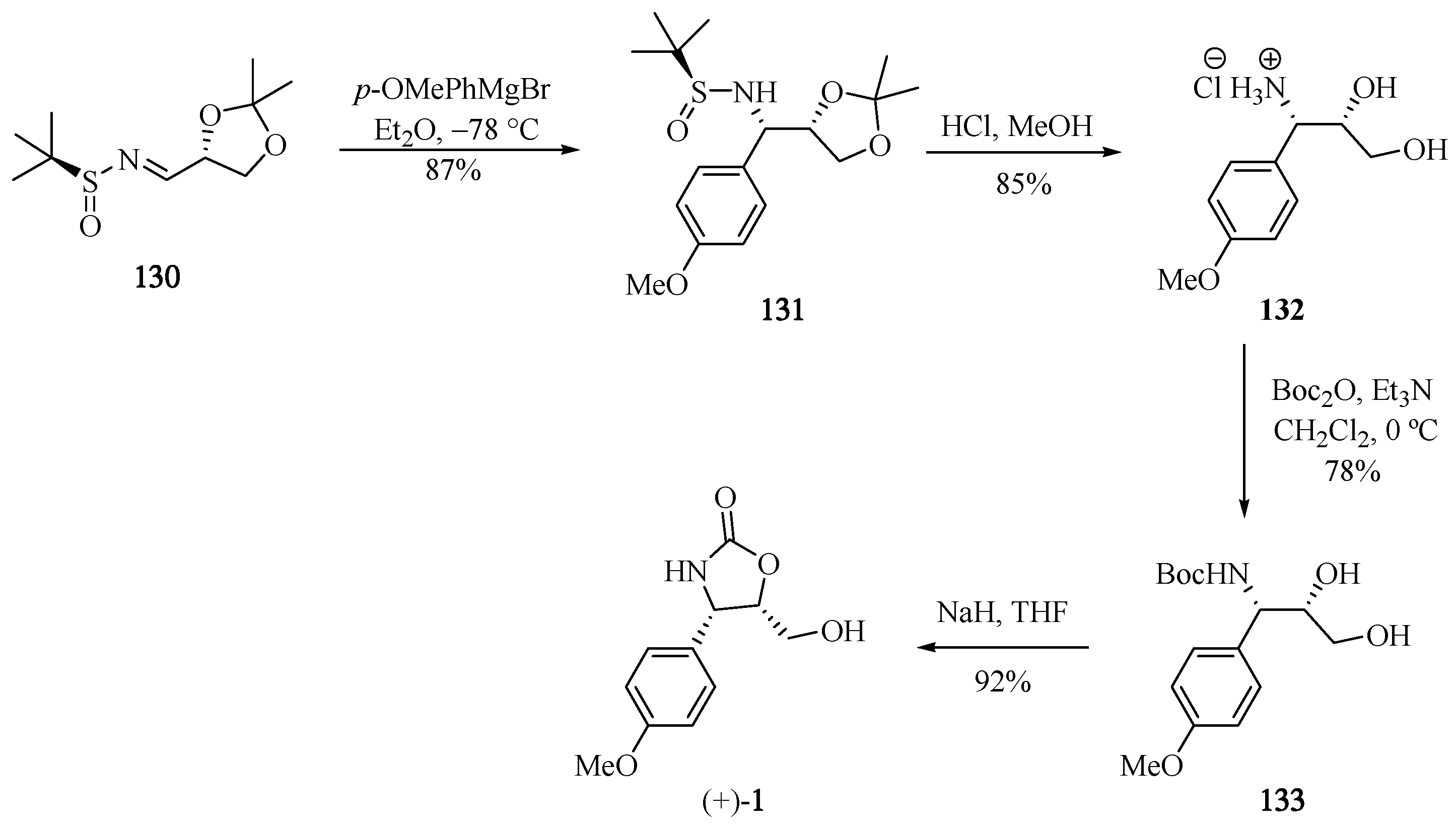
The synthetic strategy for the (+)-cytoxazone reported by Babu et al. [39] begins with the new chiral N-sulfinimine 130 (Scheme 24). The stereoselective 1,2-addition of the Grignard p-OMePhMgBr into 130 led to respective sulfonamide 131, whose t-butylsulfonyl (t-BS) and 1,3-dimethyl acetal groups were removed in a single step in acidic media, producing the corresponding syn-β-aminoalcohol 132. Finally, the N-Boc protection and heterocyclization produced (+)-cytoxazone in 53% overall yield from 130 (four steps).
