
Virtual Screening for Potential Allosteric Inhibitors of Cyclin-Dependent Kinase 2 from Traditional Chinese Medicine

Abstract
:1. Introduction
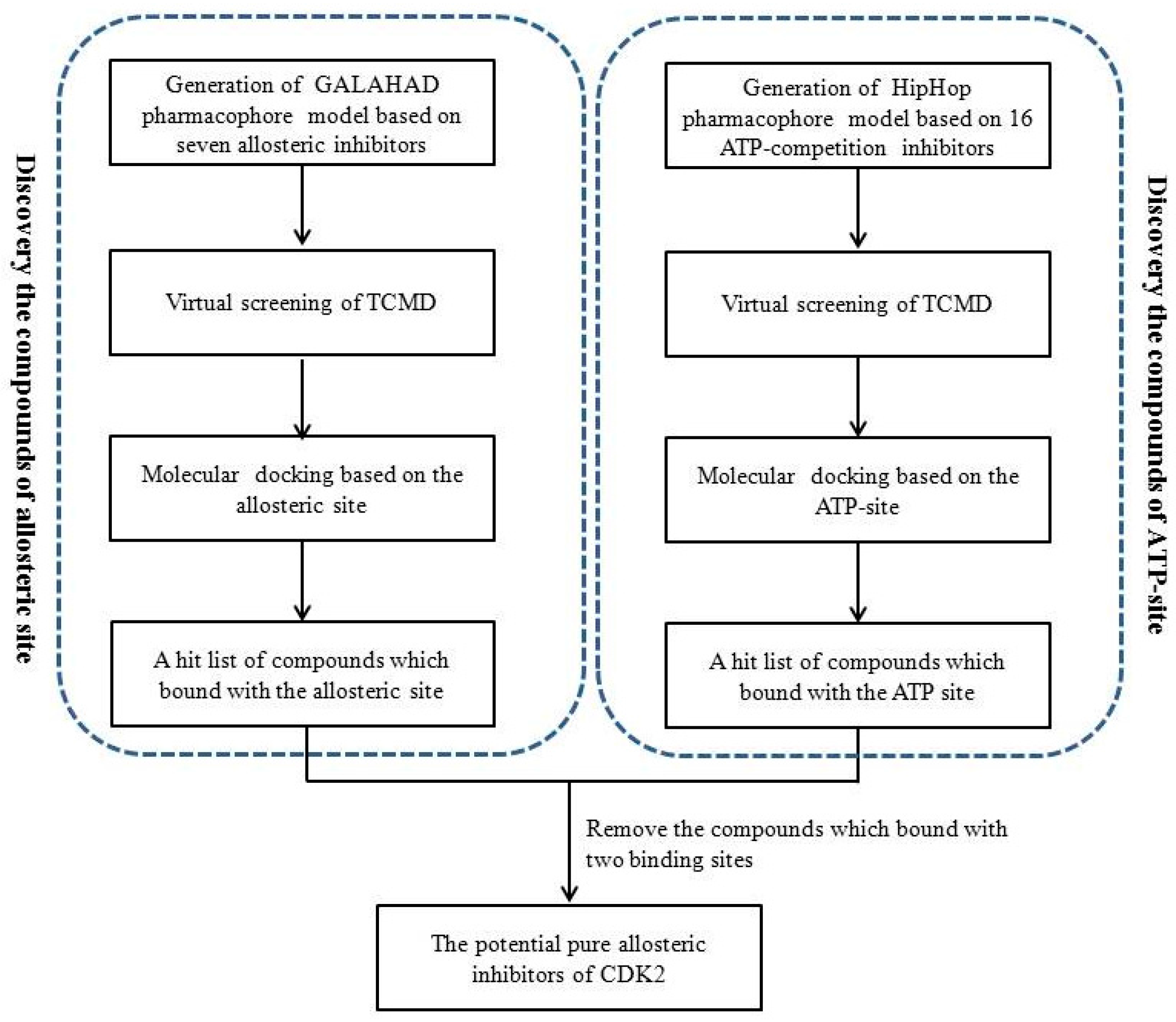
2. Results
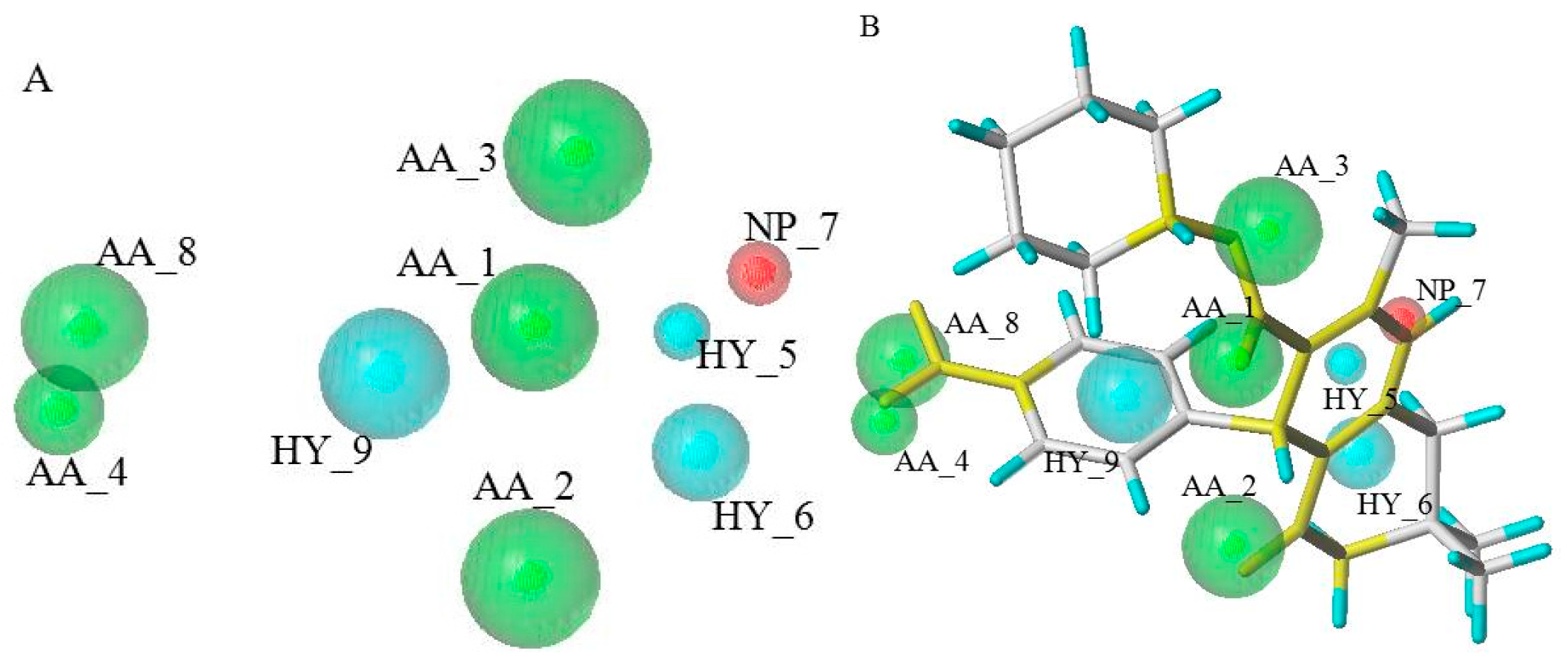
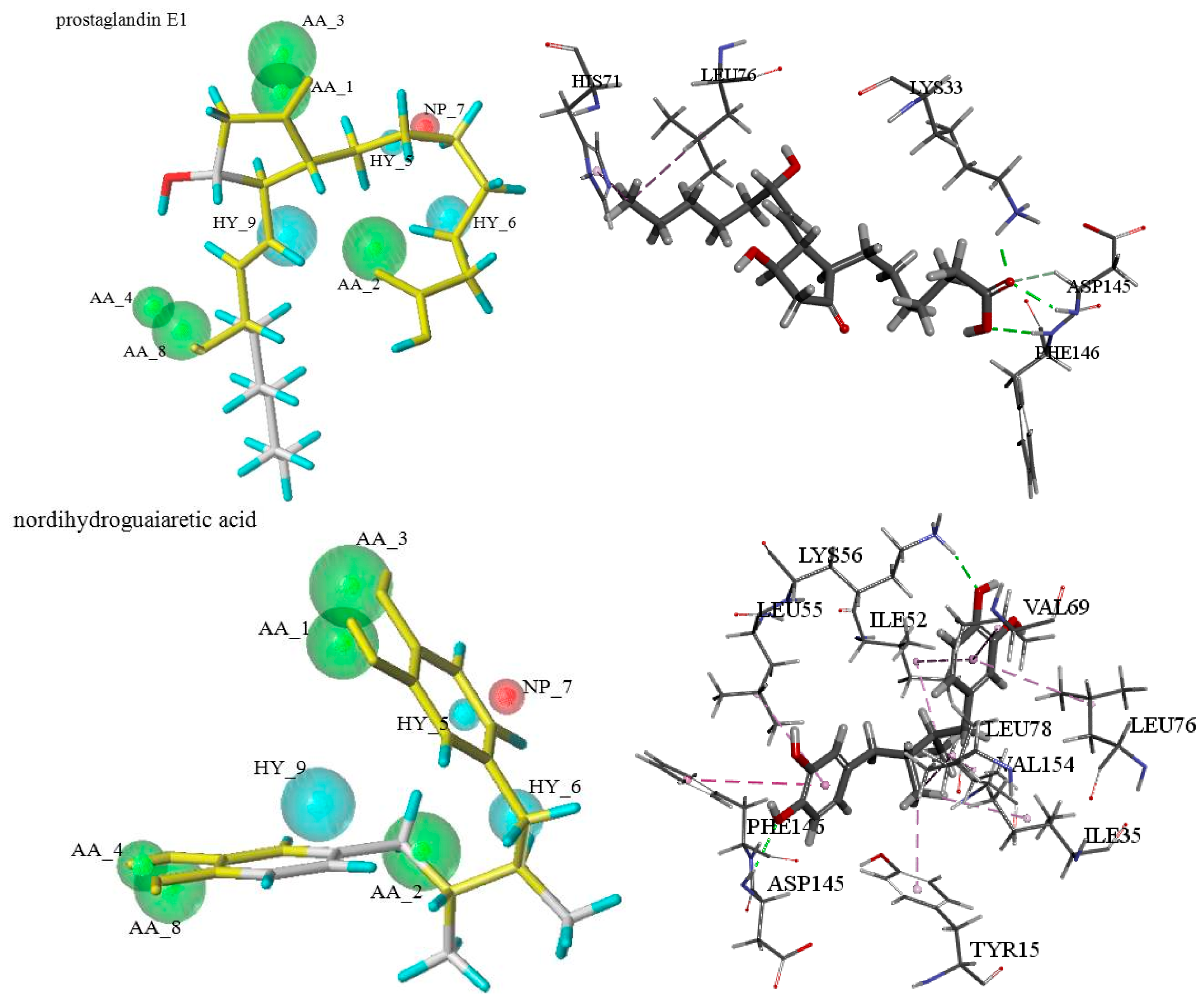
2.1. GALAHAD Pharmacophore Hypothese Construction
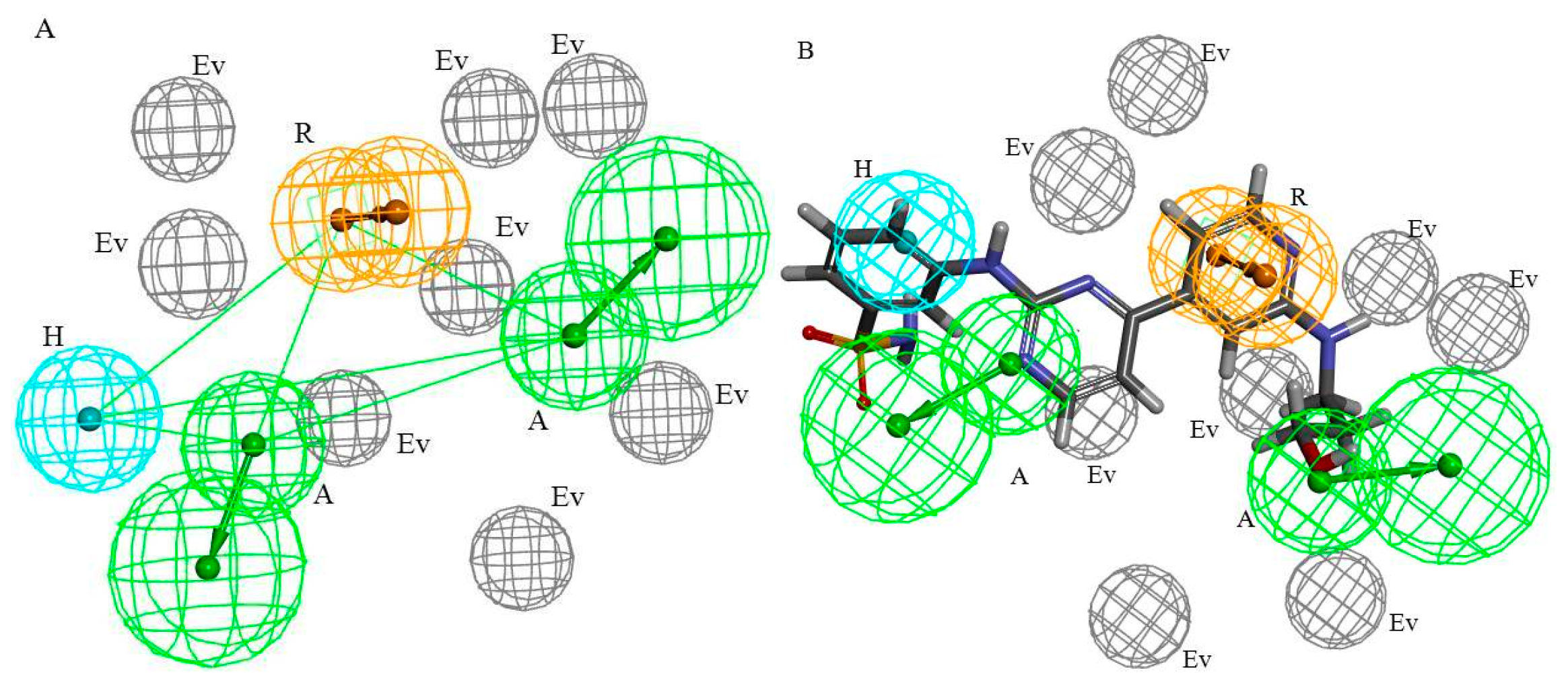
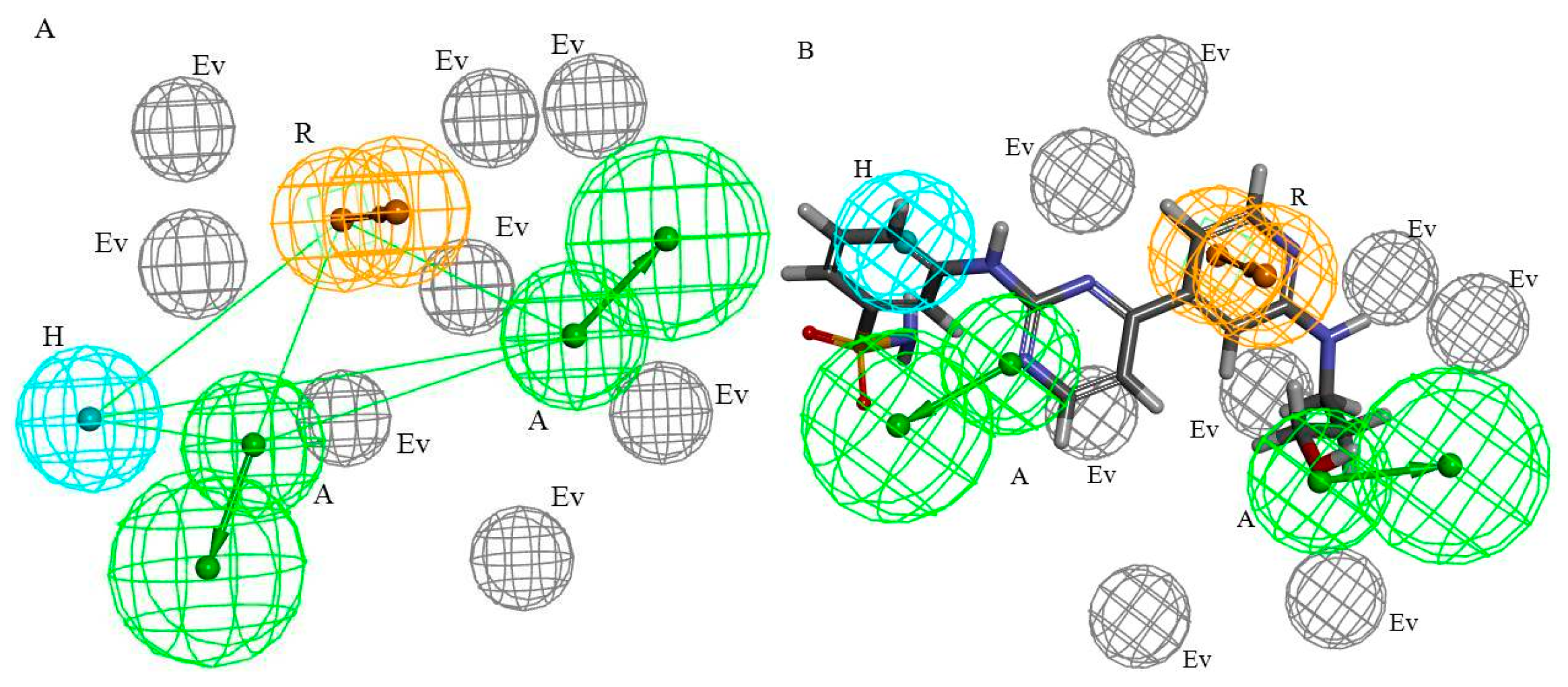
2.2. HipHop Pharmacophore Hypotheses Studies
2.2.1. HipHop Pharmacophore Hypotheses Generation
2.2.2. HipHop Pharmacophore Model Validation and Optimization
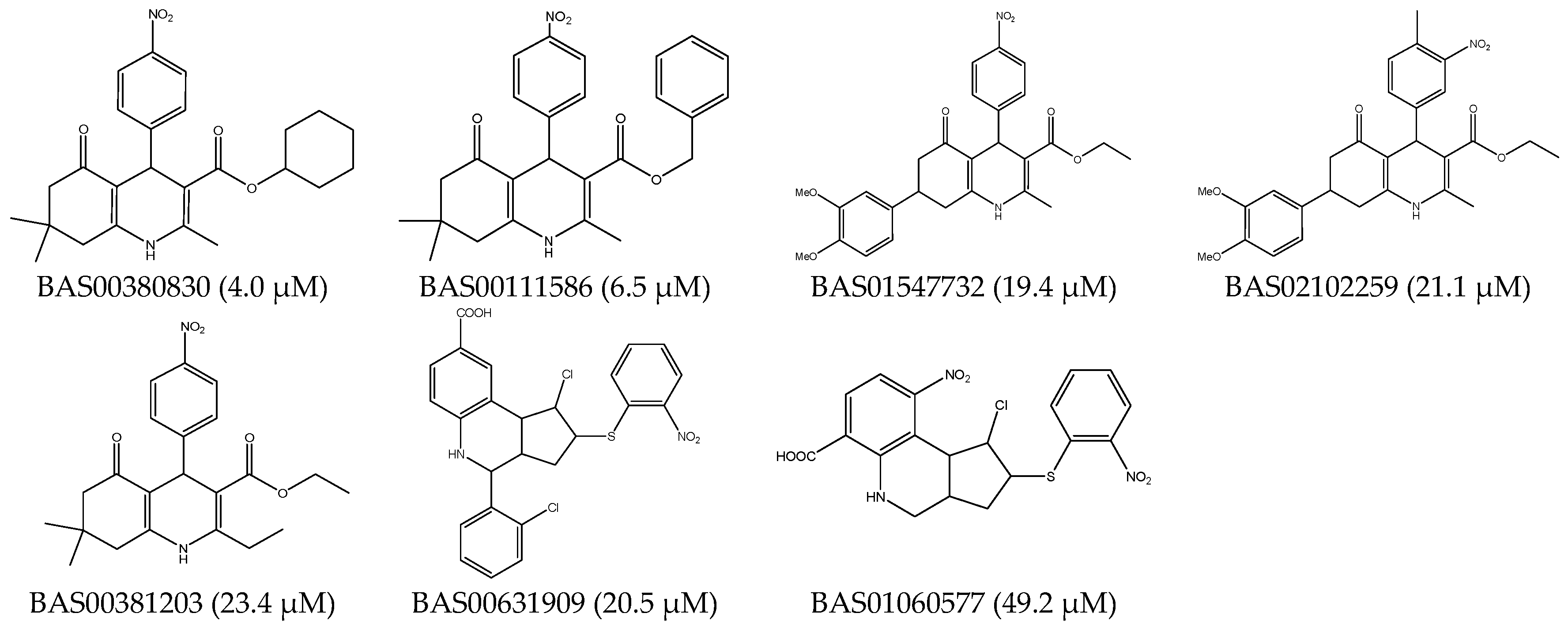
2.3. Database Searching
2.4. Molecular Docking Studies
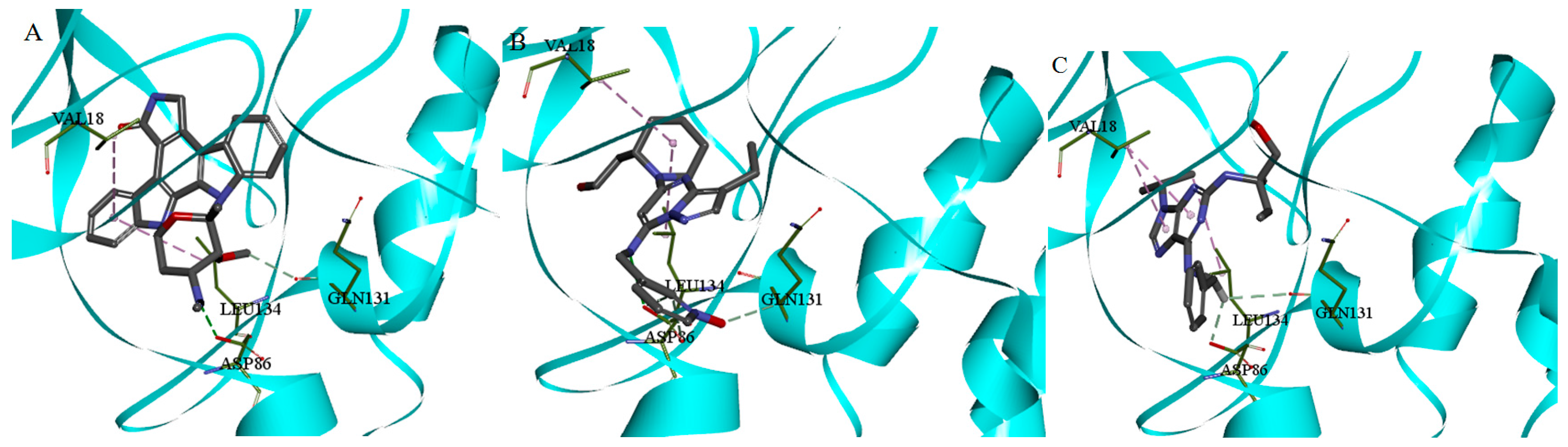
2.4.1. Molecular Docking Studies of Allosteric Site
2.4.2. Molecular Docking Studies of ATP Site
