
Synthesis and Biological Evaluation of Triazolyl 13?-Estrone–Nucleoside Bioconjugates

 ,
,
Abstract
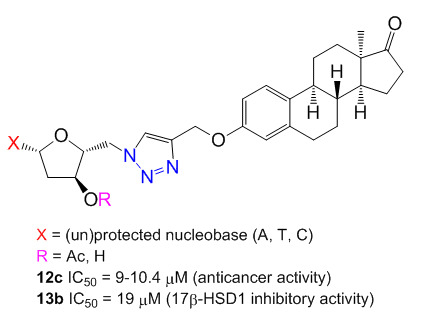
:
1. Introduction
2. Results and Discussion
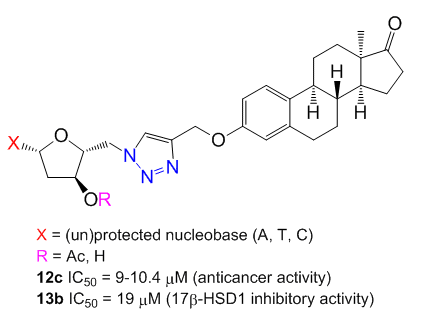
2.1. Preparation of 5′-Azido-2′,5′-dideoxynucleosides
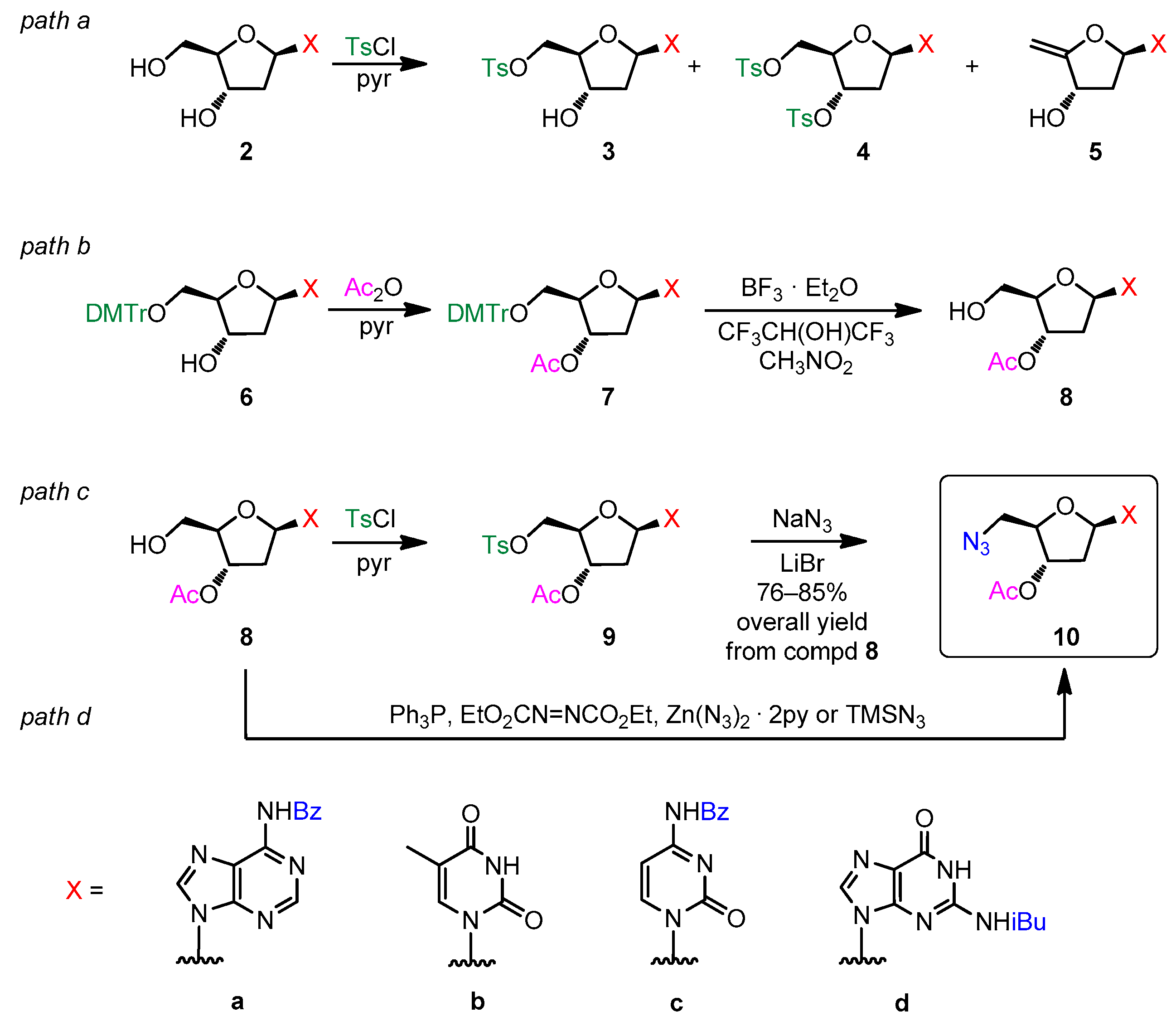
2.2. Optimization of the Click Reaction between 5′-Azido-nucleoside Derivatives and 3-O-Propargyl-13α-estrone
2.3. Optimization of the Deprotection of Conjugates (Nucleobase N-Benzoyl and 2′-Deoxy-D-ribose-3′-O-acetyl Deprotection)
2.4. Antiproliferative Activities
2.5. Inhibition of 17β-HSD1
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for Synthesis of 3′-O-Acetyl-N-acyl-protected-2′-deoxynucleosides 8a–d
3.1.2. General Procedure for Synthesis of 3′-O-Acetyl-5′-azido-N-acyl-protected-2′,5′-dideoxy-nucleosides 10a–c
3.1.3. General Procedure for Click Reactions: Preparation of 12a–c
3.1.4. General Procedure for Deprotection of Conjugates 13a–c
3.2. Cell Cultures and Antiproliferative Assays
3.3. Determination of 17β-HSD1 Inhibition
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2010, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.G.; Zeng, Q.; Tse, G.M. Estrogen and its receptors in cancer. Med. Res. Rev. 2008, 28, 954–974. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Kumar, B.S.; Negi, A.S. Current status on development of steroids as anticancer agents. J. Steroid Biochem. Mol. Biol. 2013, 137, 242–270. [Google Scholar] [CrossRef] [PubMed]
- Möller, G.; Adamski, J. Integrated view on 17beta-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2009, 301, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Marchais-Oberwinkler, S.; Henn, C.; Möller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; et al. 17beta-Hydroxysteroid dehydrogenases (17beta-HSDs) as therapeutic targets: Protein structures, functions, and recent progress in inhibitor development. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Day, J.M.; Tutill, H.J.; Purohit, A.; Reed, M.J. Design and validation of specific inhibitors of 17beta-hydroxysteroid dehydrogenases for therapeutic application in breast and prostate cancer, and in endometriosis. Endocr. Relat. Cancer 2008, 15, 665–692. [Google Scholar] [CrossRef] [PubMed]
- Poirier, D. 17beta-Hydroxysteroid dehydrogenase inhibitors: A patent review. Expert Opin. Ther. Pat. 2010, 20, 1123–1145. [Google Scholar] [CrossRef] [PubMed]
- Gobec, S.; Brozic, P.; Rizner, T.L. Inhibitors of 17beta-hydroxysteroid dehydrogenase type 1. Curr. Med. Chem. 2008, 15, 137–150. [Google Scholar] [CrossRef]
