
Synthesis Approaches to (−)-Cytoxazone, a Novel Cytokine Modulator, and Related Structures

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Compounds Containing 2-Oxazolidinone Structural Units and Biological Applications
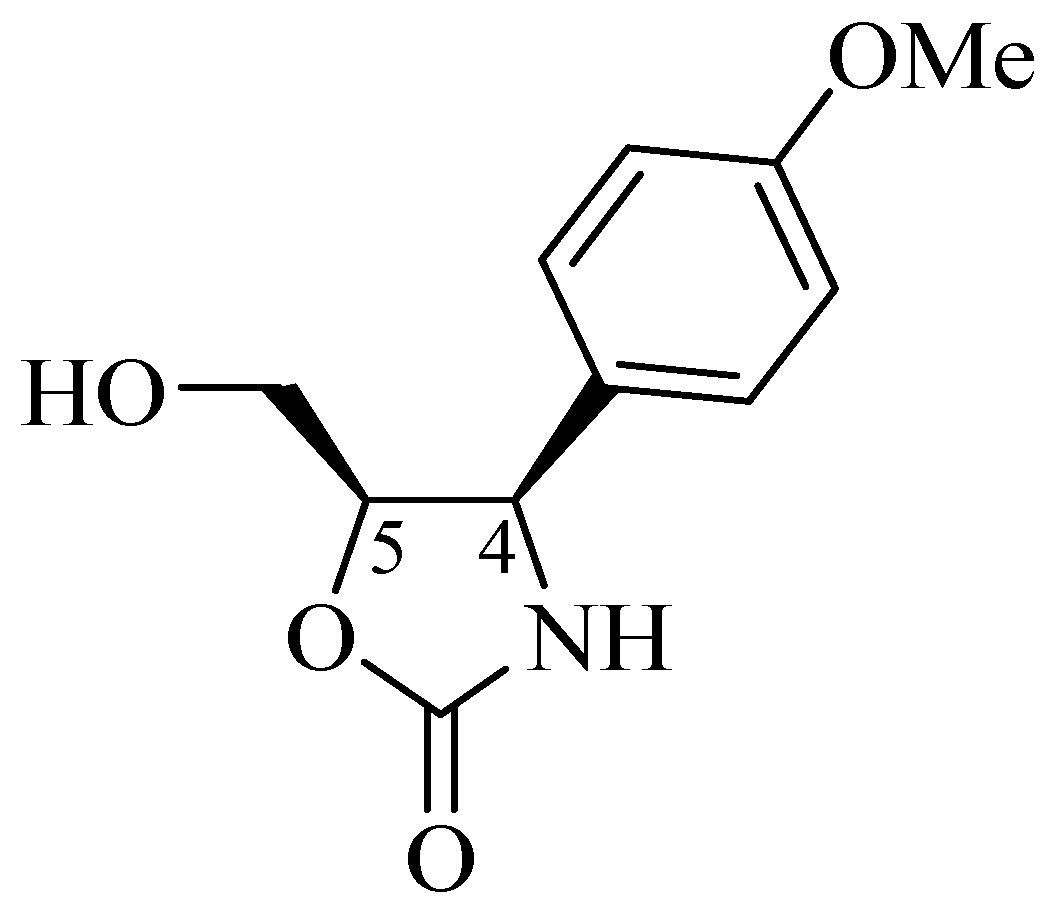
1.2. (−)-Cytoxazone
2. Total Synthesis Strategies of (−)-Cytoxazone and Congeners
2.1. Stereoselective Syntheses of (−)-Cytoxazone, a Novel Cytokine Modulator
2.2. Chemoenzymatic Synthesis of (−) and (+)-Cytoxazone Enantiomers
2.3. A New Synthesis of (−)-Cytoxazone and Its Diastereomers
2.4. Asymmetric Synthesis of (4R,5R)-Cytoxazone
2.5. Short Synthesis of Both Enantiomers of Cytoxazone using the Petasis Reaction
2.6. Imino 1,2-Wittig Rearrangement of Hydroximates and Its Application to the Synthesis of (−)-Cytoxazone
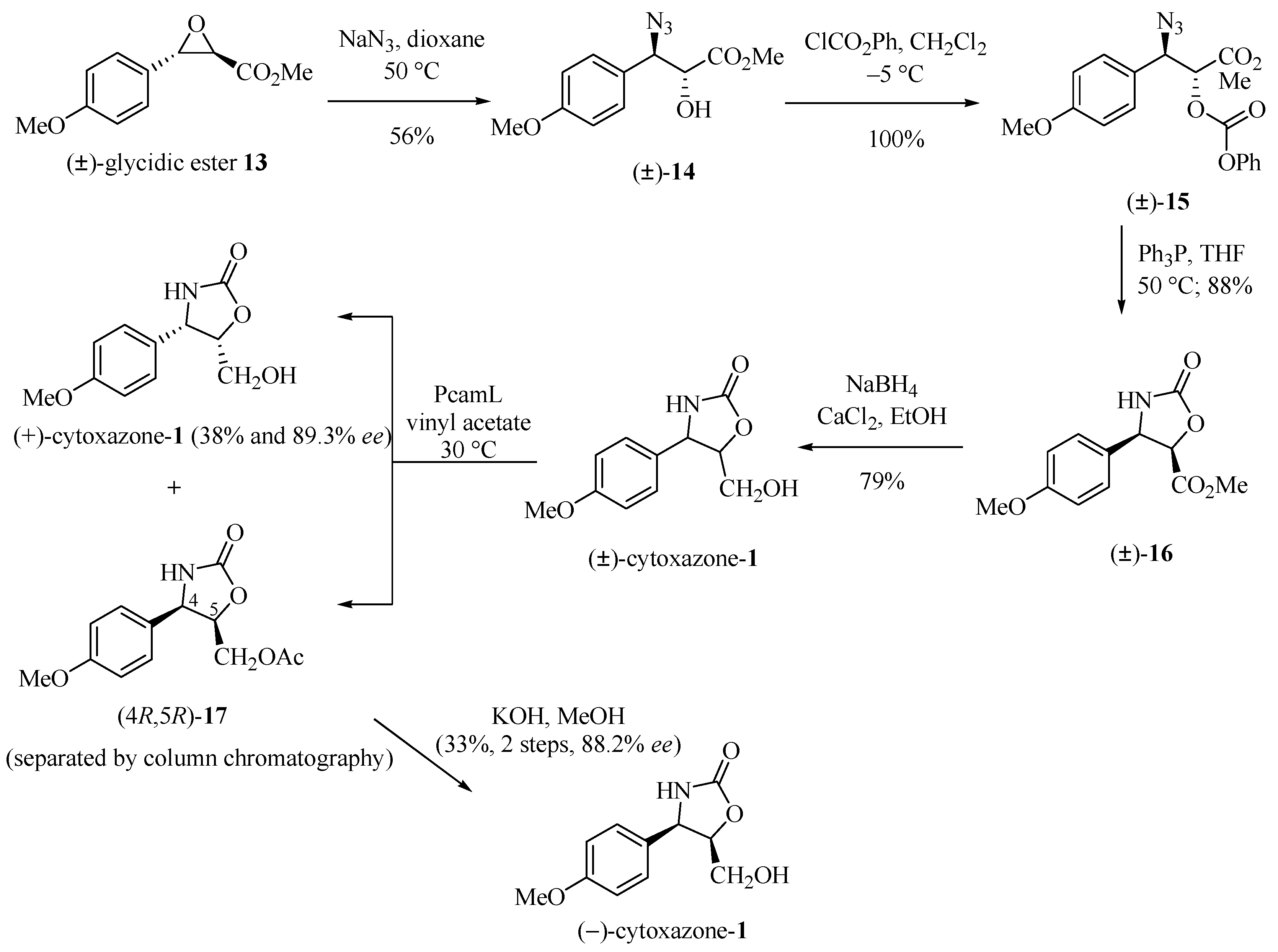
2.7. Highly Regioselective Ring Opening of Epoxides using NaN3: A Short and Efficient Synthesis of (−)-Cytoxazone
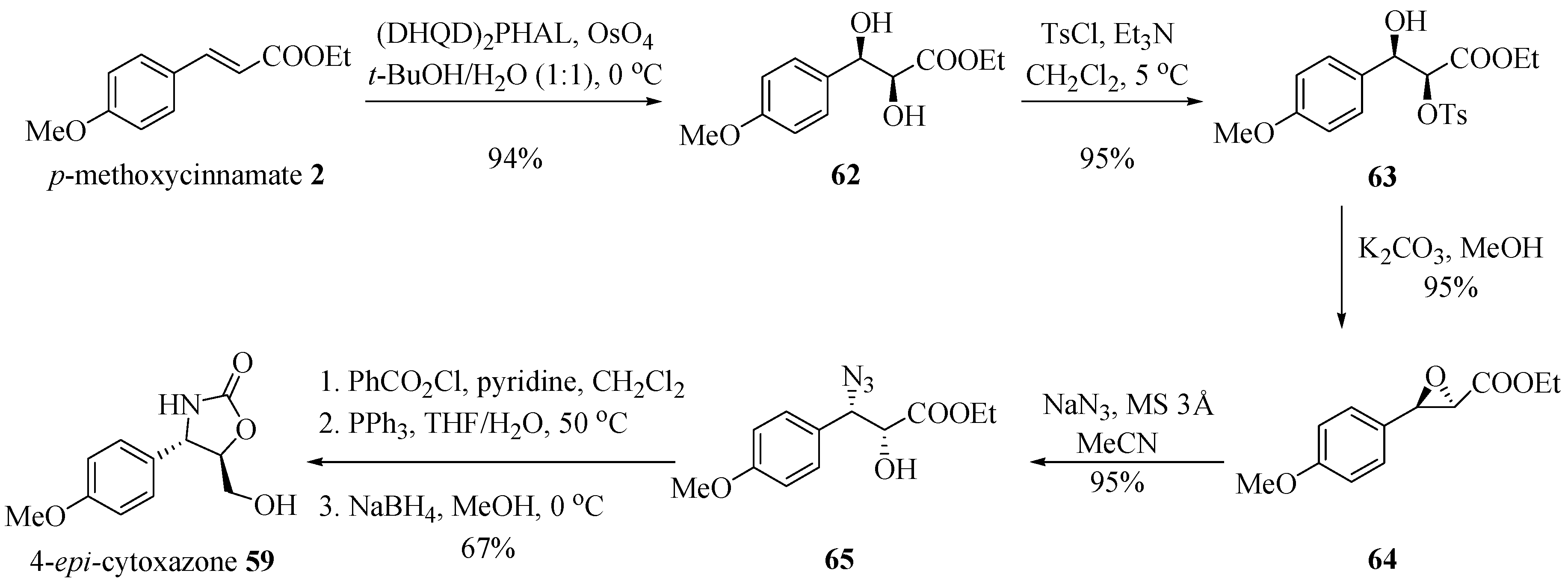
2.8. Stereoselective Synthesis of (−)-Cytoxazone and (+)-5-epi-Cytoxazone
2.9. Regioselective and Diastereoselective Amination with the use of Chlorosulfonyl Isocyanate: A Short and Efficient Synthesis of (−)-Cytoxazone
2.10. Auxiliary Strategies to Prepare β-Amino Alcohols with Reductive Cross-coupling and the Synthesis of (−)-Cytoxazone
2.11. Synthesis of (−)-Cytoxazone and (+)-epi-Cytoxazone: The Chiral Pool Approach
2.12. Short-step and Scalable Synthesis of (±)-Cytoxazone
2.13. One-pot Sequential Catalytic Asymmetric Epoxidation-regioselective Epoxide-opening Process. Application for the Synthesis of (−)-Cytoxazone
2.14. Enantioselective Synthesis of (−)-Cytoxazone and (+)-epi-Cytoxazone, Novel Cytokine Modulators via Sharpless Asymmetric Epoxidation and L-Proline Catalyzed Mannich Reaction