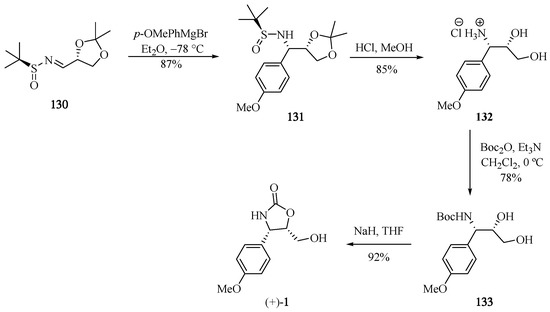
Scheme 24.
Total synthesis of (+)-cytoxazone reported by Babu and co-workers [39].
3. Conclusions
This review illustrates the various creative methods used for the synthesis of (−)-cytoxazone (1) and its analogs from the first approaches to the present day. For example, in several synthetic approaches, cinnamic acid derivatives were used as substrates and α-hydroxy-β-amino functions were introduced in a crucial step by enantioselective Sharpless epoxidation, dihydroxylation, and aminohydroxylation, or by using certain well-known chiral inducers (Evans’s oxazolidinones, N-tert-butanesulfinyl imines), as well as by other less well-known inducers. The large number of synthetic routes to this compound and its congeners evince the various methods used by chemists to prepare this class of compounds, stimulated by its relatively simple structure in addition to its biological importance as a potent cytokine modulator and its possible use as an immunotherapeutic agent.
Acknowledgments
The authors are grateful to CAPES for a scholarship awarded to I.L.M., FAPEMIG and PRPq-UFMG for the financial support.
Author Contributions
All authors have contributed substantially: I.K.B.L. and M.A.N.D. to preparation of all the graphics, I.L.M. literature research, G.D. corresponding author to write the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grajewska, A.; Rozwadowska, D. Stereoselective synthesis of cytoxazone and analogues. Tetrahedron: Asymmetry 2007, 18, 803–813. [Google Scholar] [CrossRef]
- Zappia, G.; Gacs-Baitz, E.; Monache, G.D.; Misiti, D.; Nevola, L.; Botta, B. Oxazolidin-2-one ring, a popular framework in synthetic organic chemistry: Part 1. The construction of the oxazolidin-2-one ring. Curr. Org. Synth. 2007, 4, 81–135. [Google Scholar] [CrossRef]
- Muller, M.; Schimz, K.L. Oxazolidinones: A novel class of antibiotics. Cell. Mol. Life Sci. 1999, 56, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Schito, G.C. The oxazolidinones as a new family of antimicrobial agente. Clin. Microbiol. Infect. 2001, 7, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Danielmeier, K.; Steckhan, E. Efficient pathways to (R)- and (S)-hydroxymethyl-2-oxazolidinone and some derivatives. Tetrahedron: Assymmetry 1995, 6, 1181–1190. [Google Scholar] [CrossRef]
- Dailey, C.F.; Dileto-Fang, C.L.; Buchanan, L.V.; Oramas-Shirey, M.P.; Batts, D.H.; Ford, C.W.; Gibson, J.K. Efficacy of linezolid in treatment of experimental endocarditis caused by methicillin-resistant Staphylococcus. Antimicobrial Agents Chemother. 2001, 45, 2304–2308. [Google Scholar] [CrossRef] [PubMed]
- Naresh, A.; Rao, M.V.; Kotapalli, S.S.; Ummanni, R.; Rao, B.V. Oxazolidinone derivatives: Cytoxazone-linezolid hybrids induces apoptosis and senescence in DU145 prostate cells. Eur. J. Med. Chem. 2014, 80, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Kakeya, H.; Morishita, M.; Kobinata, K.; Osono, M.; Ishizuka, M.; Osada, H. Isolation and biological activity of a novel cytokine modulator, cytoxazone. J. Antibiot. 1998, 51, 1126–1128. [Google Scholar] [CrossRef] [PubMed]