- Minorics, R.; Bózsity, N.; Wölfling, J.; Mernyák, E.; Schneider, G.; Márki, Á.; Falkay, G.; Ocsovszki, I. Zupkó Antiproliferative effect of normal and 13-epi-d-homoestrone and their 3-methyl ethers on human reproductive cancer cell lines. J. Steroid Biochem. Mol. Biol. 2012, 132, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Wölfling, J.; Mernyák, E.; Frank, É.; Falkay, G.; Márki, Á.; Minorics, R.; Schneider, G. Synthesis and receptor-binding examinations of the normal and 13-epi-d-homoestrones and their 3-methyl ethers. Steroids 2003, 68, 277–288. [Google Scholar] [CrossRef]
- Cushman, M.; He, H.-M.; Katzenellenbogen, J.A.; Varma, R.K.; Hamel, E.; Lin, C.M.; Ram, S.; Sachdeva, Y.P. Synthesis of analogs of 2-methoxyestradiol with enhanced inhibitory effects on tubulin polymerization and cancer cell growth. J. Med. Chem. 1997, 40, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Möller, G.; Deluca, D.; Gege, C.; Rosinus, A.; Kowalik, D.; Peters, O.; Droescher, P.; Elger, W.; Adamski, J.; Hillisch, A. Structure-based design, synthesis and in vitro characterization of potent 17beta-hydroxysteroid dehydrogenase type 1 inhibitors based on 2-substitutions of estrone and d-homo-estrone. Bioorg. Med. Chem. Lett. 2009, 19, 6740–6744. [Google Scholar] [CrossRef] [PubMed]
- Hillisch, A.; Peters, O.; Gege, C.; Siemeister, G.; Unger, E.; Menzenbach, B. Antitumoral d-homoestra-1,3,5(10)-trien-3-yl 2-substituted sulfamates. US Patent 7,244,762, 2007. [Google Scholar]
- Ayan, D.; Roy, J.; Maltais, R.; Poirier, D. Impact of estradiol structural modifications (18-methyl and/or 17-hydroxy inversion of configuration) on the in vitro and in vivo estrogenic activity. J. Steroid Biochem. Mol. Biol. 2011, 127, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Schönecker, B.; Lange, C.; Kötteritzsch, M.; Gunther, W.; Weston, J.; Anders, E.; Görls, H. Conformational design for 13alpha-steroids. J. Org. Chem. 2000, 65, 5487–5497. [Google Scholar] [CrossRef] [PubMed]
- Jovanović-Santa, S.; Petrović, J.; Andrić, S.; Kovačević, R.; Đurendić, E.; Sakač, M.; Lazar, D.; Stanković, S. Synthesis, structure, and screening of estrogenic and antiestrogenic activity of new 3, 17-substituted-16, 17-seco-estratriene derivatives. Bioorg. Chem. 2003, 31, 475–484. [Google Scholar]
- Yaremenko, F.G.; Khvat, A.V. A new one-pot synthesis of 17-oxo-13alpha-steroids of the androstane series from their 13beta-analogues. Mendeleev Commun. 1994, 4, 187–188. [Google Scholar] [CrossRef]