2.15. A Concise Synthesis of (−)-Cytoxazone and (−)-4-epi-Cytoxazone using Chlorosulfonyl Isocyanate
2.16. Diastereoselective and Enantioselective Henry (Nitroaldol) Reaction using a Guanidine-thiourea Bifunctional Organocatalyst. Application to the Synthesis of (4S,5R)-epi-Cytoxazone
2.17. NaIO4-mediated Asymmetric Bromohydroxylation of α,β-Unsaturated Carboxamides with High Diastereoselectivity: A Short Route to (−)-Cytoxazone
2.18. Enantioselective Synthesis of (−)-Cytoxazone and (+)-epi-Cytoxazone via Rh-catalyzed Diastereoselective Oxidative C–H Aminations
2.19. Iridium(I)-catalyzed Regio- and Enantioselective Allylic Amidation
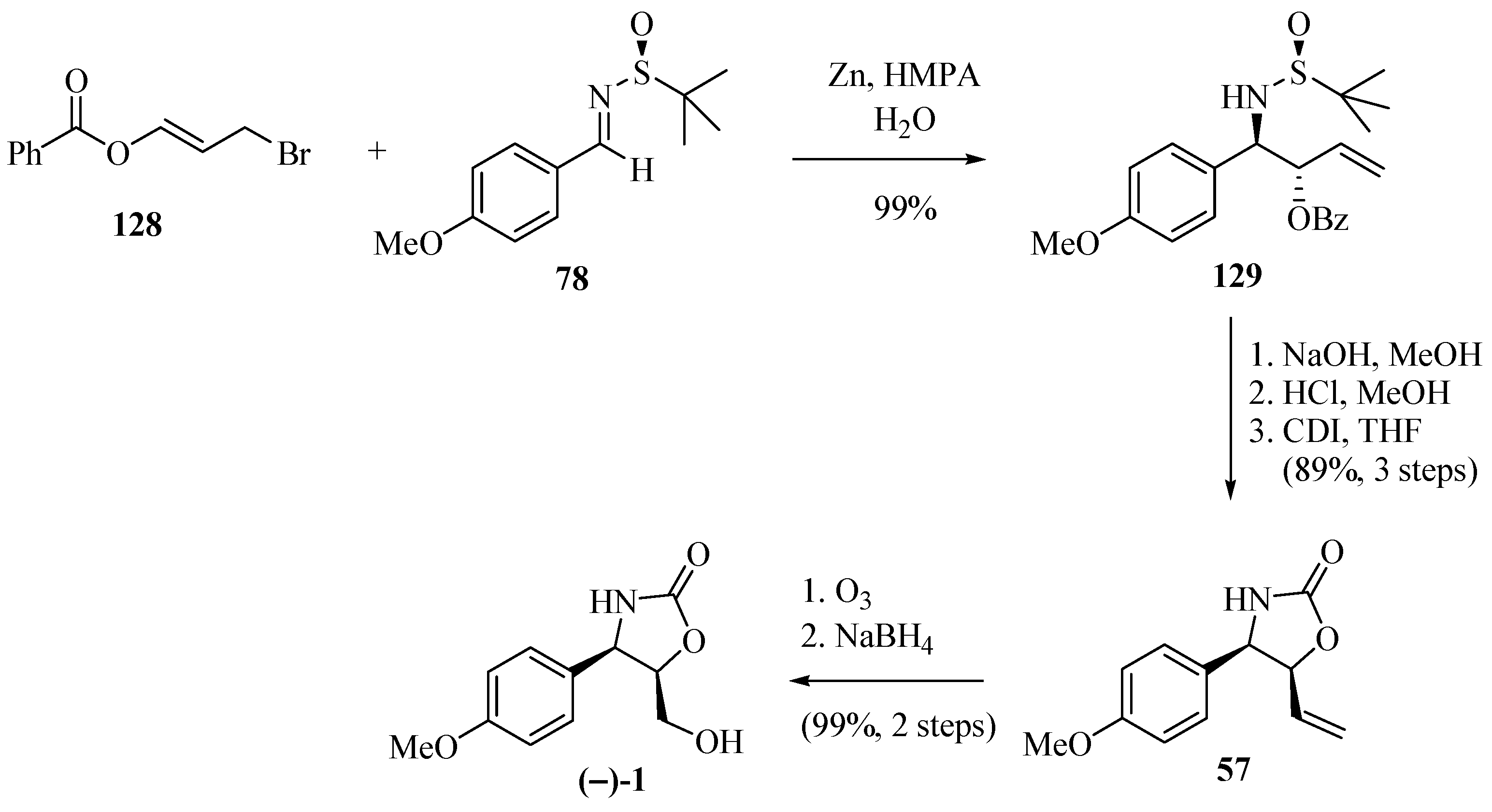
2.20. An Advance on Exploring N-tert-Butanesulfinyl Imines in Asymmetric Synthesis of Chiral Amines
2.21. Asymmetric Synthesis of Chiral Amines by Highly Diastereoselective 1,2-Additions of Organometallic Reagents to N-tert-Butanesulfinyl Imines
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grajewska, A.; Rozwadowska, D. Stereoselective synthesis of cytoxazone and analogues. Tetrahedron: Asymmetry 2007, 18, 803–813. [Google Scholar] [CrossRef]
- Zappia, G.; Gacs-Baitz, E.; Monache, G.D.; Misiti, D.; Nevola, L.; Botta, B. Oxazolidin-2-one ring, a popular framework in synthetic organic chemistry: Part 1. The construction of the oxazolidin-2-one ring. Curr. Org. Synth. 2007, 4, 81–135. [Google Scholar] [CrossRef]