2.5. Selection of the CDK2 Allosteric Inhibitors
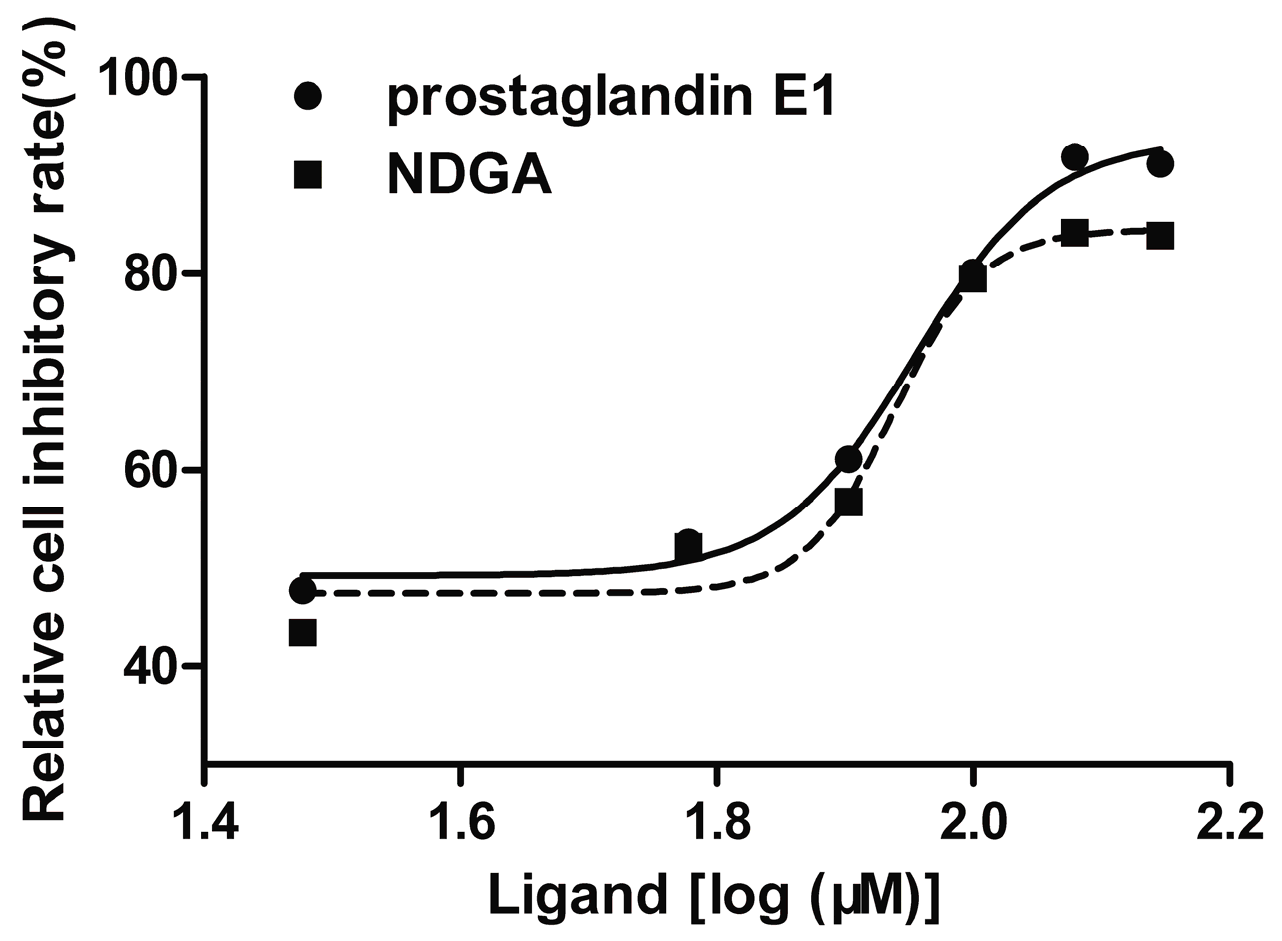
2.6. Cell Proliferation Assay
3. Materials and Methods
3.1. Pharmacophore Modeling Studies
3.1.1. GALAHAD Pharmacophore Hypotheses Construction for the CDK2 Allosteric Inhibitors
3.1.2. HipHop Pharmacophore Hypotheses Generation for CDK2 ATP-Competitive Inhibitors
HipHop Pharmacophore Hypotheses Generation
HipHop Pharmacophore Hypotheses Validation and Optimization
3.2. Database Search
3.3. Docking Studies
3.3.1. Docking Studies of Allosteric Site
3.3.2. Docking Studies of ATP Site
Modulate the Protein Conformation to the ATP-Competitive Inhibitor Binding State
Docking Strategy of the ATP Binding Site