- Mernyák, E.; Kovács, I.; Minorics, R.; Sere, P.; Czégány, D.; Sinka, I.; Wölfling, J.; Schneider, G.; Újfaludi, Z.; Boros, I.; et al. Synthesis of trans-16-triazolyl-13alpha-methyl-17-estradiol diastereomers and the effects of structural modifications on their in vitro antiproliferative activities. J. Steroid Biochem. Mol. Biol. 2015, 150, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Szabó, J.; Jerkovics, N.; Schneider, G.; Wölfling, J.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Synthesis and in vitro antiproliferative evaluation of C-13 epimers of triazolyl-D-secoestrone alcohols: The first potent 13α-d-secoestrone derivative. Molecules 2016, 21, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Szabó, J.; Pataki, Z.; Wölfling, J.; Schneider, G.; Bózsity, N.; Minorics, R.; Zupkó, I.; Mernyák, E. Synthesis and biological evaluation of 13alpha-estrone derivatives as potential antiproliferative agents. Steroids 2016, 113, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Zupkó, I.; Varga, M.; Herman, B.E.; Kalmár, L.; Szécsi, M.; Mernyák, E. Synthesis and in vitro pharmacological evaluation of N-[(1-benzyl-1,2,3-triazol-4-yl)methyl]-carboxamides on D-secoestrone scaffolds. J. Enzyme Inhib. Med. Chem. 2016, 31, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Herman, B.E.; Szabó, J.; Bacsa, I.; Wölfling, J.; Schneider, G.; Bálint, M.; Hetényi, C.; Mernyák, E.; Szécsi, M. Comparative investigation of the in vitro inhibitory potencies of 13-epimeric estrones and d-secoestrones towards 17beta-hydroxysteroid dehydrogenase type 1. J. Enzyme Inhib. Med. Chem. 2016, Early Online, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Polster, J.; Engles, J.M.; Hilton, J.; Abraham, E.H.; Wahl, R.L. In vitro evaluation of adenosine 5′-monophosphate as an imaging agent of tumor metabolism. J. Nucl. Med. 2006, 47, 837–845. [Google Scholar] [PubMed]
- Pastor-Anglada, M.; Cano-Soldado, P.; Molina-Arcas, M.; Lostao, M.P.; Larráyoz, I.; Martínez-Picado, J.; Casado, F.J. Cell entry and export of nucleoside analogues. Virus Res. 2005, 107, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torras, S.; García-Manteiga, J.; Mercadéd, E.; Casado, F.J.; Carbó, N.; Pastor-Anglada, M.; Mazo, A. Adenoviral-mediated overexpression of human equilibrative nucleoside transporter 1 (hENT1) enhances gemcitabine response in human pancreatic cancer. Biochem. Pharmacol. 2008, 76, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Fournier, D.; Poirier, D.; Mazumdar, M.; Lin, S.X. Design and synthesis of bisubstrate inhibitors of type 1 17beta-hydroxysteroid dehydrogenase: Overview and perspectives. Eur. J. Med. Chem. 2008, 43, 2298–2306. [Google Scholar] [CrossRef] [PubMed]
- An, S.H.; West, C.R.; Hong, C.I. Nucleoside conjugates. 8. The preparation of 5-fluoro-2′-deoxyuridine conjugates of corticosteroids. Steroids 1986, 47, 413–420. [Google Scholar] [CrossRef]