- Muller, M.; Schimz, K.L. Oxazolidinones: A novel class of antibiotics. Cell. Mol. Life Sci. 1999, 56, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Schito, G.C. The oxazolidinones as a new family of antimicrobial agente. Clin. Microbiol. Infect. 2001, 7, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Danielmeier, K.; Steckhan, E. Efficient pathways to (R)- and (S)-hydroxymethyl-2-oxazolidinone and some derivatives. Tetrahedron: Assymmetry 1995, 6, 1181–1190. [Google Scholar] [CrossRef]
- Dailey, C.F.; Dileto-Fang, C.L.; Buchanan, L.V.; Oramas-Shirey, M.P.; Batts, D.H.; Ford, C.W.; Gibson, J.K. Efficacy of linezolid in treatment of experimental endocarditis caused by methicillin-resistant Staphylococcus. Antimicobrial Agents Chemother. 2001, 45, 2304–2308. [Google Scholar] [CrossRef] [PubMed]
- Naresh, A.; Rao, M.V.; Kotapalli, S.S.; Ummanni, R.; Rao, B.V. Oxazolidinone derivatives: Cytoxazone-linezolid hybrids induces apoptosis and senescence in DU145 prostate cells. Eur. J. Med. Chem. 2014, 80, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Kakeya, H.; Morishita, M.; Kobinata, K.; Osono, M.; Ishizuka, M.; Osada, H. Isolation and biological activity of a novel cytokine modulator, cytoxazone. J. Antibiot. 1998, 51, 1126–1128. [Google Scholar] [CrossRef] [PubMed]
- Kakeya, H.; Morishita, H.; Koshino, T.; Morita, K.; Kobayashi, K.; Osada, H. Cytoxazone: a novel cytokine modulator containing a 2-oxazolidinone ring produced by Streptomyces sp. J. Org. Chem. 1999, 64, 1052–1053. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Shiraishi, A.; Seonhee, J.; Nakata, T. Stereoselective syntheses of cytoxazone, a novel cytokine modulator, and its stereoisomers. Tetrahedron Lett. 1999, 40, 4203–4206. [Google Scholar] [CrossRef]
- Seki, M.; Mori, K. Synthesis of (–)-cytoxazone, a novel cytokine modulator isolated from Streptomyces sp. Eur. J. Org. Chem. 1999, 11, 2965–2967. [Google Scholar] [CrossRef]