- Kakeya, H.; Morishita, H.; Koshino, T.; Morita, K.; Kobayashi, K.; Osada, H. Cytoxazone: a novel cytokine modulator containing a 2-oxazolidinone ring produced by Streptomyces sp. J. Org. Chem. 1999, 64, 1052–1053. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Shiraishi, A.; Seonhee, J.; Nakata, T. Stereoselective syntheses of cytoxazone, a novel cytokine modulator, and its stereoisomers. Tetrahedron Lett. 1999, 40, 4203–4206. [Google Scholar] [CrossRef]
- Seki, M.; Mori, K. Synthesis of (–)-cytoxazone, a novel cytokine modulator isolated from Streptomyces sp. Eur. J. Org. Chem. 1999, 11, 2965–2967. [Google Scholar] [CrossRef]
- Hamersak, Z.; Ljubovi, E.; Mercep, M.; Mesic, M.; Sunjic, V. Chemoenzymatic synthesis of all four cytoxazone stereoisomers. Synthesis 2001, 13, 1989–1992. [Google Scholar] [CrossRef]
- Smith, J.G. Synthetically useful reactions of epoxides. Synthesis 1984, 8, 629–656. [Google Scholar] [CrossRef]
- Madhan, A.; Kumar, A.R.; Rao, V. Stereoselective synthesis of (−)-cytoxazone. Tetrahedron: Asymmetry 2001, 12, 2009–2011. [Google Scholar] [CrossRef]
- Carda, M.; González, F.; Sánchez, R.; Marco, J.A. Stereoselective synthesis of (−)-cytoxazone. Tetrahedron: Asymmetry 2002, 13, 1005–1010. [Google Scholar] [CrossRef]
- Carter, P.H.; Laporte, J.R.; Scherle, P.A.; Decicco, C.P. A new synthesis of cytoxazone and its diastereomers provides key initial SAR information. Bioorg. Med. Chem. Lett. 2003, 13, 1237–1239. [Google Scholar] [CrossRef]
- Gage, J.R.; Evans, D.A. Diasteroselective aldol condensation using a chiral oxazolidinone auxiliary: (2S,3R)-3-hydroxy-3-phenyl-2-methylpropanoic acid. Org. Syn. 1989, 68, 83–91. [Google Scholar]
- Kumar, R.A.; Bhaskar, G.; Madhan, A.; Rao, B.V. Stereoselective synthesis of (R)-cytoxazone and (Q)-5-epi-cytoxazone. Synth. Commun. 2003, 33, 2907–2916. [Google Scholar] [CrossRef]
- Davies, S.G.; Hughes, D.; Nicholson, R.L.; Smith, A.D.; Wright, A. Asymmetric synthesis of (4R,5R)-cytoxazone and (4R,5S)-epi-cytoxazone. Org. Biomol. Chem. 2004, 2, 1549–1553. [Google Scholar] [CrossRef] [PubMed]
- Petasis, N.A. Method for the Synthesis of Amines and Amino Acids with Organoboran Derivatives. US6232467 B1, 15 May 2001. [Google Scholar]
- Sugiyama, S.; Arais, S.; Ishii, K. Short synthesis of both enantiomers of cytoxazone using the Petasis reaction. Tetrahedron: Assymmetry 2004, 15, 3149–3153. [Google Scholar] [CrossRef]
- Miyata, O.; Koizumi, T.; Asai, H.; Iba, R.; Naito, T. Imino 1,2-Wittig rearrangement of hidroxymates and its aplication to synthesis of cytoxazone. Tetrahedron 2004, 60, 3893–3914. [Google Scholar] [CrossRef]
- Boruwa, J.; Borah, J.C.; Kalita, B.; Barua, N.C. Highly regioselective ring opening of epoxides using NaN3: A short and efficient synthesis of (−)-cytoxazone. Tetrahedron Lett. 2004, 45, 7355–7358. [Google Scholar] [CrossRef]
- Kolb, H.C.; VanNieuwenhze, M.S.; Sharpless, K.B. Catalytic asymmetric dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
- Milicevic, S.; Matovic, R.; Saicic, R.M. Stereoselective synthesis of (−)-cytoxazone and (+)-epi-cytoxazone. Tetrahedron Lett. 2004, 45, 955–957. [Google Scholar] [CrossRef]
- Kim, J.D.; Kim, I.S.; Jin, C.H.; Zee, O.P.; Jung, Y.H. Regioselective and diastereoselective amination with use of chlorosulfonyl Isocyanate: a short and efficient synthesis of (−)-cytoxazone. Org. Lett. 2005, 7, 4025–4028. [Google Scholar] [CrossRef] [PubMed]