3.4. Cell Proliferation Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Ortega, S.; Malumbres, M.; Barbacid, M. Cyclin d-dependent kinases, INK4 inhibitors and cancer. Biochim. Biophys. Acta 2002, 1602, 73–87. [Google Scholar] [CrossRef]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Echalier, A.; Endicott, J.A.; Noble, M.E.M. Recent developments in cyclin-dependent kinase biochemical and structural studies. Biochim. Biophys. Acta 2010, 1804, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Alam, R.; Betzi, S.; Ingles, D.J.; Zhu, J.-Y.; Schoenbrunn, E. A novel approach to the discovery of small-molecule ligands of CDK2. Chembiochem 2012, 13, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Eglen, R.; Reisine, T. Drug discovery and the human kinome: Recent trends. Pharmacol. Ther. 2011, 130, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Betzi, S.; Alam, R.; Martin, M.; Lubbers, D.J.; Han, H.; Jakkaraj, S.R.; Georg, G.I.; Schoenbrunn, E. Discovery of a potential allosteric ligand binding site in CDK2. ACS Chem. Biol. 2011, 6, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.-C.; Chen, C.Y.-C. Drug design of cyclin-dependent kinase 2 inhibitor for melanoma from traditional chinese medicine. Biomed. Res. Int. 2014, 2014, 798742. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, G.; Li, X.; Zhang, Y.; Yang, J.; Chang, J.; Sun, X.; Zhou, X.; Guo, Y.; Xu, Y.; et al. Traditional chinese medicine in cancer care: A review of controlled clinical studies published in chinese. PLoS ONE 2013, 8, e60338. [Google Scholar]