- Bérubé, M.; Poirier, D. Improved synthesis of EM-1745, preparation of its C17-ketone analogue and comparison of their inhibitory potency on 17beta-hydroxysteroid dehydrogenase type 1. J. Enzyme Inhib. Med. Chem. 2009, 24, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Poirier, D. Contribution to the development of inhibitors of 17beta-hydroxysteroid dehydrogenase types 1 and 7: Key tools for studying and treating estrogen-dependent diseases. J. Steroid Biochem. Mol. Biol. 2011, 125, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Poirier, D.; Boivin, R.P.; Tremblay, M.R.; Bérubé, M.; Qiu, W.; Lin, S.-X. Estradiol-adenosine hybrid compounds designed to inhibit type 1 17beta-hydroxysteroid dehydrogenase. J. Med. Chem. 2005, 48, 8134–8147. [Google Scholar] [CrossRef] [PubMed]
- Iyer, V.K.; Butler, W.B.; Horwitz, J.P.; Rozhin, J.; Brooks, S.C.; Corombos, J.; Kessel, D. Some adenine and adenosine methylene-bridged estrogens. J. Med. Chem. 1983, 26, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Meldal, M.; Tornøe, C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Herdewijn, P.; Balzarini, J.; Pauwels, R.; Janssen, G.; Aerschot, A.V.; Clercq, E.D. Synthesis and biological activity of the mono-and diamino analogues of 2′-deoxyadenosine, cordycepin, 9-(3-deoxy-alpha-d-threo-pentofuranosyl)adenine (a structural component of Agrocin 84) and 9-(2-deoxy-alpha-d-threo-pentofuranosyl)adenine. Nucleos. Nucleot. 1989, 8, 1231–1257. [Google Scholar] [CrossRef]
- Chen, J.-B.; Liu, E.M.; Chern, T.-R.; Yang, C.-W.; Lin, C.I.; Huang, N.-K.; Lin, Y.-L.; Chern, Y.; Lin, J.-H.; Fang, J.-M. Design and synthesis of novel dual-action compounds targeting the adenosine A2A receptor and adenosine transporter for neuroprotection. ChemMedChem 2011, 6, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Murat, P.; Gennaro, B.; Garcia, J.; Spinelli, N.; Dumy, P.; Defrancq, E. The use of a peptidic scaffold for the formation of stable guanine tetrads: Control of a H-bonded pattern in water. Chem. Eur. J. 2011, 17, 5791–5795. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Liu, J.; Sun, H.; Xie, J. Synthesis of nucleoside conjugates as potential inhibitors of glycogen phosphorylase. Synthesis 2010, 1046–1052. [Google Scholar] [CrossRef]
- Mattarella, M.; Berstis, L.; Baldridge, K.K.; Siegel, J.S. Synthesis of bioconjugated sym-pentasubstituted corannulenes: Experimental and theoretical investigations of supramolecular architectures. Bioconj. Chem. 2014, 25, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Točík, Z.; Dvořáková, I.; Liboska, R.; Buděšínský, M.; Masojídková, M.; Rosenberg, I. Electrophile-promoted addition of hydroxymethylphosphonate to 4′, 5′-didehydronucleosides: A way to novel isosteric analogues of 5′-nucleotides. Tetrahedron 2007, 63, 4516–4534. [Google Scholar] [CrossRef]