- Ellman, J.A.; Owens, T.D.; Tang, T.P. N-tert-Butanesulfinyl imines: Versatile intermediates for the asymmetric synthesis of amines. Acc. Chem. Res. 2002, 35, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Bentley, P.A.; Xie, H. Auxiliary strategies for the preparation of β-amino alcohols with reductive cross-coupling and a synthesis of (−)-cytoxazone. Tetrahedron Lett. 2005, 46, 7849–7852. [Google Scholar] [CrossRef]
- Tokic-Vujosevic, Z.; Petrovic, G.; Bojana, R.; Matovic, K.; Saicic, R.N. Synthesis of (−)-cytoxazone and (+)-epi-cytoxazone: The chiral pool approach. Synth. Commun. 2005, 35, 435–447. [Google Scholar] [CrossRef]
- Asano, M.; Nagasawa, C.; Suzuki, M.; Nishiyama, S.; Sugai, T. Short-step and scalable synthesis of (±)-cytoxazone. Biosci. Biotechnol. Biochem. 2005, 69, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Tosaki, S.; Tsuji, R.; Ohshima, T.; Shibasaki, M. Dynamic ligand exchange of the lanthanide complex leading to structural and functional transformation: one-pot sequential catalytic asymmetric epoxidation-regioselective epoxide-opening process. J. Am. Chem. Soc. 2005, 127, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- Paraskar, A.S.; Sudalai, A. Enantioselective synthesis of (−)-cytoxazone and (+)-epi-cytoxazone, novel cytokine modulators via Sharpless asymmetric epoxidation and L-proline catalyzed Mannich reaction. Tetrahedron 2006, 62, 5756–5762. [Google Scholar] [CrossRef]
- Kim, I.S.; Ryu, C.B.; Zee, O.P.; Jung, Y.H. A concise synthesis of (−)-cytoxazone and (−)-4-epi-cytoxazone using chlorosulfonyl isocyanate. Tetrahedron 2006, 62, 9349–9358. [Google Scholar] [CrossRef]
- Sohtome, Y.; Hashimoto, Y.; Nagasawa, K. Diastereoselective and enantioselective Henry (nitroaldol) reaction utilizing a guanidine-thiourea bifunctional organocatalyst. Eur. J. Org. Chem. 2006, 13, 2894–2897. [Google Scholar] [CrossRef]
- George, S.; Narina, S.V.; Sudalai, A. NaIO4-mediated asymmetric bromohydroxylation of α,β-unsaturated carboxamides with high diastereoselectivity: A short route to (−)-cytoxazone and droxidopa. Tetrahedron Lett. 2007, 48, 1375–1378. [Google Scholar] [CrossRef]
- Narina, S.V.; Kumar, T.S.; George, S.; Sudalai, A. Enantioselective synthesis of (−)-cytoxazone and (+)-epi-cytoxazone via Rh-catalyzed diastereoselective oxidative C–H aminations. Tetrahedron Lett. 2007, 48, 65–68. [Google Scholar] [CrossRef]
- Singh, O.V.; Han, H. Iridium(I)-catalyzed regio- and enantioselective allylic amidation. Tetrahedron Lett. 2007, 48, 7094–7098. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Xu, M.; Zhong, Y.; Sun, X. An advance on exploring N-tert-butanesulfinyl imines in asymmetric synthesis of chiral amines. Acc. Chem. Res. 2008, 41, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Babu, C.; Reddy, R.; Mukkantik, K.; Madhusudhan, G.; Srinivasulu, P. Asymmetric synthesis of chiral amines by highly diastereoselective 1,2-additions of organometallic reagents to N-tert-butanesulfinyl imines. J. Chem. Pharm. Res. 2012, 4, 4988–4994. [Google Scholar]
- Sample Availability: Samples of the compounds are not available from the authors.