- King, V.F.; Garcia, M.L.; Himmel, D.; Reuben, J.P.; Lam, Y.K.; Pan, J.X.; Han, G.Q.; Kaczorowski, G.J. Interaction of tetrandrine with slowly inactivating calcium channels. Characterization of calcium channel modulation by an alkaloid of chinese medicinal herb origin. J. Biol. Chem. 1988, 263, 2238–2244. [Google Scholar] [CrossRef]
- Alexeev, M.; Grosenbaugh, D.K.; Mott, D.D.; Fisher, J.L. The natural products magnolol and honokiol are positive allosteric modulators of both synaptic and extra-synaptic gaba(a) receptors. Neuropharmacology 2012, 62, 2507–2514. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Griffith, R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef] [PubMed]
- Eyunni, S.K.V.K.; Gangapuram, M.; Redda, K.K. In-vitro antiproliferative activity of new tetrahydroisoquinolines (thiqs) on ishikawa cells and their 3D pharmacophore models. Lett. Drug Des. Discov. 2014, 11, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ren, Z.; He, Y.; Xiang, Y.; Zhang, Y.; Qiao, Y. A combination of pharmacophore modeling, molecular docking and virtual screening for inos inhibitors from chinese herbs. Bio-Med. Mater. Eng. 2014, 24, 1315–1322. [Google Scholar]
- Huang, S.-Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein-ligand docking: Recent advances and future directions. Phys.Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Anighoro, A.; Chripkova, M.; Carrassa, L.; Broggini, M. Structure-based discovery of the first allosteric inhibitors of cyclin-dependent kinase 2. Cell Cycle 2014, 13, 2296–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Skolnick, J. Tm-align: A protein structure alignment algorithm based on the tm-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Liu, P.; Li, Q.; Xu, Y.; Gao, J. 3D-Qsar studies on a class of IKK-2 inhibitors with galahad used to develop molecular alignment models. QSAR Comb. Sci. 2008, 27, 1113–1119. [Google Scholar] [CrossRef]
- He, Y.; Jiang, L.; Yang, Z.; Qiao, Y.; Zhang, Y. A combination of pharmacophore modeling, molecular docking, and virtual screening for P2y(12) receptor antagonists from chinese herbs. Can. J. Chem. 2015, 93, 311–316. [Google Scholar] [CrossRef]
- Holland, J.H. Genetic algorithms. Sci. Am. 1992, 267, 66–72. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, Y.; Wang, X.; Qiao, Y. Pharmacophore model generation of p2y12 inhibitor. In Proceedings of the International Conference on Biomedical Engineering & Biotechnology, Macau, China, 28–30 May 2012; pp. 396–399.