- Hamersak, Z.; Ljubovi, E.; Mercep, M.; Mesic, M.; Sunjic, V. Chemoenzymatic synthesis of all four cytoxazone stereoisomers. Synthesis 2001, 13, 1989–1992. [Google Scholar] [CrossRef]
- Smith, J.G. Synthetically useful reactions of epoxides. Synthesis 1984, 8, 629–656. [Google Scholar] [CrossRef]
- Madhan, A.; Kumar, A.R.; Rao, V. Stereoselective synthesis of (−)-cytoxazone. Tetrahedron: Asymmetry 2001, 12, 2009–2011. [Google Scholar] [CrossRef]
- Carda, M.; González, F.; Sánchez, R.; Marco, J.A. Stereoselective synthesis of (−)-cytoxazone. Tetrahedron: Asymmetry 2002, 13, 1005–1010. [Google Scholar] [CrossRef]
- Carter, P.H.; Laporte, J.R.; Scherle, P.A.; Decicco, C.P. A new synthesis of cytoxazone and its diastereomers provides key initial SAR information. Bioorg. Med. Chem. Lett. 2003, 13, 1237–1239. [Google Scholar] [CrossRef]
- Gage, J.R.; Evans, D.A. Diasteroselective aldol condensation using a chiral oxazolidinone auxiliary: (2S,3R)-3-hydroxy-3-phenyl-2-methylpropanoic acid. Org. Syn. 1989, 68, 83–91. [Google Scholar]
- Kumar, R.A.; Bhaskar, G.; Madhan, A.; Rao, B.V. Stereoselective synthesis of (R)-cytoxazone and (Q)-5-epi-cytoxazone. Synth. Commun. 2003, 33, 2907–2916. [Google Scholar] [CrossRef]
- Davies, S.G.; Hughes, D.; Nicholson, R.L.; Smith, A.D.; Wright, A. Asymmetric synthesis of (4R,5R)-cytoxazone and (4R,5S)-epi-cytoxazone. Org. Biomol. Chem. 2004, 2, 1549–1553. [Google Scholar] [CrossRef] [PubMed]
- Petasis, N.A. Method for the Synthesis of Amines and Amino Acids with Organoboran Derivatives. US6232467 B1, 15 May 2001. [Google Scholar]
- Sugiyama, S.; Arais, S.; Ishii, K. Short synthesis of both enantiomers of cytoxazone using the Petasis reaction. Tetrahedron: Assymmetry 2004, 15, 3149–3153. [Google Scholar] [CrossRef]