- Brear, P.; Freeman, G.R.; Shankey, M.C.; Trmčić, M.; Hodgson, D.R.W. Aqueous methods for the preparation of 5′-substituted guanosine derivatives. Chem. Commun. 2009, 4980–4981. [Google Scholar] [CrossRef] [PubMed]
- Paredes, E.; Das, S.R. Click chemistry for rapid labeling and ligation of RNA. Chem. Bio. Chem. 2011, 12, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, I.; Sekine, M.; Hata, T. One-step synthesis of 5′-azido-nucleosides. J. Chem. Soc. Perkin Trans. 1 1980, 306–310. [Google Scholar] [CrossRef]
- Hui, W.B.; Sherman, J.C. Self-assembly of a thymine quartet and quadruplex via an organic template. Tetrahedron Lett. 2014, 55, 1479–1485. [Google Scholar] [CrossRef]
- Seio, K.; Miyashita, T.; Sato, K.; Sekine, M. Synthesis and properties of new nucleotide analogues possessing squaramide moieties as new phosphate isosters. Eur. J. Org. Chem. 2005, 5163–5170. [Google Scholar] [CrossRef]
- Madhuri, V.; Kumar, V.A. Design and synthesis of dephosphono DNA analogues containing 1, 2, 3-triazole linker and their UV-melting studies with DNA/RNA. Nucleos. Nucleot. Nucl. Acids 2012, 31, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Austin, D.J. A general synthesis of 5′-azido-5′-deoxy-2′,3′-O-isopropylidene nucleosides. J. Org. Chem. 2001, 66, 8643–8645. [Google Scholar] [CrossRef] [PubMed]
- Nuzzi, A.; Massi, A.; Dondoni, A. Model studies toward the synthesis of thymidine oligonucleotides with triazole internucleosidic linkages via iterative Cu(I)-promoted azide-alkyne ligation chemistry. QSAR Comb. Sci. 2007, 26, 1191–1199. [Google Scholar] [CrossRef]
- Cui, H.; Carrero-Lérida, J.; Silva, A.P.; Whittingham, J.L.; Brannigan, J.A.; Ruiz-Pérez, L.M.; Read, K.D.; Wilson, K.S.; González-Pacanowska, D.; Gilbert, I.H. Synthesis and evaluation of alpha-thymidine analogues as novel antimalarials. J. Med. Chem. 2012, 55, 10948–10957. [Google Scholar] [CrossRef] [PubMed]
- Mitsunobu, O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformations of natural products. Synthesis 1981, 1–28. [Google Scholar] [CrossRef]
- Hughes, D.L. Progress in the Mitsunobu reaction. A review. Org. Prep. Proc. Int. 1996, 28, 127–164. [Google Scholar] [CrossRef]
- Kumara Swamy, K.C.; Bhuvan Kumar, N.N.; Balaraman, E.; Pavan Kumar, K.V.P. Mitsunobu and related reactions: Advances and applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Viaud, M.C.; Rollin, P. Zinc azide mediated Mitsunobu substitution. An expedient method for the one-pot azidation of alcohols. Synthesis 1990, 130–132. [Google Scholar] [CrossRef]