- Traditional Chinese Medicine Database (TCMD); Accelrys Inc.: Beijing, China, 2009.
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Li, Y.; Qiao, L.; Chen, X.; He, Y.; Zhang, Y.; Li, G. Discovery of potential negative allosteric modulators of mGLuR5 from natural products using pharmacophore modeling, molecular docking, and molecular dynamics simulation studies. Can. J. Chem. 2015, 93, 1199–1206. [Google Scholar] [CrossRef]
- Shi, X.-N.; Li, H.; Yao, H.; Liu, X.; Li, L.; Leung, K.-S.; Kung, H.-F.; Lu, D.; Wong, M.-H.; Lin, M.C.-M. In silico identification and in vitro andin vivo validation of anti-psychotic drug fluspirilene as a potential CDK2 inhibitor and a candidate anti-cancer drug. PLoS ONE 2015, 10, e0132072. [Google Scholar]
- Sample Availability: Samples of the compounds are not available from the authors.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Specificity | N_hits | Features | PARETO | Energy |
|---|---|---|---|---|---|
| MODEL_001 | 3.718 | 7 | 6 | 0 | 21.12 |
| MODEL_002 | 4.049 | 7 | 5 | 0 | 1688.09 |
| MODEL_003 | 4.638 | 7 | 7 | 0 | 1280.52 |
| MODEL_004 | 2.085 | 7 | 8 | 0 | 11.17 |
| MODEL_005 | 3.016 | 7 | 9 | 0 | 37.30 |
| MODEL_006 | 4.587 | 7 | 8 | 0 | 11.08 |
| MODEL_007 | 5.291 | 7 | 9 | 0 | 38.13 |
| MODEL_008 | 5.093 | 7 | 6 | 0 | 21.67 |
| Hypo | Features a | Rank Score b | TA c | TD d | Ha e | Ht f | HRA g | IEI h | CAI i |
|---|---|---|---|---|---|---|---|---|---|
| 1 | RHAAEv5 | 133.141 | 23 | 92 | 21 | 40 | 91.30% | 2.10 | 1.92 |
| 2 | RHAAEv5 | 132.625 | 23 | 92 | 20 | 38 | 86.96% | 2.11 | 1.83 |
| 3 | RHAAEv5 | 132.431 | 23 | 92 | 21 | 39 | 91.30% | 2.15 | 1.97 |
| 4 | RHAAEv5 | 131.957 | 23 | 92 | 18 | 34 | 78.26% | 2.12 | 1.66 |
| 5 | RHDAEv5 | 131.796 | 23 | 92 | 22 | 43 | 95.65% | 2.05 | 1.96 |
| 6 | RHAAEv5 | 131.641 | 23 | 92 | 21 | 39 | 91.30% | 2.15 | 1.97 |
| 7 | RHAAEv5 | 131.180 | 23 | 92 | 19 | 35 | 82.6% | 2.17 | 1.79 |
| 8 | RHAAEv5 | 131.114 | 23 | 92 | 20 | 37 | 86.96% | 2.16 | 1.88 |
| 9 | RHAAEv5 | 130.381 | 23 | 92 | 20 | 37 | 86.96% | 2.16 | 1.88 |
| 10 | RHAAEv5 | 130.168 | 23 | 92 | 21 | 39 | 91.30% | 2.15 | 1.97 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, F.; Luo, G.; Qiao, L.; Jiang, L.; Li, G.; Zhang, Y. Virtual Screening for Potential Allosteric Inhibitors of Cyclin-Dependent Kinase 2 from Traditional Chinese Medicine. Molecules 2016, 21, 1259. https://doi.org/10.3390/molecules21091259
Lu F, Luo G, Qiao L, Jiang L, Li G, Zhang Y. Virtual Screening for Potential Allosteric Inhibitors of Cyclin-Dependent Kinase 2 from Traditional Chinese Medicine. Molecules. 2016; 21(9):1259. https://doi.org/10.3390/molecules21091259
Chicago/Turabian StyleLu, Fang, Ganggang Luo, Liansheng Qiao, Ludi Jiang, Gongyu Li, and Yanling Zhang. 2016. "Virtual Screening for Potential Allosteric Inhibitors of Cyclin-Dependent Kinase 2 from Traditional Chinese Medicine" Molecules 21, no. 9: 1259. https://doi.org/10.3390/molecules21091259
APA StyleLu, F., Luo, G., Qiao, L., Jiang, L., Li, G., & Zhang, Y. (2016). Virtual Screening for Potential Allosteric Inhibitors of Cyclin-Dependent Kinase 2 from Traditional Chinese Medicine. Molecules, 21(9), 1259. https://doi.org/10.3390/molecules21091259