- Miyata, O.; Koizumi, T.; Asai, H.; Iba, R.; Naito, T. Imino 1,2-Wittig rearrangement of hidroxymates and its aplication to synthesis of cytoxazone. Tetrahedron 2004, 60, 3893–3914. [Google Scholar] [CrossRef]
- Boruwa, J.; Borah, J.C.; Kalita, B.; Barua, N.C. Highly regioselective ring opening of epoxides using NaN3: A short and efficient synthesis of (−)-cytoxazone. Tetrahedron Lett. 2004, 45, 7355–7358. [Google Scholar] [CrossRef]
- Kolb, H.C.; VanNieuwenhze, M.S.; Sharpless, K.B. Catalytic asymmetric dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
- Milicevic, S.; Matovic, R.; Saicic, R.M. Stereoselective synthesis of (−)-cytoxazone and (+)-epi-cytoxazone. Tetrahedron Lett. 2004, 45, 955–957. [Google Scholar] [CrossRef]
- Kim, J.D.; Kim, I.S.; Jin, C.H.; Zee, O.P.; Jung, Y.H. Regioselective and diastereoselective amination with use of chlorosulfonyl Isocyanate: a short and efficient synthesis of (−)-cytoxazone. Org. Lett. 2005, 7, 4025–4028. [Google Scholar] [CrossRef] [PubMed]
- Ellman, J.A.; Owens, T.D.; Tang, T.P. N-tert-Butanesulfinyl imines: Versatile intermediates for the asymmetric synthesis of amines. Acc. Chem. Res. 2002, 35, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Bentley, P.A.; Xie, H. Auxiliary strategies for the preparation of β-amino alcohols with reductive cross-coupling and a synthesis of (−)-cytoxazone. Tetrahedron Lett. 2005, 46, 7849–7852. [Google Scholar] [CrossRef]
- Tokic-Vujosevic, Z.; Petrovic, G.; Bojana, R.; Matovic, K.; Saicic, R.N. Synthesis of (−)-cytoxazone and (+)-epi-cytoxazone: The chiral pool approach. Synth. Commun. 2005, 35, 435–447. [Google Scholar] [CrossRef]