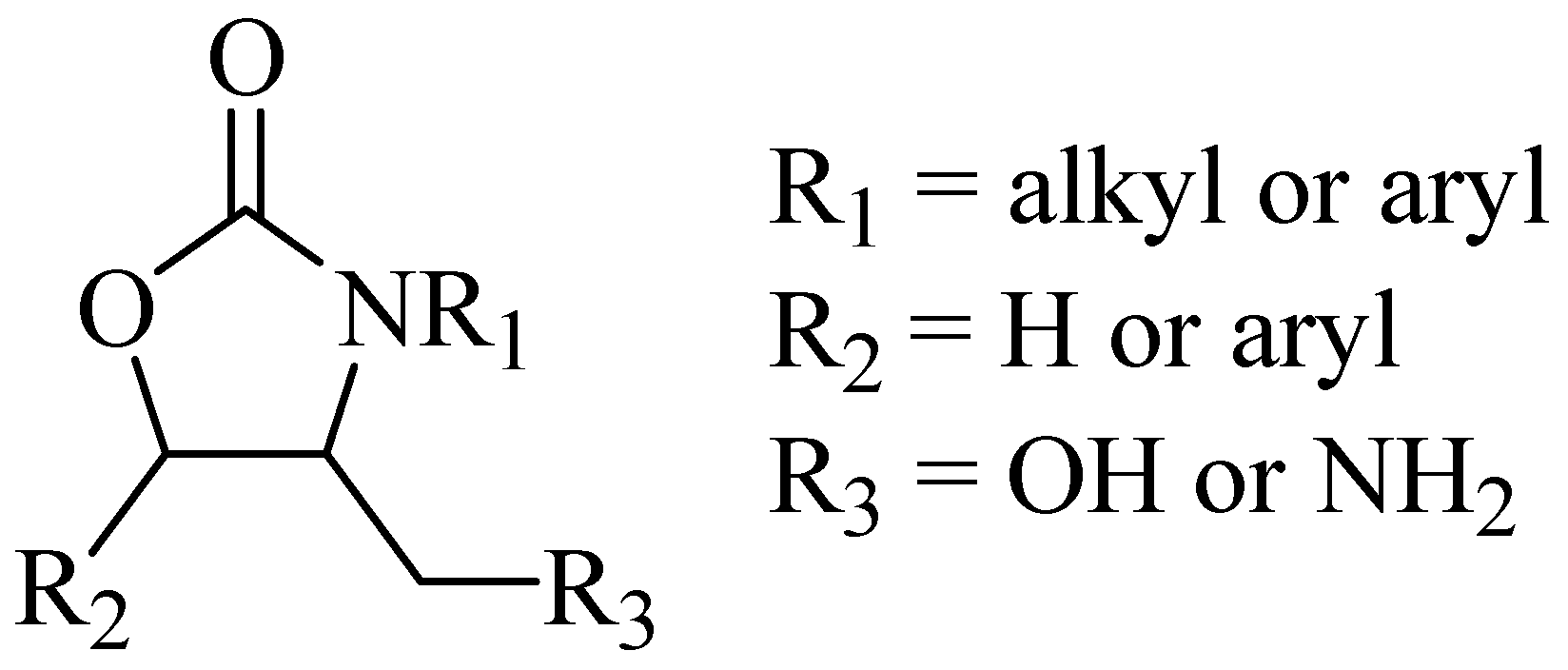
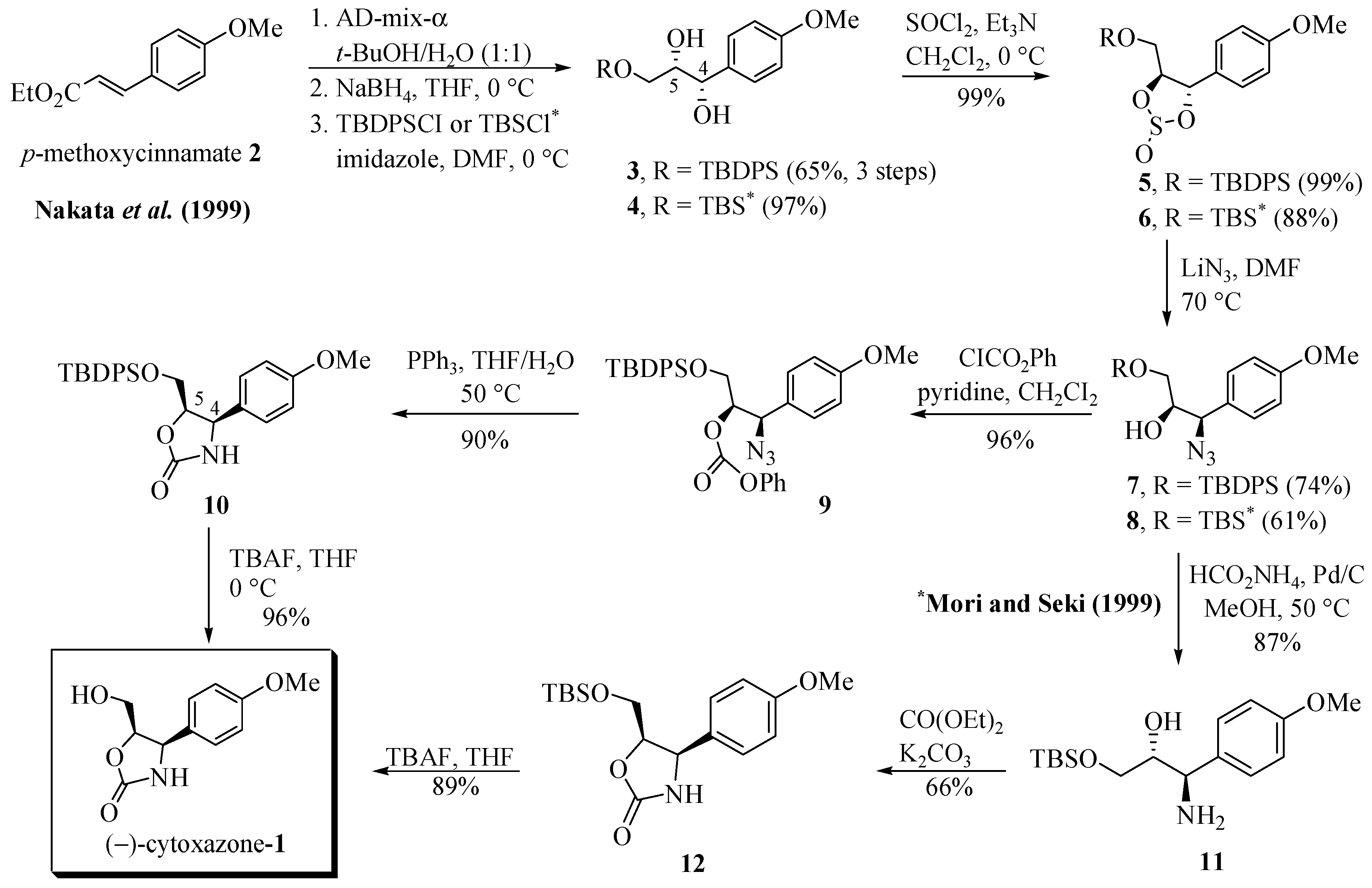
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).