- Jawalekar, A.M.; Meeuwenoord, N.; Cremers, J.G.O.; Overkleeft, H.S.; van der Marel, G.A.; Rutjes, F.P.J.T.; van Delft, F.L. Conjugation of nucleosides and oligonucleotides by [3 + 2] cycloaddition. J. Org. Chem. 2008, 73, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Lolk, L.; Pøhlsgaard, J.; Jepsen, A.S.; Hansen, L.H.; Nielsen, H.; Steffansen, S.I.; Sparving, L.; Nielsen, A.B.; Vester, B.; Nielsen, P. A click chemistry approach to pleuromutilin conjugates with nucleosides or acyclic nucleoside derivatives and their binding to the bacterial ribosome. J. Med. Chem. 2008, 51, 4957–4967. [Google Scholar] [CrossRef] [PubMed]
- Casaschi, A.; Grigg, R.; Sansano, J.M. Palladium catalysed tandem cyclisation-anion capture. Part 6: synthesis of sugar, nucleoside, purine, benzodiazepinone and beta-lactam analogues via capture of in situ generated vinylstannanes. Tetrahedron 2000, 56, 7553–7560. [Google Scholar] [CrossRef]
- Tjarks, W.; Anisuzzaman, A.K.; Liu, L.; Soloway, A.H.; Barth, R.F.; Perkins, D.J.; Adams, D.M. Synthesis and in vitro evaluation of boronated uridine and glucose derivatives for boron neutron capture therapy. J. Med. Chem. 1992, 35, 1628–1633. [Google Scholar] [CrossRef] [PubMed]
- Fernicola, S.; Torquati, I.; Paiardini, A.; Giardina, G.; Rampioni, G.; Messina, M.; Leoni, L.; Del Bello, F.; Petrelli, R.; Rinaldo, S.; et al. Synthesis of triazole-linked analogues of c-di-GMP and their interactions with diguanylate cyclase. J. Med. Chem. 2015, 58, 8269–8284. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Teste, K.; Zerrouki, R.; Champavier, Y.; Guilloton, M. Chelation-controlled regioselective alkylation of pyrimidine 2′-deoxynucleosides. Carbohydr. Res. 2010, 345, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Kicsák, M.; Bege, M.; Bereczki, I.; Csávás, M.; Herczeg, M.; Kupihár, Z.; Kovács, L.; Borbás, A.; Herczegh, P. A three-component reagent system for rapid and mild removal of O-, N- and S-trityl protecting groups. Org. Biomol. Chem. 2016, 14, 3190–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kádár, Z.; Molnár, J.; Schneider, G.; Zupkó, I.; Frank, É. A facile ‘click’ approach to novel 15beta-triazolyl-5alpha-androstane derivatives, and an evaluation of their antiproliferative activities in vitro. Bioorg. Med. Chem. 2012, 20, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Bacsa, I.; Jójárt, R.; Schneider, G.; Wölfling, J.; Maróti, P.; Herman, B.E.; Szécsi, M.; Mernyák, E. Synthesis of A-ring halogenated 13alpha-estrone derivatives as potential 17beta-HSD1 inhibitors. Steroids 2015, 104, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions and Yields of CuAAC Reaction | ||||||
|---|---|---|---|---|---|---|
| Ref. [32] 1 | Refs. [36,38,61] 2 | This work 3 | ||||
| Product Code | Product Yield (%) | Recovered Nucleoside (%) 4 | Product Yield (%) | Recovered Nucleoside (%) 4 | Product Yield (%) | Recovered Nucleoside (%) 5 |
| 12a | 0 | 93 | 18–22 | 63–68 | 68 | - |