- Asano, M.; Nagasawa, C.; Suzuki, M.; Nishiyama, S.; Sugai, T. Short-step and scalable synthesis of (±)-cytoxazone. Biosci. Biotechnol. Biochem. 2005, 69, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Tosaki, S.; Tsuji, R.; Ohshima, T.; Shibasaki, M. Dynamic ligand exchange of the lanthanide complex leading to structural and functional transformation: one-pot sequential catalytic asymmetric epoxidation-regioselective epoxide-opening process. J. Am. Chem. Soc. 2005, 127, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- Paraskar, A.S.; Sudalai, A. Enantioselective synthesis of (−)-cytoxazone and (+)-epi-cytoxazone, novel cytokine modulators via Sharpless asymmetric epoxidation and L-proline catalyzed Mannich reaction. Tetrahedron 2006, 62, 5756–5762. [Google Scholar] [CrossRef]
- Kim, I.S.; Ryu, C.B.; Zee, O.P.; Jung, Y.H. A concise synthesis of (−)-cytoxazone and (−)-4-epi-cytoxazone using chlorosulfonyl isocyanate. Tetrahedron 2006, 62, 9349–9358. [Google Scholar] [CrossRef]
- Sohtome, Y.; Hashimoto, Y.; Nagasawa, K. Diastereoselective and enantioselective Henry (nitroaldol) reaction utilizing a guanidine-thiourea bifunctional organocatalyst. Eur. J. Org. Chem. 2006, 13, 2894–2897. [Google Scholar] [CrossRef]
- George, S.; Narina, S.V.; Sudalai, A. NaIO4-mediated asymmetric bromohydroxylation of α,β-unsaturated carboxamides with high diastereoselectivity: A short route to (−)-cytoxazone and droxidopa. Tetrahedron Lett. 2007, 48, 1375–1378. [Google Scholar] [CrossRef]
- Narina, S.V.; Kumar, T.S.; George, S.; Sudalai, A. Enantioselective synthesis of (−)-cytoxazone and (+)-epi-cytoxazone via Rh-catalyzed diastereoselective oxidative C–H aminations. Tetrahedron Lett. 2007, 48, 65–68. [Google Scholar] [CrossRef]
- Singh, O.V.; Han, H. Iridium(I)-catalyzed regio- and enantioselective allylic amidation. Tetrahedron Lett. 2007, 48, 7094–7098. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Xu, M.; Zhong, Y.; Sun, X. An advance on exploring N-tert-butanesulfinyl imines in asymmetric synthesis of chiral amines. Acc. Chem. Res. 2008, 41, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Babu, C.; Reddy, R.; Mukkantik, K.; Madhusudhan, G.; Srinivasulu, P. Asymmetric synthesis of chiral amines by highly diastereoselective 1,2-additions of organometallic reagents to N-tert-butanesulfinyl imines. J. Chem. Pharm. Res. 2012, 4, 4988–4994. [Google Scholar]
- Sample Availability: Samples of the compounds are not available from the authors.
























© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda, I.L.; Lopes, Í.K.B.; Diaz, M.A.N.; Diaz, G. Synthesis Approaches to (−)-Cytoxazone, a Novel Cytokine Modulator, and Related Structures. Molecules 2016, 21, 1176. https://doi.org/10.3390/molecules21091176
Miranda IL, Lopes ÍKB, Diaz MAN, Diaz G. Synthesis Approaches to (−)-Cytoxazone, a Novel Cytokine Modulator, and Related Structures. Molecules. 2016; 21(9):1176. https://doi.org/10.3390/molecules21091176
Chicago/Turabian StyleMiranda, Izabel L., Ítala K. B. Lopes, Marisa A. N. Diaz, and Gaspar Diaz. 2016. "Synthesis Approaches to (−)-Cytoxazone, a Novel Cytokine Modulator, and Related Structures" Molecules 21, no. 9: 1176. https://doi.org/10.3390/molecules21091176
APA StyleMiranda, I. L., Lopes, Í. K. B., Diaz, M. A. N., & Diaz, G. (2016). Synthesis Approaches to (−)-Cytoxazone, a Novel Cytokine Modulator, and Related Structures. Molecules, 21(9), 1176. https://doi.org/10.3390/molecules21091176