| 12b | 0 | 92 | 23–28 | 64–70 | 76 | - |
| 12c | 0 | >95 | 8–11 | 78–82 | 61 | - |
| Structure | Compd Code or Name [ref.] | Conc. (µM) | Inhibition (%) ± SEM [Calculated IC50, µM] 1 | ||
|---|---|---|---|---|---|
| A2780 | HeLa | MCF-7 | |||
 | 12a | 10 | 39.4 ± 2.4 | - 2 | - 2 |
| 30 | 70.2 ± 1.6 | 55.5 ± 0.6 | 49.6 ± 1.4 | ||
| [10.9] | [16.3] | [>30] 3 | |||
 | 12b | 10 | 29.8 ± 0.2 | - | 29.2 ± 2.9 |
| 30 | 35.1 ± 2.7 | - | 26.7 ± 2.0 | ||
| [>30] | [>30] | [>30] | |||
 | 12c | 10 | 63.2 ± 1.5 | 53.5 ± 1.0 | 47.4 ± 2.4 |
| 30 | 66.6 ± 1.6 | 61.9 ± 1.2 | 57.3 ± 1.8 | ||
| [9.0] | [9.0] | [10.4] | |||
 | 13a | 10 | - | 26.6 ± 1.8 | - |
| 30 | 41.9 ± 1.7 | 60.1 ± 0.7 | 36.6 ± 1.0 | ||
| [>30] | [23.5] | [>30] | |||
 | 13b | 10 | - | 25.9 ± 0.8 | - |
| 30 | 32.4 ± 2.0 | 38.2 ± 2.1 | - | ||
| [>30] | [>30] | [>30] | |||
 | 13c | 10 | 31.8 ± 2.5 | 41.4 ± 1.5 | - |
| 30 | 31.6 ± 3.6 | 46.1 ± 2.6 | 26.4 ± 2.2 | ||
| [>30] | [>30] | [>30] | |||
 | 1 [20] | 10 | 77.5 ± 0.4 | 90.9 ± 0.3 | 85.8 ± 1.3 |
| 30 | 78.4 ± 0.9 | 93.3 ± 0.2 | 85.0 ± 0.2 | ||
| [0.5] | [0.9] | [0.6] | |||
 | cisplatin | 10 | 83.6 ± 1.2 | 42.6 ± 2.3 | 66.9 ± 1.8 |
| 30 | 95.0 ± 0.3 | 99.9 ± 0.3 | 96.8 ± 0.4 | ||
| [1.3] | [12.4] | [5.8] | |||
| Structure | Compd Code | Relative conversion ± SD at 10 µM (%) [IC50 ± SD (µM) NADPH] |
|---|---|---|
 | 12a | 90 ± 13 |
 | 12b | 89 ± 7 |
 | 12c | 98 ± 14 |
 | 13a | 113 ± 6 |
 | 13b | 65 ± 3 [IC50 = 19 ± 10] |
 | 13c | 91 ± 4 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bodnár, B.; Mernyák, E.; Wölfling, J.; Schneider, G.; Herman, B.E.; Szécsi, M.; Sinka, I.; Zupkó, I.; Kupihár, Z.; Kovács, L. Synthesis and Biological Evaluation of Triazolyl 13?-Estrone–Nucleoside Bioconjugates. Molecules 2016, 21, 1212. https://doi.org/10.3390/molecules21091212
Bodnár B, Mernyák E, Wölfling J, Schneider G, Herman BE, Szécsi M, Sinka I, Zupkó I, Kupihár Z, Kovács L. Synthesis and Biological Evaluation of Triazolyl 13?-Estrone–Nucleoside Bioconjugates. Molecules. 2016; 21(9):1212. https://doi.org/10.3390/molecules21091212
Chicago/Turabian StyleBodnár, Brigitta, Erzsébet Mernyák, János Wölfling, Gyula Schneider, Bianka Edina Herman, Mihály Szécsi, Izabella Sinka, István Zupkó, Zoltán Kupihár, and Lajos Kovács. 2016. "Synthesis and Biological Evaluation of Triazolyl 13?-Estrone–Nucleoside Bioconjugates" Molecules 21, no. 9: 1212. https://doi.org/10.3390/molecules21091212
APA StyleBodnár, B., Mernyák, E., Wölfling, J., Schneider, G., Herman, B. E., Szécsi, M., Sinka, I., Zupkó, I., Kupihár, Z., & Kovács, L. (2016). Synthesis and Biological Evaluation of Triazolyl 13?-Estrone–Nucleoside Bioconjugates. Molecules, 21(9), 1212. https://doi.org/10.3390/molecules21091212